
Well-defined nickel(II) tetrazole-saccharinate complex as homogeneous catalyst on the reduction of aldehydes: scope and reaction mechanism
-
Luís M. T. Frija
 ,
Bruno G. M. Rocha
,
Bruno G. M. Rocha

Abstract
A new (tetrazole-saccharin)nickel complex is shown to be a valuable catalyst for the hydrosilative reduction of aldehydes under microwave radiation at low temperatures. With typical 1 mol% content of the catalyst (microwave power range of 5–15 W) most reactions are complete within 30 min. The Ni(II)-catalyzed reduction of aldehydes, with a useful scope, was established for the first time by using this catalyst, and is competitive with the most effective transition-metal catalysts known for such transformation. The catalyst reveals tolerance to different functional groups, is air and moisture stable, and is readily prepared in straightforward synthetic steps. Supported by experimental data and DFT calculations, a plausible reaction mechanism involving the new catalytic system is outlined.
Introduction
Cumulative environmental concerns have led chemists to develop cleaner and sustainable reactions for chemical transformations. One of the essential questions in synthetic organic chemistry is to develop effective, selective and atom-economical reactions which can be performed under safe and mild conditions with the aid of transition metal catalysts.
Specifically, the reduction of numerous organic compounds is an essential and pervasive reaction, assuming equivalent relevance on a laboratory or industrial scale [1], [2], [3]. Sodium borohydride is one of the most used and effective reducing agents for organic compounds, massively applied since its discovery [4], [5]. Because of its popularity, thousands of tons of sodium borohydride are produced and used every year. On the other hand, hydrosilanes are equally versatile reducing agents for a diversity of organic functional groups, in particular for aldehydes and ketones [6], [7], [8], [9], [10], [11], [12], [13]. Analogous to the borohydrides, the Si–H bond is polarized towards hydrogen permitting silanes to act as mild sources of hydride. In addition, silanes are readily accessible, as silicon is the second most abundant element in the earth’s crust.
Despite the fact that direct and transfer hydrogenation [1], [2], [3], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26] usually requires severe reaction conditions such as high pressure and temperature and strong base as additive, hydrosilylation is classically performed under more smooth conditions [27], [28], [29], [30], [31]. Besides, in catalytic hydrosilylation, both the reduction of carbonyl functionality and protection of the resulting alcohol in the form of silyl ethers, can be accomplished in a single step with atom economy [1], [32]. Up till now, most of the hydrosilylation reactions reported have been catalyzed by precious metals, such as gold [33], [34], [35], [36], platinum [37], [38], silver [34], [39], iridium [40], [41], [42] and ruthenium [30], [43]. As an alternative to these metals, Cu [44], [45], [46], Fe [47], [48] and Ni [31], [49], [50], [51], [52] have recently been applied in the reduction of the carbonyl group by hydrosilylation, typically in presence of a base or PPh3 [34], [53], [54], [55].
In spite of the important advantages in terms of environmental and cost benefits, as compared to borohydrides, the synthetic community has not yet developed a broadly applied, simple and cheap method, for the reduction of aldehydes using silanes. The main reason for this relates to the silane reactivity which is hard to adjust [56].
Nevertheless, in this regard, alkylsilanes are usually easy to handle [57], [58] while they require forceful activation in the form of a Brønsted acid [59], Lewis acid [60], [61], Lewis base [62] or transition metal [63], [64], [65], [66] in the reaction mixture. The catalytic processes using these additives differ, but their presence is essential to increase the hydridic nature of the hydrosilane [67]. On the other hand, silanes bearing more electronegative substituents (as halide or alkoxy) or multiple hydrides are more reactive, permitting the development of excellent protocols, yet simultaneously making them difficult to manipulate [68], [69], [70], [71].
The first example of hydrosilylation catalyzed by a N-heterocyclic carbene (NHC) complex was reported by Herrmann in 1996 using a rhodium complex comprising a naphthyl-derived carbene ligand [72]. The synthesis of NHC-metal complexes has contributed for an increase in catalyst performance in different topics, particularly on transfer hydrogenation [73], [74], [75], [76], oxidation [77], [78], [79], [80], [81], C–C cross coupling [82], [83], [84], [85], [86], [87] and polymerization reactions [88], [89], [90], [91], [92], [93]. Since then, much effort has been devoted to the development of robust and efficient catalysts mainly based on noble metal–NHC complexes for hydrosilylation of ketones, alkynes and alkenes [94], [95], [96].
In an effort to exploit Earth-abundant base metals [97], [98], [99], [100], [101], [102], [103], [104], a series of NHC complexes of copper(I) [105], [106], manganese(I) [107], and iron [108], [109], [110] were recently developed for catalyzing the hydrosilylation of carbonyl compounds. Ritleng and co-workers reported the first well-defined nickel(II) complexes bearing an imidazolylidene ligand [111], which efficiently catalyze the hydrosilylation of aldehydes and ketones at room temperature with a maximum turnover frequency of 2300 h−1 at 0.5 mol% catalyst loading. Successive investigations involved in particular carbene wingtip modifications afforded catalysts for the hydrosilylation of nitroarenes [112], olefins [113] and imines [114].
Besides, very recently, already in the period in which this investigation was proceeding, Albrecht and co-workers reported the synthesis of triazolylidene nickel complexes and their catalytic application in selective aldehyde hydrosilylation [115]. Even more recently, Trovitch and co-workers published an article describing the hydrosilylation of aldehydes, ketones and ester C–O bonds using κ4-diimine nickel catalysts [116]. Homogeneous nickel compounds are recognized for the mediation of alkene hydrosilylation [117], [118], [119], [120], [121], [122], [123]; nonetheless, their trend to catalyze carbonyl hydrosilylation remains understudied. Despite everything, some nickel catalysts for aldehyde hydrosilylation have appeared in the last years showing remarkable results (see Fig. 1) [124], [125], [126].
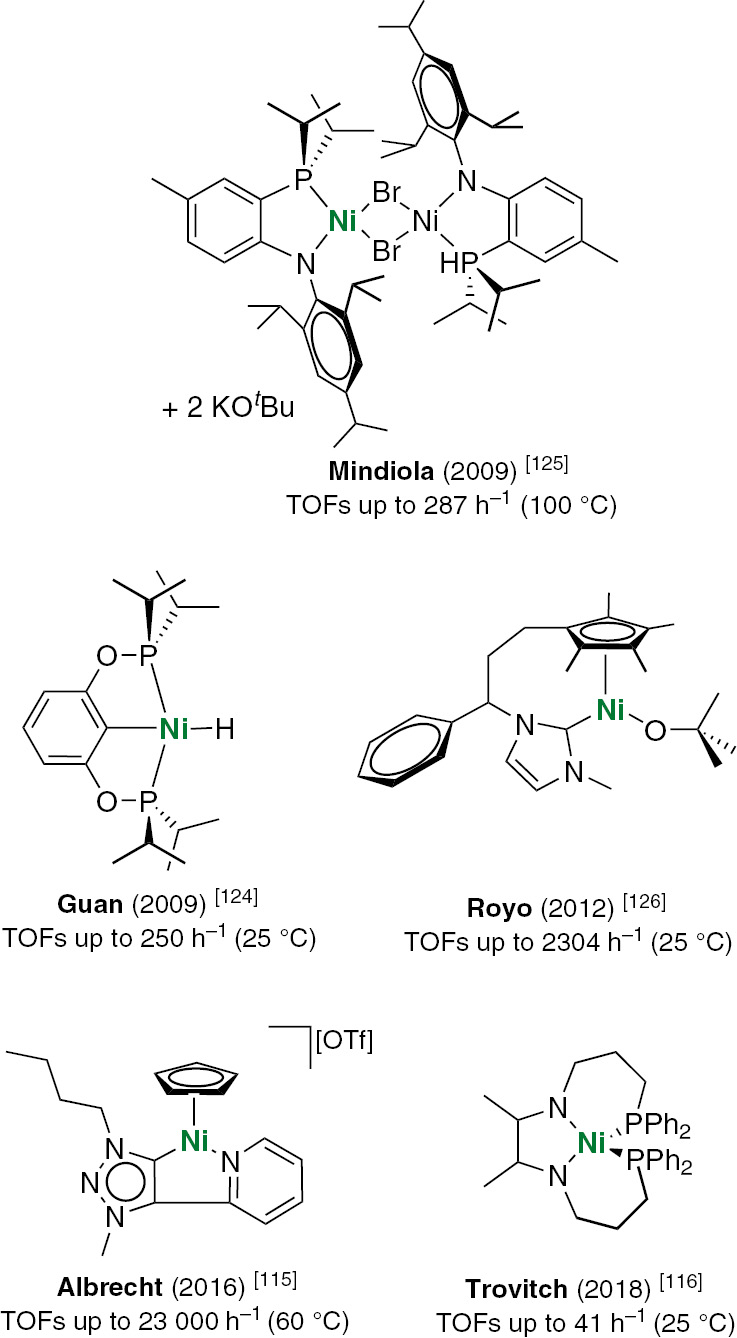
Selected prominent nickel catalysts for aldehyde hydrosilylation.
Herein, succeeding our more recent research covering catalytic applications of tetrazole-saccharin based ligands and some of their related metal complexes [127], [128], [129], [130], we report the discovery of a specific nickel(II) tetrazole-saccharinate complex as an excellent homogeneous catalyst for the reduction of aldehydes via hydrosilylation. Detailed density functional theory (DFT) calculations were also completed, supporting a probable reaction mechanism involving the new catalytic system.
Results and discussion
Tetrazolyl-type compounds can coordinate through four nitrogen electron-donating atoms and may therefore act as multidentate ligands or as a bridging building block in supramolecular assemblies. Similarly, the saccharinate anion (1,2-benzisothiazole-3-one 1,1-dioxide anion; deprotonated saccharin) interacts with metal centers in different ways, generating relatively strong interactions in crystalline environments, mostly through hydrogen bonding. As a polyfunctional ligand, it can be engaged in N, O(C=O) or O(SO2)-metal coordination, and can also act as a bidentate amidato-like bridging agent [131], [132], [133], [134], [135], [136], [137], [138], [139], [140], [141].
In a previous work on copper- and cobalt-catalyzed alcohol oxidation reactions, the influence of a tetrazole-saccharin based ligand over the oxidation products was recognized [127]. The catalysts based on the 3-((2-methyl-2H-tetrazol-5-yl)amino)-benzisothiazole 1,1-dioxide (5) ligand were identified for efficient and selective conversion of a variety of secondary alcohols into the corresponding ketones. Short reaction times, absence of a solvent and usage of low power microwave irradiation are factors that contributed to the effectiveness of the process. In addition, molecule 5 was successfully applied as an organocatalyst on anaerobic oxidation reactions of benzyl alcohols [130].
