A rare FBXO25–SEPT14 fusion in a patient with chronic myeloid leukemia treatment to tyrosine kinase inhibitors: a case report
-
Yun Liao


Abstract
Objectives
Exploring the pathogenesis of resistance to tyrosine kinase inhibitors in patients with chronic myeloid leukemia.
Case presentation
This case report describes a rare fusion of FBXO25 and SEPT14 genes in a 58-year-old male patient with chronic myeloid leukemia. The patient had been treated with tyrosine kinase inhibitors for one year. After 6 months of imatinib treatment, the patient's symptoms improved significantly and the complete blood count returned to normal, but the optimal ratio of BCR::ABL transcripts to ABL transcripts is greater than 10 % indicating treatment failure. Then we switched to a second generation TKIs to continue treatment, During the Flumatinib treatment period, the patient developed severe bone marrow suppression and exhibited additional cytogenetic abnormalities involving chromosome aberration: 47, XY,+8[5]/47, idem, inv(Y)(p11.2q11.23)[15]. By adjusting the drug dose and elevating blood cells, the patient’s BCR::ABL P210/ABL was 2.56 % after six months of Flumatinib treatment. The patient’s BCR::ABL P210/ABL consistently remained above 1 % throughout the treatment, and additional cytogenetic abnormalities were present. Next-generation sequencing revealed the recombination of exon 4 of the FBXO25 and exon 10 of the SEPT14, and this mutation has not been previously reported.
Conclusions
Our findings suggest that the FBXO25-SEPT14 fusion may be associated with tyrosine kinase inhibitors resistance in chronic myeloid leukemia.
Introduction
Chronic myeloid leukemia (CML) is a type of myeloproliferative neoplasm that affects pluripotent hematopoietic stem cells. The main clinical features of CML include the Philadelphia chromosome (Ph), which results from the long arms of chromosomes 9 and 22, and the t(9;22)(q34;q11) translocation that gives rise to a BCR::ABL1 fusion gene in the affected cell lineage. The BCR::ABL1 fusion gene encodes an oncoprotein, p210BCR::ABL, which is a highly active constitutive tyrosine kinase that activates multiple downstream signaling pathways, leading to aberrant proliferation, inhibition of apoptosis, regulation of cell adhesion, and survival of growth factors in CML cell [1]. The natural course of CML is divided into three phases: chronic phase (CP), accelerated phase (AP), and blastic phase (BP).
Tyrosine kinase inhibitors (TKIs) specifically block the binding site of ATP on ABL kinase, making tyrosine residues unphosphorylatable and thus inhibiting the proliferation of BCR::ABL-positive cells. The first-generation TKIs Imatinib achieves an 83 % complete cytogenetic remission rate, and the overall survival rate at 10 years is 83 % in CML patients [2], Second-generation TKIs, such as Flumatinib and Dasatinib, can achieve faster and deeper molecular responses in CML and are gradually becoming optional first-line treatment options for CML. Efficacy monitoring should be conducted at months 3, 6, and 12 after starting TKI therapy. Patients who are judged to have failed treatment should be examined for mutations in the ABL kinase region, and depending on the form of the mutation and the patient’s response to the drug, TKI replacement or hematopoietic stem cell transplantation should be considered.
We describe a male patient who was resistant to TKIs and whose FBXO25–SEPT14 fusion gene was discovered using next-generation sequencing (NGS). This is a novel pathogenic mutation that has never been reported before.
Case presentation
A 58-year-old man was admitted to Southwest Medical University’s Affiliated Hospital due to abdominal pain. During physical examination, splenomegaly was observed (spleen 5 cm below costal). Blood tests revealed a white blood cell (WBC) count of 243.81 × 109/L (reference value: 3.5–9.5 × 109/L) and a neutrophil count (NEU) of 187.73 × 109/L (reference value: 1.8–6.3 × 109/L). The patient’s hemoglobin (HB) level was 99 g/L (reference value: 115–150/L) and platelet (PLT) count was 713 × 109/L (reference value: 125–350 × 109/L). Further examination of the bone marrow smear showed nucleated cell proliferation extremely active (Figure 1). Chromosome analysis of the bone marrow showed a result of 46, XY, t(9;22)(q34;q11)[5] (Figure 2A). The BCR::ABL genotyping test was positive for P210 and the BCR::ABL P210 typing quantitative test revealed a 77.15 % ratio of BCR::ABL transcripts to ABL transcript. Based on these findings, a diagnosis of chronic myeloid leukemia was made.