On this basis, we started to investigate the feasibility of the synthesis of new nickel complexes based on the tetrazole-saccharin ligand 5, with the purpose of their use as catalysts in reduction reactions. Compound 5 was prepared in three steps using a convergent synthetic strategy (Scheme 1) as previously stated by us [127]. Succeeding, the synthesis of nickel complex 6 (Scheme 1) proceeded at room temperature by reaction of ligand 5 with Ni(OAc)2·4H2O salt. Complex 6 was isolated as a mononuclear compound comprising two monoanionic bidentate N,N-(tetrazole-saccharin) ligands, one water and one DMSO molecule coordinated to Ni(II) ion. The complex crystallizes in the monoclinic system with space group P21/c and the central nickel atom is hexacoordinated with a distorted octahedral N4O2 coordination. Detailed structural data for complex 6 can be consulted in Table 1 and Tables S1–S2 in Electronic supplementary information (ESI).
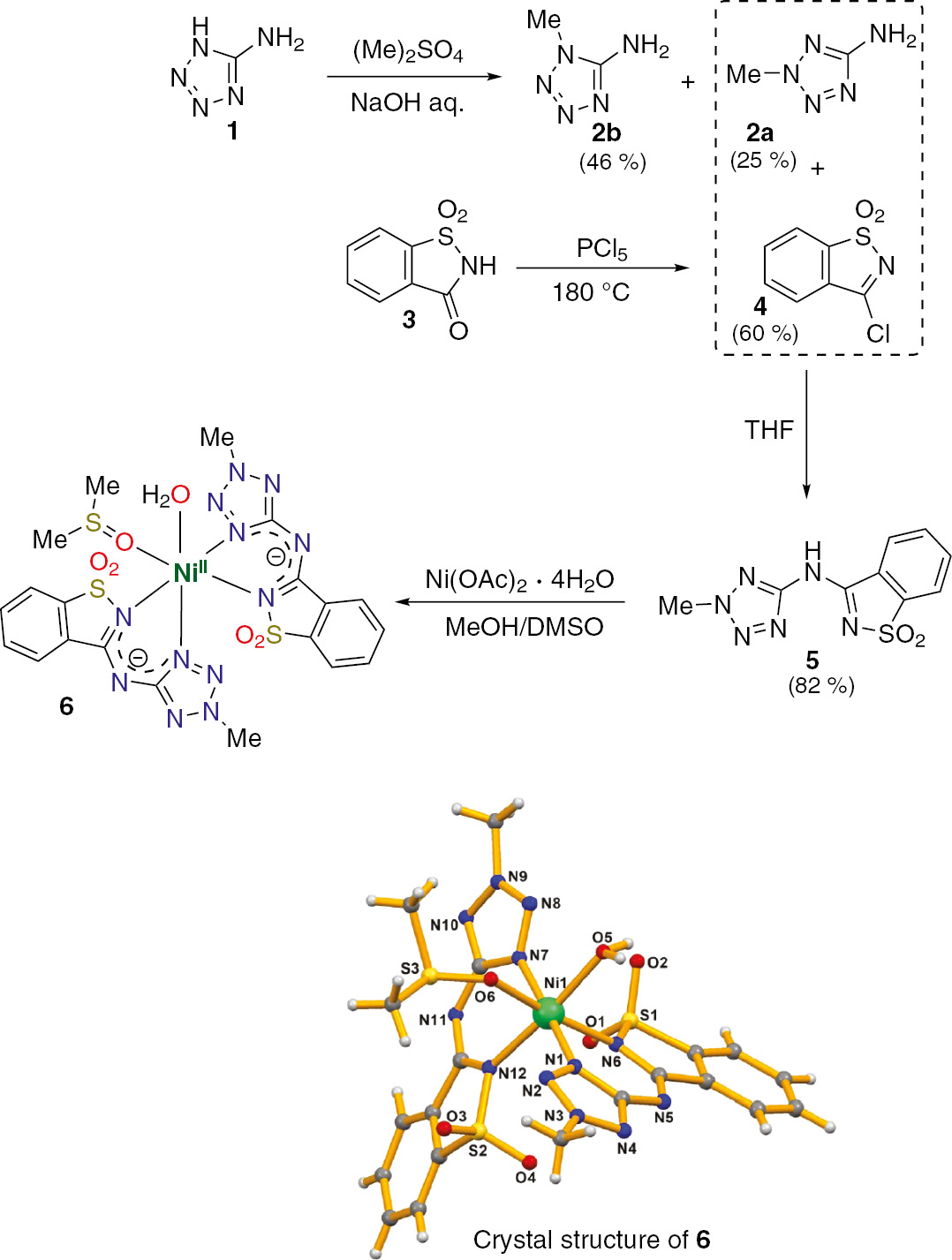
Synthetic approach used for the production of nickel(II) complex 6.
Crystal data and structure refinement details for complex 6.
| Empirical formula | C20H22N12O6S3Ni |
| Molecular weight/g mol−1 | 681.38 |
| Crystal system | Monoclinic |
| Temperature/K | 297(2) |
| Space group | P21/c |
| a/Å | 19.9384 (12) |
| b/Å | 8.9196 (5) |
| c/Å | 15.5131 (8) |
| α/° | 90 |
| β/° | 100.244 (2) |
| γ/° | 90 |
| V (Å3) | 2714.9 (3) |
| Z | 4 |
| Dcalc (g cm−3) | 1.667 |
| μ(Mo Kα)/mm−1 | 1.01 |
| Rfls. collected/unique/observed | 44 131/5005/4013 |
| Rint | 0.057 |
| Final R1a, wR2b (I≥2σ) | 0.032, 0.079 |
| Goodness-of-fit on F2 | 1.03 |
aR=Σ||Fo|–|Fc||/Σ|Fo|; bwR(F2)=[Σw(|Fo|2–|Fc|2)2/Σw|Fo|4]½.
We began by investigating the influence of the solvent, reaction time, temperature and catalyst amount on the hydrosilylation of benzaldehyde (7a) using PhSiH3 as the silane reagent (Table 2). Likewise, in sequence to other successful chemical protocols hitherto developed by us, microwave radiation was used as energy source [127,130]. During the optimization of the reaction conditions we were pleased to find that the benzyl alcohol (8a) generated by our method could be obtained in good yields for different solvents and temperatures in a short period of time.
Solvent, reaction time, temperature and catalyst amount screening for the hydrosilylation of benzaldehyde with PhSiH3a.
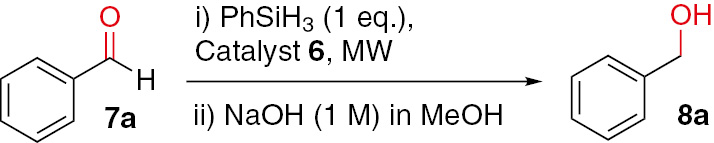 |
|||||
|---|---|---|---|---|---|
| Entry | Solvent | t (min) | T (°C) | Catalyst amount (mol%) | Yield (%)b |
| 1 | DMF | 60 | 80 | 1.0 | 89 |
| 2 | 30 | 80 | 1.0 | 85 | |
| 3 | 30 | 80 | 1.5 | 91 | |
| 4 | 15 | 80 | 1.0 | 78 | |
| 5 | THF | 60 | 80 | 1.0 | 61 |
| 6 | 30 | 80 | 1.5 | 53 | |
| 7 | CH3CN | 60 | 80 | 1.0 | 55 |
| 8 | 30 | 80 | 1.0 | 30 | |
| 9 | 60 | 80 | 1.0 | 96 | |
| 10 | 60 | 50 | 1.0 | 91 | |
| 11 | DCM | 30 | 50 | 1.0 | 92 |
| 12 | 30 | 50 | 1.5 | 95 | |
| 13 | 15 | 50 | 1.0 | 78 | |
| 14 | 60 | 80 | 1.0 | 90c | |
| 15 | 360 | 80 | 0.25 | 81 | |
| 16 | 60 | 80 | – | 23d | |
| 17 | 60 | 80 | 1.0 | 78 | |
| 18 | 60 | 50 | 1.5 | 80 | |
| 19 | DCE | 30 | 50 | 1.0 | 81 |
| 20 | 30 | 50 | 2.0 | 77 | |
| 21 | 15 | 50 | 1.0 | 55 | |
| 22 | n-Hexane | 60 | 80 | 1.0 | 54 |
| 23 | 30 | 80 | 1.0 | 51 | |
| 24 | EtOH | 60 | 80 | 1.0 | 46 |
| 25 | 30 | 80 | 1.0 | 41 | |
aReaction conditions: aldehyde 7a (2.0 mmol), PhSiH3 (2.0 mmol), solvent (1 mL) and 6, in a microwave reactor (5–15 W) at a determined temperature. bConversion into 8a determined by use of calibrated GC methods. cReaction performed with 1.5 eq. of PhSiH3 (3.0 mmol). dReaction performed in absence of catalyst.
Even, although with lower yields, the hydrosilylation of aldehyde 7a catalyzed by complex 6 can be implemented in EtOH, a protic polar solvent (Table 2, entries 24 and 25). Increasing the stoichiometry of the silane did not provide any further beneficial effect in terms of reaction time or conversion (Table 2, entry 14).
Interestingly, 7a undergoes reduction to 8a with as little as 0.25 mol% (approx.) of complex 6, although a longer reaction time is necessary (Table 2, entry 15). Nevertheless, in view of the operational easiness for small-scale reactions, the use of 1.0 mol% of 6 was considered as an optimal catalyst amount. Besides, when the reaction was done in the absence of catalyst 6, a 23 % yield was achieved in 1 h for the transformation of benzaldehyde in DCM (Table 2, entry 16).
Thereby, based on the best results from the screening completed for delineation of optimal reaction conditions, a specific protocol was adopted for a series of aldehydes (Table 3). Typical experimental conditions used for the reduction of aldehydes via hydrosilylation comprised the use of one equivalent of PhSiH3 and 1 mol% of the nickel catalyst (6) in a dichloromethane solution. The established reaction temperatures for the sort of aldehydes considered varied from 50 to 80 °C corresponding to low-power microwave radiation (5–15 W). Besides, we should note that the reactions proceeded in a sealed tube into a microwave reactor and the pressure in course of the process varied in the range of 1–10 bar.
Scope of the microwave-assisted hydrosilylation of aldehydes catalyzed by nickel complex 6a.
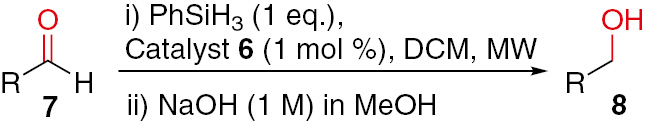 |
|||||
|---|---|---|---|---|---|
| Entry | Aldehyde | t (min) | T (°C) | Yield (%)b | TOF (h−1)c |
| 1 | 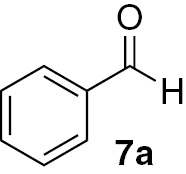 |
60 | 50 | 85 | 89 |
| 2 | 60 | 80 | 94 | 94 | |
| 3 | 30 | 50 | 83 | 184 | |
| 4 | 15 | 50 | 70 | 312 | |
| 5d | 960d | Reflux | 91 | 6 | |
| 6 | 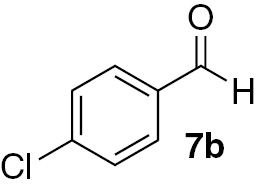 |
30 | 50 | 79 | 158 |
| 7 | 60 | 50 | 85 | 85 | |
| 8 | 60 | 80 | 75 | 75 | |
| 9 | 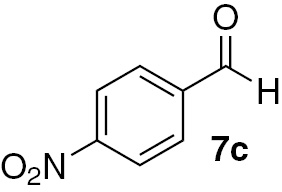 |
30 | 50 | 64 | 128 |
| 10 | 60 | 50 | 67 | 67 | |
| 11 | 90 | 80 | 71 | 47 | |
| 12 | 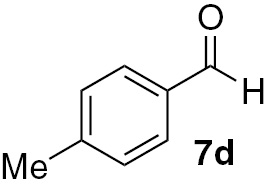 |
30 | 50 | 95 | 190 |
| 13 | 15 | 50 | 85 | 340 | |
| 14 | 10 | 80 | 91 | 546 | |
| 15 | 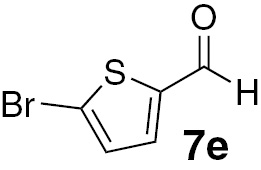 |
60 | 50 | 67 | 67 |
| 16 | 30 | 50 | 61 | 122 | |
| 17 | 30 | 80 | 71 | 142 | |
| 18 | 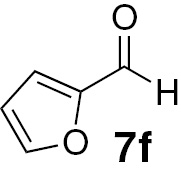 |
60 | 50 | 61 | 61 |
| 19 | 30 | 50 | 52 | 104 | |
| 20 | 30 | 80 | 64 | 128 | |
| 21 | 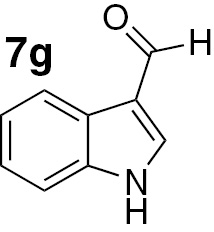 |
60 | 50 | 78 | 78 |
| 22 | 30 | 50 | 67 | 134 | |
| 23 | 30 | 80 | 92 | 184 | |
| 24 | 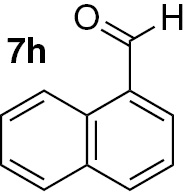 |
60 | 50 | 85 | 85 |
| 25 | 30 | 50 | 76 | 152 | |
| 26 | 30 | 80 | 92 | 184 | |
| 27 | 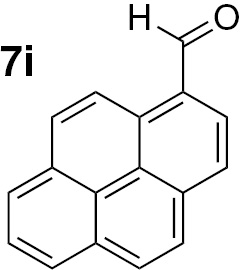 |
90 | 50 | 64 | 43 |
| 28 | 60 | 50 | 53 | 53 | |
| 29 | 30 | 80 | 70 | 140 | |
| 30 | 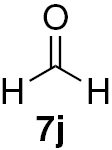 |
60 | 50 | 94 | 94 |
| 31 | 30 | 50 | 87 | 174 | |
| 32 | 30 | 80 | 97 | 194 | |
aReaction conditions: aldehyde 7 (2.0 mmol), PhSiH3 (2.0 mmol), dichloromethane (1 mL) and 6 (1 mol%) in a microwave reactor (5–15 W) at a determined temperature. bIsolated yields. cTOF=TON·h−1. dReaction mixture heated in an oil bath.
Under our chosen conditions (Table 3), 6 proved effective in enabling the high-yielding and selective reduction of various aldehydes to the corresponding primary alcohols. The para-substituted benzaldehyde analogues 7b–d featured reduction in good to excellent yields for different times of reaction (Table 3, entries 6–14). For these three aldehydes, the electronic nature of the substituent group in the para position does not seem to be effectively striking for the reactivity shown. Nevertheless, we can infer that the presence of the electron-donating methyl group in the para position on aldehyde 7d may have some effect in the formation of the silyl ether intermediary on the origin of the alcohol 8a, since somewhat higher yields were attained in the case of such substrate (Table 3, entry 12).
Attending to the reaction yields, even though a slight decrease is noticeable, the reactivity of heterocyclic aldehydes 7e–g (Table 3, entries 15–23) is quite similar to the benzaldehyde derivatives 7b–d. Similarly, the reduction of aldehydes comprising extended π systems, such as naphthalene (7h) or pyrene (7i), proceeded successfully (Table 3, entries 24–29). Lastly, it was found the production of methanol by reduction of formaldehyde in almost quantitative yields (Table 3, entries 30–32).
Mechanistic studies
The knowledge of some electronic and structural properties of the nickel complex 6, acquired by crystallographic data, allowed us to conjecture a reaction mechanism for the reduction of aldehydes via hydrosilylation promoted by such a metal compound. Complex 6 owns a metal oxidation state of +2, definitely the most common one for such a kind of nickel structures. The highest oxidation state of +4 is relatively infrequent in nickel intermediates [142], suggesting that the hydrosilylation possibly should not proceed by oxidative addition pathways, i.e. through formation of intermediate species based on the insertion of the aldehyde C=O bond, both at a Ni–H bond (Chalk–Harrod mechanism type) and at a Ni–Si bond (modified Chalk–Harrod mechanism type) [143], [144].
Besides, the presence of labile ligands, such as water and DMSO, in the sterically hindered complex 6, supports a potential ligand pseudo-substitution on the molecule. Herein, the term pseudo-substitution is used to distinguish it from the classical ligand substitution. The former covalent bond among the metal centre and the ligand is really cleaved and the free coordination position is used in the formation of a non-covalent interaction between the metal centre and the silane substrate (Si–H activation), without any change in the primary nickel oxidation state.
Sustained by DFT results (discussed in more detail below) a mechanistic proposal for the reduction of aldehydes promoted by 6 was assembled and is depicted in Scheme 2 (formaldehyde appears as model substrate).
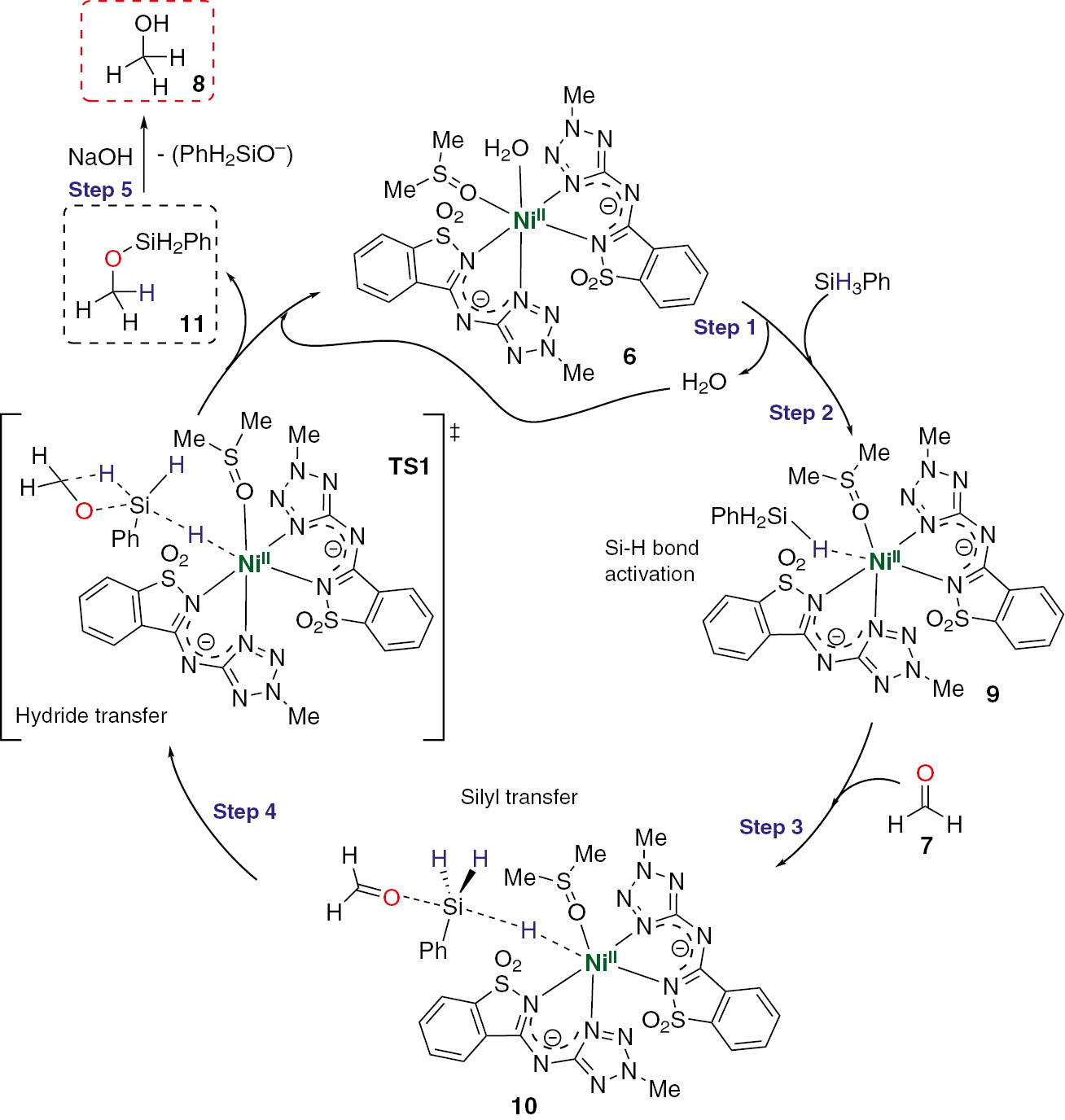
Proposed mechanism for aldehyde reduction catalyzed by nickel complex 6.
Bearing in mind that mechanisms of the type Chalk–Harrod or modified Chalk–Harrod should involve the formation of Ni(IV) species, quite unstable, these were discarded, considering to study a concerted-type outer sphere mechanism.
(i) Initial ligand elimination
The first step of the catalytic cycle should involve the elimination of a ligand, presumably water and DMSO, from the sterically hindered nickel complex (6). In this context, thermodynamics parameters of potential possibilities for ligand elimination were calculated (see Table S4, entries 1–9).