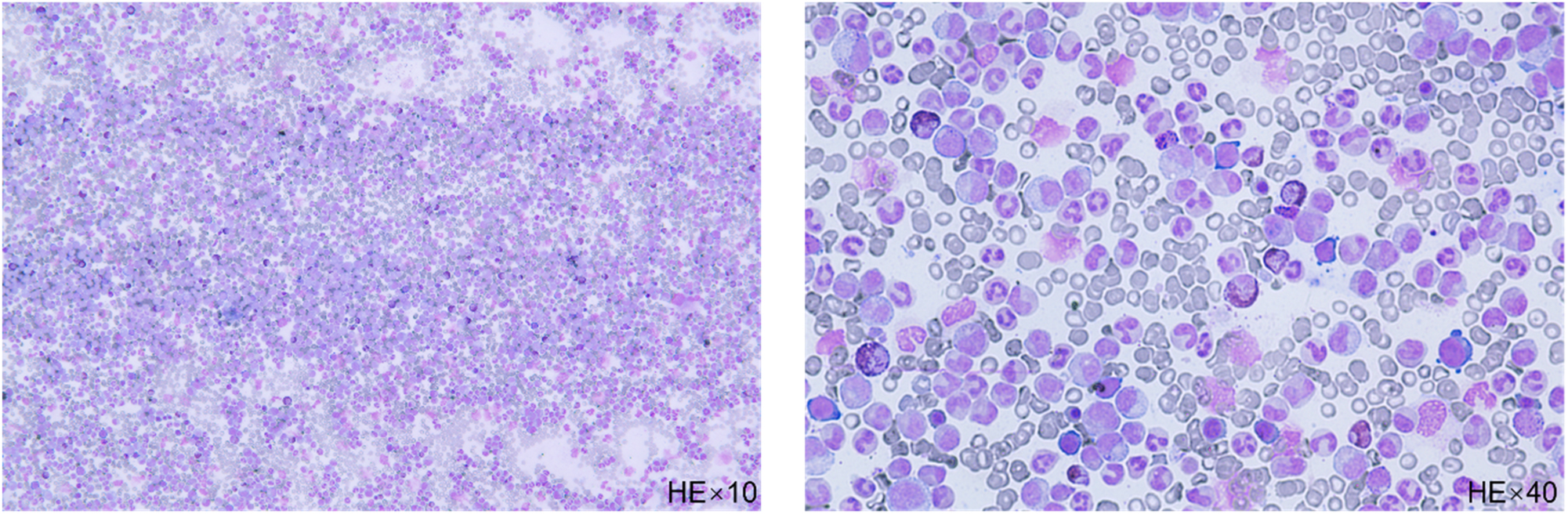
The bone marrow smear shows nucleated cell proliferation extremely active, late neutrophils, and rod nucleated cells are the most common, their cytosol size varies and the nucleoplasmic development is unbalanced (HE stain).
![Figure 2:
The patient’s chromosome analysis. (A) 46, XY, t(9;22)(q34;q11)[5], before treatment. (B) 46, XY, t(9,22)(q34,q11)[12]/46, XY[8], after 6 months of Imatinib treatment. (C) 47, XY, +8[10], after 3 months of Flumatinib treatment. (D) 47, XY, +8[5]/47, idem, inv(Y)(p11.2q11.23)[15], after 6 months of Flumatinib treatment.](/document/doi/10.1515/oncologie-2023-0217/asset/graphic/j_oncologie-2023-0217_fig_002.jpg)
The patient’s chromosome analysis. (A) 46, XY, t(9;22)(q34;q11)[5], before treatment. (B) 46, XY, t(9,22)(q34,q11)[12]/46, XY[8], after 6 months of Imatinib treatment. (C) 47, XY, +8[10], after 3 months of Flumatinib treatment. (D) 47, XY, +8[5]/47, idem, inv(Y)(p11.2q11.23)[15], after 6 months of Flumatinib treatment.