Although the unsaturated complexes 6′ (water elimination), 6″ (DMSO elimination) and 6‴ (water+DMSO elimination), were found to be less stable than the saturated one (6) (Table S3, entries 1, 4–6), water elimination is the only spontaneous process (ΔG<0) (see Table S4, entries 1, 4 and 7). Besides, the DMSO elimination is thermodynamically non-favourable, being much less favourable than the analogous process with water (Table S4, entries 4–6 and 1–3, respectively). The conjugated elimination of water and DMSO in the same step is the less favourable among all, being the product 6‴ the less stable one as well (Table S4, entry 7).
The possibility of the first step ligand elimination from 6 to a second coordination sphere was also considered for all the discussed processes, not being favourable in any case (Table S4, entries 2, 5 and 9).
(ii) Phenylsilane activation
After the elimination of one ligand, the presence of a free coordination place in 6 leads to phenylsilane activation. Despite the results obtained in the first step of the catalytic cycle, the establishment of a non-covalent interaction between the phenylsilane and the nickel, with consequent activation of a Si–H bond, was theoretically studied for all the plausible pathways. That comprehends the water and/or DMSO elimination (Table S4, entries 10–19).
Specifically, a ligand pseudo-substitution from 6 to result either in the complex with the ligand in the 2nd sphere of coordination or in its elimination, as well as the reaction starting from 6 without ligand or with it at the 2nd sphere of coordination, were considered. It was verified that the processes involving the water pseudo-substitution are thermodynamically more favourable, comparing with those with DMSO (Table S4, entries 10–19). In fact, the stability of the intermediates in this second step follows the trend observed in the first step, where the more stables structures keep the DMSO bonded to the metal centre.
(iii) Aldehyde activation
The Si–H bond activation by nickel promotes subsequent activation of C=O bond of the formaldehyde to form the intermediate 10 (Scheme 2). From this point, and taking into account the tendency observed in the previous steps, calculations were performed only for intermediates starting from the water substitution and/or elimination in the first step. For instance, the reaction of 12 (see optimized structure at ESI) with phenylsilane is unfavourable from both a thermodynamic and a kinetic point-of-view (Table S4, entry 14). It was observed that there are no significant differences between the reaction with the water present or absent in the 2nd coordination sphere. Thus, the presence of water molecule in the 2nd coordination sphere does not facilitate the aldehyde activation, and its role is expressed at previous and later stages of the reaction.
(iv) Silyl ether formation
The interaction of C=O and Si–H bonds passes by the creation of a 4-member ring transition state (TS1, Scheme 2) with consequent formation of the corresponding silyl ether. At this stage, the interaction of phenylsilane with the metal centre is broken and the catalyst 6 is regenerated. The genesis of TS1 structure was found to be the limiting step of the overall reaction with a calculated activation barrier of 23.4 kcal·mol−1 (Scheme 3).
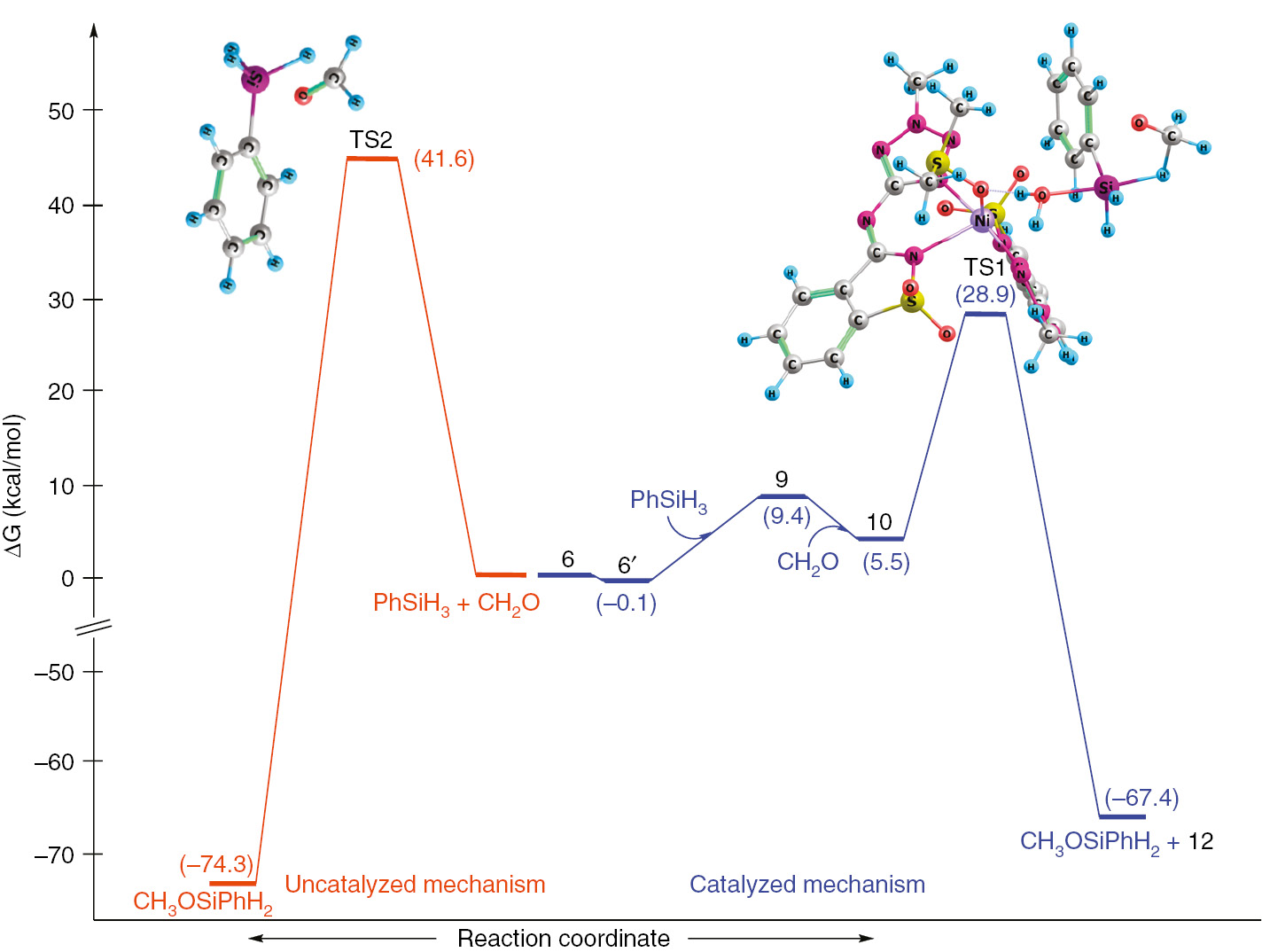
Energy profile for the catalyzed (blue line) and uncatalyzed (orange line) reduction of formaldehyde via hydrosilylation.
(v) Silyl ether hydrolysis
This step corresponds to the formation of the desired product (alcohol) from silyl ether, that is, in fact, the product of the catalytic cycle. The silyl ether hydrolysis (using NaOH as base) does not belong to the catalytic cycle itself, once it occurs after the ether isolation. However, this step was also studied in order to enable an energetic comparison of all the reaction pathways (catalyzed or not). At this stage, the solvent used in both experimental and theoretical studies is water, where the base (OH−) attacks the silicon nucleus to liberate PhH2SiO− and produce methanol. The calculated overall reaction activation barrier for the silyl ether hydrolysis is 11.9 kcal·mol−1 (Table S4, entry 28) and occurs via TS3 transition state formation (see optimized structure at ESI).
(vi) Uncatalyzed reaction
In order to compare the catalyzed hydrosilylation with the correspondent uncatalyzed reaction, several calculations were performed in absence of catalyst. Generically, the uncatalyzed process follows a similar behaviour of the catalyzed one, with formation of a 4-member ring transition state (TS2, see Scheme 3) comprising the phenylsilane and the aldehyde. The genesis of TS2 (the rate limiting step for the uncatalyzed reaction) has an activation barrier of 41.6 kcal·mol−1 (Table S4, entry 31). This value is substantially higher than that for the catalysed reaction (23.4 kcal·mol−1 Scheme 3, formation of TS1), demonstrating the pronounced effect of our Ni(II) catalyst. In fact, experimentally, the hydrosilylation of formaldehyde in the absence of the nickel catalyst proceeded in very low yield (see Table 2, entry 16).
Conclusions
The novel (tetrazole-saccharin)nickel complex (6) discovered in this study is shown to be an efficient catalyst for the hydrosilative reduction of aldehydes using microwave radiation under smooth experimental conditions. This catalyst is easily synthetized, air and moisture stable and shows a visible functional group tolerance. These characteristics further demonstrate the potential of the present nickel complex as catalyst, being competitive with analogous systems containing precious metals, provided the base metal is placed in a suitable ligand environment.
Experimental section
Materials and methods
Unless indicated otherwise, solvents and starting materials were obtained from Aldrich. All chemicals used were of reagent grade without further purification before use. Column chromatography was performed using silica gel 60 MN and aluminium-backed silica gel Merck 60 F254 plates were used for analytical thin layer chromatography (TLC). Melting points were recorded and are uncorrected. 1H and 13C NMR spectra were recorded at room temperature on a Bruker Avance II 300 (UltraShield™ Magnet) spectrometer operating at 300 MHz (1H) and 75 MHz (13C). The chemical shifts are reported in ppm using TMS as internal standard. Carbon, hydrogen and nitrogen elemental analyses were carried out by the Microanalytical Service of the Instituto Superior Técnico – University of Lisbon. FT-IR spectra (4000–400 cm−1) were recorded on a VERTEX 70 (Bruker) spectrometer using KBr pellets. Mass spectra were obtained on a VG 7070E mass spectrometer by electron ionization (EI) at 70 eV. Chromatographic analyses were performed in a Fisons Instruments GC 8000 series gas chromatograph with a DB-624 (J&W) capillary column (DB-WAX, column length: 30 m; internal diameter: 0.32 mm), FID detector and the Jasco-Borwin v.1.50 software; GC conditions: Tinjection=240 °C, Tinitial=140 °C (1 min) raised 10 °C min−1 to 220 °C (1 min), carrier gas: He.