After admission, we administered rehydration fluids to hydrate and alkalize the patient’s blood. We also used Hydroxyurea for 10 days to reduce the patient’s peripheral blood leukocytes. Once the patient’s diagnosis was clear, we instructed the patient to start taking Imatinib mesylate (400 mg po qd) outside the hospital. Before discharge, we reviewed the patient’s blood work, which showed a WBC count of 9.47 × 109/L, NEU count of 7.31 × 109/L, HB level of 99 g/L, and PLT count of 664 × 109/L. From September 2021 to December 2021, the patient received 3 months of Imatinib (400 mg po qd) treatment. We re-examined the patient’s bone marrow and found that the granulocytic lineage accounted for 65 %, with 32.5 % fractionated nucleated cells. No abnormalities in cell morphology and size were seen in any phase, and no pathological hematopoietic changes were observed in any cell lineage. The BCR::ABL P210/ABL was 10.29 %, so we continued the patient’s Imatinib treatment. However, in March 2022, a review of the bone marrow results revealed chromosome: 46, XY, t(9,22)(q34,q11)[12]/46, XY[8], and BCR::ABL P210/ABL 10.12 % (Figure 2B). According to the European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia [3], the optimal ratio of BCR::ABL transcripts to ABL transcripts after 6 months of treatment should be less than or equal to 1 %, and greater than 10 % indicates treatment failure. However, ABL mutation gene testing did not detect any mutations such as T315I, P-Loop, or A-Loop in the patient’s peripheral blood, and the patient’s complete blood count (CBC) showed WBC count of 3.72 × 109/L, NEU count of 1.79 × 109/L, HB level of 123 g/L, and PLT count of 109 × 109/L. Then, we switched the patient’s medication from Imatinib to Flumatinib (600 mg po qd) to continue treatment. During treatment with Flumatinib, the patient developed severe bone marrow suppression, and the patient’s platelet count dropped to 8 × 109/L. We adjusted the dose of Flumatinib to 400 mg and administered platelet transfusions. Three months later, a bone marrow examination showed chromosome: 47, XY, +8[10] (Figure 2C) and BCR::ABL P210/ABL 2.50 %. We deemed the treatment to be effective, so the patient continued to take Flumatinib for three more months. At the next bone marrow review, the chromosome results were 47, XY, +8[5]/47, idem, inv(Y)(p11.2q11.23)[15] (Figure 2D), and the BCR::ABL P210/ABL was 2.56 %, and CBC showed WBC count 3.31 × 109/L, NEU count 1.71 × 109/L, HB level 122 g/L, and PLT count 58 × 109/L. Throughout the treatment, the patient’s BCR::ABL P210/ABL consistently remained above 1 %, and additional cytogenetic abnormalities were present.
The patient underwent next-generation sequencing (NGS), using the Ribozero method to remove ribosomal RNA from total RNA. The resulting cDNA was used to construct a library for sequencing on an Illumina platform, allowing for whole transcriptome level detection of RNA from patient samples. This approach facilitated the analysis of gene fusions and SNV variants at the transcriptional level. Bioinformatics analysis of mutations and gene fusions involved aligning the sequenced fragments with the UCSChg9 reference genome using STAR software. VarDict software was used for mutation detection, which included PCR repeat removal, InDels position and quality score correction, as well as SNVs and InDels variant discovery and typing. Gene fusion prediction was performed using STAR-Fusion, with fusion gene databases such as chimerdb, PubMed, CCLE, Klijn CellLines, chimerdb omim, ChimerSe, ChimerKB, FA CancerSupp, Cosmic, Mitelman, and ChimerPub consulted. The analysis revealed the presence of an FBXO25/SEPT14 fusion gene in the peripheral blood sample, formed by the recombination of exon 4 of the FBXO25 and exon 10 of the SEPT14 (Figure 3). To our knowledge, this fusion gene has not been previously reported.

Schematic of the results of NGS: The left gene represents the FBXO25 gene and the right gene represents the SEPT14 gene. Next-generation sequencing revealed the recombination of exon 4 of the FBXO25 and exon 10 of the SEPT14.
The patient was diagnosed with Chronic myeloid leukemia (CML) accelerated phase (AP), P210 (+), with FBXO25/SEPT14 fusion gene. He was prescribed Imatinib and Flumatinib for a year, and his symptoms improved. As of now, his white blood cell and platelet counts remain normal, and his spleen has significantly reduced in size.