X-ray measurements
X-ray quality single crystals of complex 6 were immersed in cryo-oil, mounted in Nylon loops and measured at a temperature of 296–297 K. Intensity data were collected using a Bruker AXS-KAPPA APEX II PHOTON 100 diffractometer with graphite monochromated Mo–Kα (λ=0.71073) radiation. Data were collected using omega scans of 0.5° per frame and full sphere of data were obtained. Cell parameters were retrieved using Bruker SMART [145] software and refined using Bruker SAINT [146] on all the observed reflections. Absorption corrections were applied using SADABS [124]. Structures were solved by direct methods by using SIR-97 [147] and refined with SHELXL-2014 [148]. Calculations were performed using the WinGX-Version 2014.01 [149]. The hydrogen atoms attached to carbon atoms were inserted at geometrically calculated positions and included in the refinement using the riding-model approximation; Uiso(H) were defined as 1.2 Ueq of the parent carbon atoms for phenyl residues and 1.5 Ueq of the parent carbon atoms for the methyl groups. The hydrogen atoms of water molecules were located from the final difference Fourier map and the isotropic thermal parameters were set at 1.5 times the average thermal parameters of the belonging oxygen atoms. Least square refinements with anisotropic thermal motion parameters for all the non-hydrogen atoms and isotropic for the remaining atoms were employed.
Synthesis of tetrazole-saccharin 5 and precursor compounds 2a and 4
2-Methyl-(2H)-tetrazole-5-amine (2a)
The molecules, 2-methyl-(2H)-tetrazole-5-amine (2a), 3-Chloro-1,2-benzisothiazole-1,1-dioxide (saccharyl chloride, (4) and 3-((2-methyl-2H-tetrazol-5-yl)amino)benzisothiazole 1,1-dioxide (5) were synthesised following previously reported protocols [127], [150].
Synthesis of the nickel(II) complex 6
To a 10 mL methanol/dimethyl sulfoxide (DMSO) solution (1:1) of 2-methyltetrazole-saccharinate 5 (100 mg; 0.4 mmol) was added Ni(OAc)2·4H2O (100 mg; 0.4 mmol) and the reaction mixture was stirred for 1 h at room temperature until complete dissolution of the solids. The resulting greenish solution was kept in open air at room temperature for crystallization. After 11 days, single blue-green crystals suitable for X-ray diffraction were isolated [130 mg; 48 % yield (95 % yield based on ligand 5)]. Elemental analysis (%): C, 35.15, H, 3.24, N, 24.58 (found); C, 35.26, H, 3.25, N, 24.67 (calculated for C20H22NiN12O6S3). IR (KBr; cm−1): 3404 ν(OH), 2919 ν(C–H), 1607, 1578, 1497, 1374 δ(CH3), 1279 ν(C–N), 1159, 1013 ν(S=O), 958 ν(S=O), 799, 645, 558, 437.
Typical procedure for the hydrosilylation catalysis under MW irradiation
Experiments were conducted with 2.0 mmol of substrate, 2.2 mmol of PhSiH3, 1 mol% of catalyst 6 and 1 mL of solvent, using a focused Anton Paar Monowave 300 microwave reactor in 10 mL glass vessels with 10 mm internal diameter, sealed with rubber membranes in a stirred mode with simultaneous cooling (IR temperature detector). A targeted temperature together with a maximum microwave power was set. The targeted temperature was reached within a few minutes. During the course of the reaction, the microwave power (5–15 W) as well as the pressure (1–10 bar) varied. Once the reaction was complete, the reaction vessel was cooled down to room temperature and then the cap was carefully opened to slowly release the pressure. After, 5 mL of a NaOH solution (1 M in MeOH) were added to the mixture, which remained under stirring at room temperature for 16 h (overnight). The organic layer was extracted with Et2O (3 × 10 mL), dried over MgSO4, filtered through a short Celite plug, and concentrated under reduced pressure. The isolated crude residue, without any further purification, was analyzed by 1H-NMR.
For the particular case of the formaldehyde, after microwave reaction, the reaction solvent (dichloromethane) was primarily removed under reduced pressure. After, 0.5 mL of aqueous NaOH (1 M) were added to the mixture, which persisted under stirring at room temperature for 24 h. The methanol (reaction product) was separated from the water by distillation (Vigreux).
All of the isolated alcohols under this procedure are known compounds. Structural data for the isolated selected products are included in Electronic supplementary information (ESI).
Typical procedure for the hydrosilylation catalysis via conventional heating
To an oven-dried round-bottom flask (25 mL) equipped with a stirring bar were added DCM (5 mL) and PhSiH3 (0.33 mL; 0.290 g; 2.68 mmol). The solution was stirred for 5 min. to allow complete homogenization of the silane, after which benzaldehyde (0.250 mL; 0.259 g; 2.44 mmol) and catalyst 6 (16.6 mg; 2.44×10−2 mmol) were added. The reaction occurred under stirring at room temperature and was monitored periodically by analysing an aliquot by TLC [hexane: diethyl ether (3:2)]. Once the reaction was complete, indicated by the disappearance of the benzaldehyde, the mixture was treated with 5 mL of a NaOH solution (1 M in MeOH) continued under stirring for 16 h. The solvent was removed under vacuum and the corresponding alcohol was extracted into Et2O (3×10 mL). This solution was dried over MgSO4, filtered through Celite and concentrated under reduced pressure to afford the corresponding alcohol (0.24 g; 91 % yield) without any further purification.
Computational details
The full geometry optimization of all structures and transition states (TS) was carried out at the DFT/HF hybrid level of theory using hybrid meta-GGA density functional M06-2X [151] with the help of the Gaussian-09 [152] program package. It was shown [153], [154] that the M06-2X functional demonstrates very good performance towards the main group thermochemistry, reasonably treating the weak dispersion forces. The quasi-relativistic Stuttgart-Dresden [155] pseudopotential MDF10 and the appropriate contracted basis set [156], [157], [158] were employed for the Ni atom. The Pople basis set 6-31G(d) was applied for all other atoms (C, H, N, O, Si and Si) [159], [160], [161], [162], [163] The geometries of all reported reactants, intermediates, transition states and products were optimized without symmetry constraints, including the solvent effect correction using the SMD model [164] with dichloromethane (ε=4.7113) or water (ε=78.3553), followed by analytical frequency calculations to confirm that a minimum or a transition state had been reached. The nature of the species connected by a given transition state structure was checked by IRC. Restricted approximations for the structures with closed electron shells have been employed.
Exhaustive conformational searches were performed for all intermediates and the Hessian matrix was calculated analytically for the optimized structures in order to map out the lowest energy profile and prove the location of correct minima (no imaginary frequencies) or saddle points (only one imaginary frequency) and to estimate the thermodynamic parameters, the latter being calculated at 298 K at the ideal gas conditions. The nature of all transition states was investigated by the analysis of vectors associated with the imaginary frequency and by the calculations of the intrinsic reaction coordinates (IRC) to ensure transitions states connected the illustrated ground states, using the Gonzalez-Schlegel method [165].
Single point calculations with the 6-311G(d) basis set were then performed for the equilibrium geometries found. The enthalpies and Gibbs free energies in solution (Hs and Gs) were estimated using the expressions Hs=Es+Hg–Eg and Gs=Hs–T•Ss, where Eg and Hg are the gas-phase total energy and enthalpy. The relative energies discussed in the text are Gibbs free energies in solution if not stated otherwise. Relative Gibbs free energies (ΔG) and enthalpies (ΔH) are presented in kcal·mol−1. Cartesian coordinates and total energies of all reported structures are included in ESI.
Electronic supplementary information
Table S1, with the selected bond distances and angles in the complex 6; Table S2, with the hydrogen-bond geometry for complex 6; The infrared spectrum of complex 6; Structural NMR data for the isolated compounds; Table S3, with the total energies (E), enthalpies (H) and Gibbs free energies (G), for the calculated structures; Table S4, with the calculated thermodynamic parameters for the studied reactions; DFT-optimized geometries in XYZ file format, using the M06-2X functional for all the studied structures.
CCDC number 1899338 for complex 6 (Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif).
Article note
A collection of invited papers based on presentations at the 24th IUPAC International Conference on Physical Organic Chemistry (ICPOC 24) held in Faro, Portugal, 1–6 July 2018.
Acknowledgements
This work was partially supported by the Foundation for Science and Technology (FCT), Portugal (UID/QUI/00100/2019 and UID/MULTI/04326/2019 – CCMAR). L.M.T.F. express gratitude to FCT for the post-doc fellowship (SFRH/BPD/99851/2014: FCT Grant) and working contract n° IST-ID/115/2018: Contract. B.G.M.R. is grateful to FCT for the PhD fellowship (SFRH/BD/52370/2013: Grant; CATSUS PhD program PD/00248/2012) (UID/QUI/00100/2013: Project).
Conflicts of interest: The authors declare no conflict of interest.
References
[1] J. G. de Vries, C. J. Elsevier (Eds.). The Handbook of Homogeneous Hydrogenation. Wiley-VCH, Weinheim, Germany (2007).10.1002/9783527619382Suche in Google Scholar
[2] P. A. Chaloner, M. A. Esteruelas, F. Joó, L. A. Oro. Homogeneous Hydrogenation, p. 87, Kluwer Academic Publishers, Dordrecht (1994).10.1007/978-94-017-1791-5Suche in Google Scholar
[3] R. H. Morris. Chem. Soc. Rev. 38, 2282 (2009).10.1039/b806837mSuche in Google Scholar PubMed
[4] H. I. Schlesinger, C. H. Brown, A. E. Finholt, R. J. Gilbreath, R. H. Hoekstra, K. E. Hyde. J. Am. Chem. Soc. 75, 215 (1953).10.1021/ja01097a057Suche in Google Scholar
[5] H. I. Schlesinger, C. H. Brown, B. Abraham, A. C. Bond, N. Davidson, A. E. Finholt, R. J. Gilbreath, H. Hoekstra, L. Horvitz, K. E. Hyde. J. Am. Chem. Soc. 75, 186 (1953).10.1021/ja01097a049Suche in Google Scholar
[6] G. L. Larson, J. L. Fry. Org. React. 71, 1 (2008).10.1002/0471264180.or071.01Suche in Google Scholar
[7] Z. Jia, M. Liu, X. Li, A. S. C. Chan, C.-J. Li. Synlett 24, 2049 (2013).10.1055/s-0033-1339660Suche in Google Scholar
[8] M. Fujita, T. Hiyama. J. Org. Chem. 53, 5405 (1988).10.1021/jo00258a003Suche in Google Scholar
[9] M. P. Doyle, D. J. DeBruyn, S. J. Donnelly, D. A. Kooista, A. A. Odubeta, C. T. West, S. M. Zonnedelt. J. Org. Chem. 39, 2740 (1974).10.1021/jo00932a015Suche in Google Scholar
[10] M. Fujita, T. Hiyama. J. Am. Chem. Soc. 106, 4629 (1984).10.1021/ja00328a062Suche in Google Scholar
[11] L. Gan, M. A. Brook. Can. J. Chem. 84, 1416 (2006).10.1139/v06-116Suche in Google Scholar
[12] I. Ojima, M. Nihonyanagi, Y. Nagai. Bull. Chem. Soc. Jpn. 45, 3722 (1972).10.1246/bcsj.45.3722Suche in Google Scholar
[13] H. Mimoun, J. V. de Saint Laumer, L. Giannini, R. Scopelliti, C. Floriani. J. Am. Chem. Soc. 121, 6158 (1999).10.1021/ja990522iSuche in Google Scholar
[14] P. Gallezot, D. Richard. Catal. Rev. 40, 81 (1998).10.1080/01614949808007106Suche in Google Scholar
[15] R. Noyori, T. Ohkuma. Angew. Chem. Int. Ed. 40, 40 (2001).10.1002/1521-3773(20010105)40:1<40::AID-ANIE40>3.0.CO;2-5Suche in Google Scholar
[16] R. Noyori. Angew. Chem. Int. Ed. 41, 2008 (2002).10.1002/1521-3773(20020617)41:12<2008::AID-ANIE2008>3.0.CO;2-4Suche in Google Scholar
[17] P. Mäki-Arvela, J. Hájek, T. Salmi, D. Y. Murzin. Appl. Catal. A 292, 1 (2005).10.1016/j.apcata.2005.05.045Suche in Google Scholar
[18] J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen, E. A. Pidko. Chem. Soc. Rev. 44, 3808 (2015).10.1039/C5CS00038FSuche in Google Scholar
[19] Y. Y. Li, S. L. Yu, W. Y. Shen, J. X. Gao. Acc. Chem. Res. 48, 2587 (2015).10.1021/acs.accounts.5b00043Suche in Google Scholar
[20] G. Brieger, T. J. Nestrick. Chem. Rev. 74, 567 (1974).10.1021/cr60291a003Suche in Google Scholar
[21] G. Zassinovich, G. Mestroni, S. Gladiali. Chem. Rev. 92, 1051 (1992).10.1021/cr00013a015Suche in Google Scholar
[22] J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson, P. Brandt. Chem. Soc. Rev. 35, 237 (2006).10.1039/b515269kSuche in Google Scholar
[23] G. Chelucci, S. Baldino, W. Baratta. Coord. Chem. Rev. 300, 29 (2015).10.1016/j.ccr.2015.04.007Suche in Google Scholar
[24] J.-I. Ito, H. Nishiyama. Tetrahedron Lett. 55, 3133 (2014).10.1016/j.tetlet.2014.03.140Suche in Google Scholar
[25] D. Wang, D. Astruc. Chem. Rev. 115, 6621 (2015).10.1021/acs.chemrev.5b00203Suche in Google Scholar
[26] F. Foubelo, C. Nájera, M. Yus. Tetrahedron: Asymmetry 26, 769 (2015).10.1016/j.tetasy.2015.06.016Suche in Google Scholar
[27] M. B. Smith, J. March. March’s Advanced Organic Chemistry. Wiley-Interscience, New York (2001).Suche in Google Scholar
[28] B. Marciniec (Ed.). Hydrosilylation: A Comprehensive Review on Recent Advances. Springer, Netherlands (2009).Suche in Google Scholar
[29] L. Postigo, B. Royo. Adv. Synth. Catal. 354, 2613 (2012).10.1002/adsc.201200389Suche in Google Scholar
[30] B. Chatterjee, C. Gunanathan. Chem. Commun. 50, 888 (2014).10.1039/C3CC47593JSuche in Google Scholar
[31] S. N. MacMillan, W. H. Harman, J. C. Peters. Chem. Sci. 5, 590 (2014).10.1039/C3SC52626GSuche in Google Scholar
[32] E. J. Corey, C. J. Helal. Angew. Chem. Int. Ed. 37, 1986 (1998).10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-ZSuche in Google Scholar
[33] H. Ito, T. Yajima, J.-I. Tateiwa, A. Hosomi. Chem. Commun. 11, 981 (2000).10.1039/b001743oSuche in Google Scholar
[34] S. Díez-González, S. P. Nolan. Acc. Chem. Res. 41, 349 (2008).10.1021/ar7001655Suche in Google Scholar
[35] H. Kinoshita, R. Uemura, D. Fukuda, K. Miura. Org. Lett. 15, 5538 (2013).10.1021/ol4026952Suche in Google Scholar
[36] D. Lantos, M. A. Contel, S. Sanz, A. Bodor, I. T. Horváth. J. Organomet. Chem. 692, 1799 (2007).10.1016/j.jorganchem.2006.10.025Suche in Google Scholar
[37] A. Kinting, H.-J. Kreuzfeld, H.-P. Abicht. J. Organomet. Chem. 370, 343 (1989).10.1016/0022-328X(89)87297-3Suche in Google Scholar
[38] J. L. Martinez, H. K. Sharma, R. Arias-Ugarte, K. H. Pannell. Organometallics 33, 2964 (2014).10.1021/om500439pSuche in Google Scholar PubMed PubMed Central
[39] B. M. Wile, M. Stradiotto. Chem. Commun. 39, 4104 (2006).10.1039/b609679dSuche in Google Scholar PubMed
[40] C. Cheng, M. Brookhart. Angew. Chem. Int. Ed. 51, 9422 (2012).10.1002/anie.201205154Suche in Google Scholar PubMed
[41] S. Park, M. Brookhart. Organometallics 29, 6057 (2010).10.1021/om100818ySuche in Google Scholar PubMed PubMed Central
[42] Y. Nishibayashi, K. Segawa, H. Takada, K. Ohe, S. Uemura. Chem. Commun. 7, 847 (1996).10.1039/cc9960000847Suche in Google Scholar
[43] Y. Nishibayashi, I. Takei, S. Uemura, M. Hidai. Organometallics 17, 3420 (1998).10.1021/om980399oSuche in Google Scholar
[44] J. T. Issenhuth, S. Dagorne, S. Bellemin-Laponnaz. Adv. Synth. Catal. 348, 1991 (2006).10.1002/adsc.200606152Suche in Google Scholar
[45] B. H. Lipshutz, B. A. Frieman. Angew. Chem. Int. Ed. 44, 6345 (2005).10.1002/anie.200500800Suche in Google Scholar PubMed
[46] J. Wu, J.-X. Ji, A. S. C. Chan. Proc. Natl. Acad. Sci. USA 102, 3570 (2005).10.1073/pnas.0409043102Suche in Google Scholar PubMed PubMed Central
[47] T. Bleith, H. Wadepohl, L. H. Gade. J. Am. Chem. Soc. 137, 2456 (2015).10.1021/ja512986mSuche in Google Scholar PubMed
[48] B. A. F. Le Bailly, S. P. Thomas. RSC Adv. 1, 1435 (2011).10.1039/c1ra00476jSuche in Google Scholar
[49] F.-G. Fontaine, R.-V. Nguyen, D. Zargarian. Can. J. Chem. 81, 1299 (2003).10.1139/v03-133Suche in Google Scholar
[50] S. Chakraborty, J. A. Krause, H. Guan. Organometallics 28, 582 (2009).10.1021/om800948fSuche in Google Scholar
[51] B. L. Tran, M. Pink, D. J. Mindiola. Organometallics 28, 2234 (2009).10.1021/om801160jSuche in Google Scholar
[52] L. Wang, H. Sun, X. Li. Organometallics 34, 5175 (2015).10.1021/acs.organomet.5b00734Suche in Google Scholar
[53] R. M. Bullock (Ed.). Catalysis without Precious Metals, p. 51, Wiley-VCH, Weinheim: Germany (2010).10.1002/9783527631582.ch3Suche in Google Scholar
[54] T. Vergote, F. Nahra, A. Merschaert, O. Riant, D. Peeters, T. Leyssens. Organometallics 33, 1953 (2014).10.1021/om401097qSuche in Google Scholar
[55] K. Revunova, G. I. Nikonov. Chem. Eur. J 20, 839 (2014).10.1002/chem.201302728Suche in Google Scholar PubMed
[56] Y. F. Wei, S. X. Liu, H. Mueller-Bunz, M. Albrecht. ACS Catal. 6, 8192 (2016).10.1021/acscatal.6b02269Suche in Google Scholar
[57] S. Bhattacharyya. J. Org. Chem. 63, 7101 (1998).10.1021/jo980685+Suche in Google Scholar PubMed
[58] T. Mizuta, S. Sakaguchi, Y. Ishii. J. Org. Chem. 70, 2195 (2005).10.1021/jo0481708Suche in Google Scholar PubMed
[59] M. P. Doyle, C. T. West. J. Org. Chem. 40, 3835 (1975).10.1021/jo00914a004Suche in Google Scholar
[60] N. Gandhamsetty, J. Jeong, J. Park, S. Park, S. Chang. J. Org. Chem. 80, 7281 (2015).10.1021/acs.joc.5b00941Suche in Google Scholar PubMed
[61] S. Rendler, M. Oestreich. Angew. Chem. Int. Ed. 47, 5997 (2008).10.1002/anie.200801675Suche in Google Scholar PubMed
[62] A. Fedorov, A. Toutov, N. Swisher, R. Grubbs. Chem. Sci. 4, 1640 (2013).10.1039/c3sc22256jSuche in Google Scholar
[63] M. Rubio, J. Campos, E. Carmona. Org. Lett. 4, 5236 (2011).10.1021/ol202117tSuche in Google Scholar PubMed
[64] S. E. Denmark, J. D. Baird. Chem. Eur. J. 12, 4954 (2006).10.1002/chem.200600034Suche in Google Scholar PubMed
[65] T. T. Metsanen, M. Oestreich. Organometallics 34, 543 (2015).10.1021/om501279aSuche in Google Scholar
[66] S. Díez-González, N. M. Scott, S. P. Nolan. Organometallics 25, 2355 (2006).10.1021/om0600487Suche in Google Scholar
[67] V. Skrypai, J. J. M. Hurley, M. J. Adler. Eur. J. Org. Chem. 12, 2207 (2016).10.1002/ejoc.201501599Suche in Google Scholar
[68] H. Zhou, H. Sun, S. Zhang, X. Li. Organometallics 34, 1479 (2015).10.1021/om5011929Suche in Google Scholar
[69] K. Junge, B. Wendt, D. Addis, S. Zhou, S. Das, M. Beller. Chem. Eur. J. 16, 68 (2010).10.1002/chem.200902442Suche in Google Scholar PubMed
[70] S. A. Wells. Org. Process Res. Dev. 14, 484 (2010).10.1021/op100080vSuche in Google Scholar
[71] M. Zhao, W. Xie, C. Cui. Chem. Eur. J. 20, 9259 (2014).10.1002/chem.201403497Suche in Google Scholar PubMed
[72] W. A. Herrmann, L. J. Goossen, C. Köcher, G. R. J. Artus. Angew. Chem. Int. Ed. 35, 2805 (1996).10.1002/anie.199628051Suche in Google Scholar
[73] M. Poyatos, A. Maisse-François, S. Bellemin-Laponnaz, E. Peris, L. H. Gade. J. Organomet. Chem. 691, 2713 (2006).10.1016/j.jorganchem.2006.02.008Suche in Google Scholar
[74] W. A. Herrmann, G. D. Frey, E. Herdtweck, M. Steinbeck. Adv. Synth. Catal. 349, 1677 (2007).10.1002/adsc.200700079Suche in Google Scholar
[75] M. Yigit, B. Yigit, I. Özdemir, E. Çetinkaya, B. Çetinkaya. Appl. Organomet. Chem. 20, 322 (2006).10.1002/aoc.1054Suche in Google Scholar
[76] X. Y. Yu, H. Sun, B. O. Patrick, B. R. James. Eur. J. Inorg. Chem. 13, 1752 (2009).10.1002/ejic.200801169Suche in Google Scholar
[77] D. R. Jensen, M. J. Schultz, J. A. Mueller, M. S. Sigman. Angew. Chem. Int. Ed. 42, 3810 (2003).10.1002/anie.200351997Suche in Google Scholar PubMed
[78] F. Hanasaka, K.-I. Fujita, R. Yamaguchi. Organometallics 23, 1490 (2004).10.1021/om049918fSuche in Google Scholar
[79] C. N. Cornell, M. S. Sigman. J. Am. Chem. Soc. 127, 2796 (2005).10.1021/ja043203mSuche in Google Scholar PubMed PubMed Central
[80] F. Hanasaka, K.-I. Fujita, R. Yamaguchi. Organometallics 25, 4643 (2006).10.1021/om060475kSuche in Google Scholar
[81] M. M. Konnick, B. A. Gandhi, I. A. Guzei, S. S. Stahl. Angew. Chem. Int. Ed. 45, 2904 (2006).10.1002/anie.200600532Suche in Google Scholar PubMed
[82] G. Altenhoff, R. Goddard, C. W. Lehmann, F. Glorius. J. Am. Chem. Soc. 126, 15195 (2004).10.1021/ja045349rSuche in Google Scholar PubMed
[83] O. Navarro, R. A. Kelly, S. P. Nolan. J. Am. Chem. Soc. 125, 16194 (2003).10.1021/ja038631rSuche in Google Scholar PubMed
[84] R. Ntaganda, B. Dhudshia, C. L. B. Macdonald, N. A. Thadani. Chem. Commun. 46, 6200 (2008).10.1039/b815757jSuche in Google Scholar PubMed
[85] C. Tubaro, A. Biffis, E. Scattolin, M. Basato. Tetrahedron 64, 4187 (2008).10.1016/j.tet.2008.02.092Suche in Google Scholar
[86] V. Percec, G. M. Golding, J. Smidrkal, O. Weichold. J. Org. Chem. 69, 3447 (2004).10.1021/jo049940iSuche in Google Scholar PubMed
[87] J.-I. Kuroda, K. Inamoto, K. Hiroya, T. Doi. Eur. J. Org. Chem. 14, 2251 (2009).10.1002/ejoc.200900067Suche in Google Scholar
[88] H. M. Sun, Q. Shao, D. M. Hu, W. F. Li, Q. Shen, Y. Zhang. Organometallics 24, 331 (2005).10.1021/om049334dSuche in Google Scholar
[89] B. E. Ketz, X. G. Ottenwaelder, R. M. Waymouth. Chem. Commun. 45, 5693 (2005).10.1039/b511202hSuche in Google Scholar PubMed
[90] W.-F. Li, H.-M. Sun, M.-Z. Chen, Q. Shen, Y. Zhang. J. Organomet. Chem. 693, 2047 (2008).10.1016/j.jorganchem.2008.03.005Suche in Google Scholar
[91] X. Wang, S. Liu, G. X. Jin. Organometallics 23, 6002 (2004).10.1021/om049467zSuche in Google Scholar
[92] W. Li, H. Sun, M. Chen, Z. Wang, D. Hu, Q. Shen, Y. Zhang. Organometallics 24, 5925 (2005).10.1021/om050612ySuche in Google Scholar
[93] S. Sujith, E. K. Noh, B. Y. Lee, J. W. Han. J. Organomet. Chem. 693, 2171 (2008).10.1016/j.jorganchem.2008.03.026Suche in Google Scholar
[94] M. Poyatos, A. Maisse-François, S. Bellemin-Laponnaz, L. H. Gade. Organometallics 25, 2634 (2006).10.1021/om060166uSuche in Google Scholar
[95] H. Kaur, F. K. Zinn, E. D. Stevens, S. P. Nolan. Organometallics 23, 1157 (2004).10.1021/om034285aSuche in Google Scholar
[96] A. Zanardi, E. Peris, J. A. Mata. New J. Chem. 32, 120 (2008).10.1039/B707280ESuche in Google Scholar
[97] C. E. Tucker, J. G. de Vries. Top. Catal. 19, 111 (2002).10.1023/A:1013841518270Suche in Google Scholar
[98] B. Su, Z. C. Cao, Z. J. Shi. Acc. Chem. Res. 48, 886 (2015).10.1021/ar500345fSuche in Google Scholar PubMed
[99] M. A. Asraf, H. A. Younus, M. Yusubov, F. Verpoort. Catal. Sci. Technol. 5, 4901 (2015).10.1039/C5CY01251ASuche in Google Scholar
[100] P. Chirik, R. Morris. Acc. Chem. Res. 48, 2495 (2015).10.1021/acs.accounts.5b00385Suche in Google Scholar PubMed
[101] I. Bauer, H.-J. Knölker, in Iron Catalysis in Organic Chemistry, B. Plietker (Ed.). Wiley-VCH, Weinheim: Germany (2008).10.1002/9783527623273.ch1Suche in Google Scholar
[102] M. Albrecht, R. Bedford, B. Plietker. Organometallics 33, 5619 (2014).10.1021/om5010379Suche in Google Scholar
[103] V. Ritleng, M. Henrion, M. J. Chetcuti. ACS Catal. 6, 890 (2016).10.1021/acscatal.5b02021Suche in Google Scholar
[104] S. Chakraborty, H. Guan. Dalton Trans. 39, 7427 (2010).10.1039/c002942dSuche in Google Scholar PubMed
[105] A. Albright, R. E. Gawley. J. Am. Chem. Soc. 133, 19680 (2011).10.1021/ja209187aSuche in Google Scholar PubMed PubMed Central
[106] S. R. Roy, S. C. Sau, S. K. Mandal. J. Org. Chem. 79, 9150 (2014).10.1021/jo501505jSuche in Google Scholar PubMed
[107] J. Zheng, S. Elangovan, D. A. Valyaev, R. Brousses, V. César, J.-B. Sortais, C. Darcel, N. Lugan, G. Lavigne. Adv. Synth. Catal. 356, 1093 (2014).10.1002/adsc.201300905Suche in Google Scholar
[108] S. Demir, Y. Gökçe, N. Kaloğlu, J.-B. Sortais, C. Darcel, İ. Özdemir. Appl. Organomet. Chem. 27, 459 (2013).10.1002/aoc.3006Suche in Google Scholar
[109] D. Bézier, F. Jiang, T. Roisnel, J.-B. Sortais, C. Darcel. Eur. J. Inorg. Chem. 2012, 1333 (2012).10.1002/ejic.201100762Suche in Google Scholar
[110] D. Kumar, A. P. Prakasham, L. P. Bheeter, J.-B. Sortais, M. Gangwar, T. Roisnel, A. C. Kalita, C. Darcel, P. Ghosh. J. Organomet. Chem. 762, 81 (2014).10.1016/j.jorganchem.2014.03.016Suche in Google Scholar
[111] L. P. Bheeter, M. Henrion, L. Brelot, C. Darcel, M. J. Chetcuti, J.-B. Sortais, V. Ritleng. Adv. Synth. Catal. 354, 2619 (2012).10.1002/adsc.201200460Suche in Google Scholar
[112] G. Vijaykumar, S. K. Mandal. Dalton Trans. 45, 7421 (2016).10.1039/C6DT00470ASuche in Google Scholar PubMed
[113] L. Benı́tez Junquera, M. C. Puerta, P. Valerga. Organometallics 31, 2175 (2012).10.1021/om200937dSuche in Google Scholar
[114] L. P. Bheeter, M. Henrion, M. J. Chetcuti, C. Darcel, V. Ritleng, J.-B. Sortais. Catal. Sci. Technol. 3, 3111 (2013).10.1039/c3cy00514cSuche in Google Scholar
[115] Y. Wei, S.-X. Liu, H. Mueller-Bunz, M. Albrecht. ACS Catal. 6, 8192 (2016).10.1021/acscatal.6b02269Suche in Google Scholar
[116] C. L. Rock, T. L. Groy, R. J. Trovitch. Dalton Trans. 47, 8807 (2018).10.1039/C8DT01857JSuche in Google Scholar PubMed
[117] Y. Chen, C. Sui-Seng, S. Boucher, D. Zargarian. Organometallics 24, 149 (2005).10.1021/om0494420Suche in Google Scholar
[118] M. I. Lipschutz, T. D. Tilley. Chem. Commun. 48, 7146 (2012).10.1039/c2cc32974cSuche in Google Scholar PubMed
[119] A. Kuznetsov, V. Gevorgyan. Org. Lett. 14, 914 (2012).10.1021/ol203428cSuche in Google Scholar PubMed PubMed Central
[120] T. J. Steiman, C. Uyeda. J. Am. Chem. Soc. 137, 6104 (2015).10.1021/jacs.5b03092Suche in Google Scholar PubMed
[121] I. Buslov, J. Becouse, S. Mazza, M. Montandon-Clerc, X. Hu. Angew. Chem. Int. Ed. 54, 14523 (2015).10.1002/anie.201507829Suche in Google Scholar PubMed
[122] I. Pappas, S. Treacy, P. J. Chirik. ACS Catal. 6, 4105 (2016).10.1021/acscatal.6b01134Suche in Google Scholar
[123] Y. Nakajima, K. Sato, S. Shimada. Chem. Rec. 16, 2379 (2016).10.1002/tcr.201600056Suche in Google Scholar PubMed
[124] S. Chakraborty, J. A. Krause, H. Guan. Organometallics 28, 582 (2009).10.1021/om800948fSuche in Google Scholar
[125] B. L. Tran, M. Pink, D. J. Mindiola. Organometallics 28, 2234 (2009).10.1021/om801160jSuche in Google Scholar
[126] L. Postigo, B. Royo. Adv. Synth. Catal. 354, 2613 (2012).10.1002/adsc.201200389Suche in Google Scholar
[127] L. M. T. Frija, E. C. B. A. Alegria, M. Sutradhar, M. L. S. Cristiano, A. Ismael, M. N. Kopylovich, A. J. L. Pombeiro. J. Mol. Catal. A Chem. 425, 283 (2016).10.1016/j.molcata.2016.10.023Suche in Google Scholar
[128] A. Ismael, J. A. Paixão, R. Fausto, M. L. S. Cristiano. J. Mol. Struct. 1023, 128 (2012).10.1016/j.molstruc.2012.04.018Suche in Google Scholar
[129] L. M. T. Frija, R. Fausto, R. M. S. Loureiro, M. L. S. Cristiano. J. Mol. Catal. A Chem. 305, 142 (2009).10.1016/j.molcata.2008.12.007Suche in Google Scholar
[130] L. M. T. Frija, M. L. Kuznetsov, B. G. M. Rocha, L. Cabral, M. L. S. Cristiano, M. N. Kopylovich, A. J. L. Pombeiro. Mol. Catal. 442, 57 (2017).10.1016/j.mcat.2017.09.003Suche in Google Scholar
[131] H. Zhao, Z.-R. Qu. Chem. Soc. Rev. 37, 84 (2008).10.1039/B616738CSuche in Google Scholar PubMed
[132] E. J. Baran, V. T. Yilmaz. Coord. Chem. Rev. 250, 1980 (2006).10.1016/j.ccr.2005.11.021Suche in Google Scholar
[133] L. M. T. Frija, A. J. L. Pombeiro, M. N. Kopylovich. Coord. Chem. Rev. 308, 32 (2016).10.1016/j.ccr.2015.10.003Suche in Google Scholar
[134] M. A. M. Abu-Youssef, F. A. Mautner, A. A. Massoud, L. Öhrström. Polyhedron 26, 1531 (2007).10.1016/j.poly.2006.11.044Suche in Google Scholar
[135] R.-G. Xiong, X. Xue, H. Zhao, X.-Z. You, B. F. Abrahams, Z.-L. Xue. Angew. Chem. Int. Ed. 41, 3800 (2002).10.1002/1521-3773(20021018)41:20<3800::AID-ANIE3800>3.0.CO;2-3Suche in Google Scholar
[136] Y.-J. Kim, Y.-S. Joo, J.-T. Han, S. H. Won, W. L. Soon. J. Chem. Soc. Dalton Trans. 18, 3611 (2002).10.1039/b204179kSuche in Google Scholar
[137] A. K. Gupta, C. H. Song, C. H. Oh. Tetrahedron Lett. 45, 4113 (2004).10.1016/j.tetlet.2004.03.162Suche in Google Scholar
[138] A. S. Burukin, A. A. Vasilév, N. L. Merkulova, M. I. Struchkova, S. G. Zlotin. Russ. Chem. Bull. Int. Ed. 55, 118 (2006).10.1007/s11172-006-0224-0Suche in Google Scholar
[139] P. Naumov, G. Jovanovski. Struct. Chem. 11, 19 (2000).10.1023/A:1009264221838Suche in Google Scholar
[140] F. A. Cotton, L. R. Falvello, W. Schwotzer, C. A. Murillo, G. Valle-Bourrouet. Inorg. Chim. Acta 190, 89 (1991).10.1016/S0020-1693(00)80236-4Suche in Google Scholar
[141] G. Jovanovski, B. Šoptrajanov. J. Mol. Struct. 174, 467 (1998).10.1016/0022-2860(88)80202-3Suche in Google Scholar
[142] N. M. Camasso, M. S. Sanford. Science 347, 1218 (2015).10.1126/science.aaa4526Suche in Google Scholar
[143] R. Hofmann, M. Vlatkovic, F. Wiesbrock. Polymers 9, 534 (2017).10.3390/polym9100534Suche in Google Scholar
[144] N. Nakatani, J.-Y. Hasegawa, Y. Sunada, H. Nagashima. Dalton Trans. 44, 19344 (2015).10.1039/C5DT02767ESuche in Google Scholar
[145] APES4. Bruker AXS Inc., Madison, WI (2004).Suche in Google Scholar
[146] SAINT. Bruker AXS Inc., Madison, WI (2004).Suche in Google Scholar
[147] A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori, R. Spagna. J. Appl. Cryst. 32, 115 (1999).10.1107/S0021889898007717Suche in Google Scholar
[148] G. M. Sheldrick. Acta Crystallogr. Sect. A 64, 112 (2008).10.1107/S0108767307043930Suche in Google Scholar PubMed
[149] L. J. Farrugia. J. Appl. Crystallogr. 32, 837 (1999).10.1107/S0021889899006020Suche in Google Scholar
[150] A. F. Brigas, C. S. C. Fonseca, R. A. W. Johnstone. J. Chem. Res. 6, 299 (2002).10.3184/030823402103172077Suche in Google Scholar
[151] Y. Zhao, D. G. Truhlar. Theor. Chem. Acc. 120, 215 (2008).10.1007/s00214-007-0310-xSuche in Google Scholar
[152] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox. Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford, CT (2009).Suche in Google Scholar
[153] L. A. Burns, Á. Vazquez-Mayagoitia, B. G. Sumpter, C. D. Sherrill J. Chem. Phys. 134, 84107 (2011).10.1063/1.3545971Suche in Google Scholar PubMed
[154] M. Sumimoto, Y. Kawashima, D. Yokogawa, K. Hori, H. Fujimoto. J. Comput. Chem. 32, 3062 (2011).10.1002/jcc.21889Suche in Google Scholar PubMed
[155] M. Dolg, U. Wedig, H. Stoll, H. Preuss. J. Chem. Phys. 86, 866 (1987).10.1063/1.452288Suche in Google Scholar
[156] W. Küchle, M. Dolg, H. Stoll, H. Preuss. Mol. Phys. 74, 1245 (1991).10.1080/00268979100102941Suche in Google Scholar
[157] D. Andrae, U. Häußermann, M. Dolg, H. Stoll, H. Preuß. Theor. Chim. Acta 77, 123 (1990).10.1007/BF01114537Suche in Google Scholar
[158] A. Bergner, M. Dolg, W. Küchle, H. Stoll, H. Preuß. Mol. Phys. 80, 1431 (1993).10.1080/00268979300103121Suche in Google Scholar
[159] A. D. McLean, G. S. Chandler. J. Chem. Phys. 72, 5639 (1980).10.1063/1.438980Suche in Google Scholar
[160] R. Krishnan, J. S. Binkley, R. Seeger, J. A. Pople. J. Chem. Phys. 72, 650 (1980).10.1063/1.438955Suche in Google Scholar
[161] A. J. H. Wachters. Chem. Phys. 52, 1033 (1970).10.1063/1.1673095Suche in Google Scholar
[162] P. J. Hay. J. Chem. Phys. 66, 4377 (1977).10.1063/1.433731Suche in Google Scholar
[163] P. C. Hariharan, J. A. Pople. Theor. Chim. Acta 28, 213 (1973).10.1007/BF00533485Suche in Google Scholar
[164] G. Scalmani, M. J. Frisch. J. Chem. Phys. 132, 114110 (2010).10.1063/1.3359469Suche in Google Scholar PubMed
[165] C. Gonzalez, H. B. Schlegel. J. Chem. Phys. 95, 5853 (1991).10.1063/1.461606Suche in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/pac-2019-0220).
© 2020 IUPAC & De Gruyter, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this issue
- Preface
- The 24th IUPAC International Conference on Physical Organic Chemistry (ICPOC 24)
- Conference papers
- Binding motifs of cisplatin interaction with simple biomolecules and aminoacid targets probed by IR ion spectroscopy
- Synthesis and reactivity/stability study of double-functionalizable strained trans-cyclooctenes for tetrazine bioorthogonal reactions
- Counterion effect on sulfonatocalix[n]arene recognition
- Ping-pong tunneling reactions, part 2: boron and carbon bell-clapper rearrangement
- Insights into the photochemistry of 5-aminotetrazole derivatives with applications in coordination chemistry. Effect of the saccharyl moiety on the photostability
- A green road map for heterogeneous photocatalysis
- Computational simulation of mechanism and isotope effects on acetal heterolysis as a model for glycoside hydrolysis
- Coordination of tridentate ligands to SmI2: cooperativity and incremental effect on reduction potential and on reactivity
- An efficient and eco-friendly method for the thiol-Michael addition in aqueous solutions using amino acid ionic liquids (AAILs) as organocatalysts
- Changing the kinetic order of enantiomer formation and distinguishing between iminium ion and imine as the reactive species in the asymmetric transfer hydrogenation of substituted imines using a cyclopentadienyl iridium (III) complex
- Oxidative sulfamidation as a route to N-heterocycles and unsaturated sulfonamides
- Well-defined nickel(II) tetrazole-saccharinate complex as homogeneous catalyst on the reduction of aldehydes: scope and reaction mechanism
- Unravelling mechanistic insights in the platinum-catalysed dihydroalkoxylation of allenes
- The VES KM: a pathway for protein folding in vivo
- IUPAC Recommendations
- Nomenclature and terminology for linear lactic acid-based polymers (IUPAC Recommendations 2019)
Artikel in diesem Heft
- Frontmatter
- In this issue
- Preface
- The 24th IUPAC International Conference on Physical Organic Chemistry (ICPOC 24)
- Conference papers
- Binding motifs of cisplatin interaction with simple biomolecules and aminoacid targets probed by IR ion spectroscopy
- Synthesis and reactivity/stability study of double-functionalizable strained trans-cyclooctenes for tetrazine bioorthogonal reactions
- Counterion effect on sulfonatocalix[n]arene recognition
- Ping-pong tunneling reactions, part 2: boron and carbon bell-clapper rearrangement
- Insights into the photochemistry of 5-aminotetrazole derivatives with applications in coordination chemistry. Effect of the saccharyl moiety on the photostability
- A green road map for heterogeneous photocatalysis
- Computational simulation of mechanism and isotope effects on acetal heterolysis as a model for glycoside hydrolysis
- Coordination of tridentate ligands to SmI2: cooperativity and incremental effect on reduction potential and on reactivity
- An efficient and eco-friendly method for the thiol-Michael addition in aqueous solutions using amino acid ionic liquids (AAILs) as organocatalysts
- Changing the kinetic order of enantiomer formation and distinguishing between iminium ion and imine as the reactive species in the asymmetric transfer hydrogenation of substituted imines using a cyclopentadienyl iridium (III) complex
- Oxidative sulfamidation as a route to N-heterocycles and unsaturated sulfonamides
- Well-defined nickel(II) tetrazole-saccharinate complex as homogeneous catalyst on the reduction of aldehydes: scope and reaction mechanism
- Unravelling mechanistic insights in the platinum-catalysed dihydroalkoxylation of allenes
- The VES KM: a pathway for protein folding in vivo
- IUPAC Recommendations
- Nomenclature and terminology for linear lactic acid-based polymers (IUPAC Recommendations 2019)