Discussion
The advent of TKIs has transformed CML from a fatal malignant disease to a manageable chronic disease. Several TKIs have been synthesized to target BCR::ABL, the underlying mechanism of CML. With continued treatment, most patients can expect to live a normal life. Imatinib mesylate (IM) is the gold standard of first-line therapy and has greatly improved the overall survival (OS) of patients with CML in the chronic phase (CP). It is now available as a generic drug in many countries [4]. However, over time, many patients develop resistance to TKIs. Clinical trials have estimated that 20–30 % of patients do not achieve the desired effect or develop resistance to TKIs after initial efficacy, and most of these patients have a high probability of entering the accelerated or blastic phase [5]. Second and third-generation TKIs, such as Nilotinib, Flumatinib and Ponatinib, have more targets and better efficacy. However, there are still patients with CML who do not respond to all TKIs. Moreover, about 60 % of patients in complete molecular remission experience relapse after treatment interruption [6]. This may be due to residual TKIs-resistant CML stem cells that contribute to disease relapse. Therefore, there is a need to further investigate the molecular resistance mechanisms and find new targets to overcome drug resistance.
At the cytogenetic level, t(9;22)(q34;q11) is the primary abnormality found in over 90 % of CML patients in the chronic phase (CP) [7]. Secondary chromosomal aberrations in CML patients are non-random and often include +8 (34 %), +Ph (30 %), −Y (8 % of males) [8]. The World Health Organization (WHO) criteria for the accelerated phase of CML, recently updated, considers any new clonal chromosomal abnormality in t(9;22) positive cells that occur during therapy as a criterion for AP [9]. In our patient, two CML-APs were identified, both of which were caused by trisomy 8. The first accelerated phase revealed trisomy 8, and the second revealed trisomy 8 and inv(Y)(p11.2q11.23), indicating a poor prognosis. Chromosome 8 is a medium-length pair of 23 human chromosomes containing approximately 1,450,000 base pairs, and its instability is linked to the development of several tumors [10]. The Myc gene on 8q24, MOS, and RUNX1T1 genes on 8q22 [11] are associated with AML prognosis. The Myc gene is a pro-oncogene, promoting cell proliferation and apoptosis inhibition, and its overexpression may promote telomerase activity, leading to excessive cell proliferation and a poor disease prognosis [12].
FBXO25 gene is located on human chromosome 8p23.3 and encodes a 40-amino acid motif F-box protein, which belongs to the F-box protein family. This protein is one of four subunits that make up the SKP1-cullin-F-box (SCFs) ubiquitin-protein ligase complex, involved in phosphorylation-dependent ubiquitination. The expression and biological functions of FBXO25 in human tumors remain unknown. However, Aleksandar Kuzmanov et al. [13] reported that FBXO25 promotes cutaneous squamous cell carcinoma growth and metastasis via Cyclin D1. The roles of F-box proteins in cancer are complex, and whether they act as tumor suppressors or oncogenes is still debated. In non-small-cell lung cancer (NSCLC), FBXO25 has been shown to promote cell proliferation, invasion, and migration [14]. However, Baumann et al. [15] demonstrated that disrupting the PRKCD-FBXO25-HAX-1 axis reduces apoptosis and promotes lymphomagenesis, implying that FBXO25 has tumor-suppressive properties. In this case, FBXO25 appeared as the disease progressed, indicating its association with tumor cell proliferation in CML. Nonetheless, more research is needed to confirm this hypothesis.
SEPT14 gene encodes a GTP-binding cytoskeletal protein that participates in various cellular processes, such as membrane trafficking, apoptosis, cell polarity, cell cycle regulation, and cell division. The most frequent fusion partner gene of SEPT14 is EGFR. The EGFR-SEPT14 fusion has been identified in glioblastoma using a large RNA sequencing dataset of primary glioblastomas and glioma sphere cultures [16]. Moreover, a patient with colorectal adenocarcinoma was found to have an EGFR-SEPT14 fusion using NGS analysis [17]. The mechanism underlying the presence of the SEPT14 gene in this case of chronic granulocytic leukemia is unknown, but it may be associated with tyrosine kinase inhibitor resistance and disease progression. Drug resistance and disease progression have been the subject of numerous research studies over the last two decades, aimed at identifying dysregulated molecules, validating new druggable targets, and optimizing treatment decision algorithms. Further research is necessary to fully understand the clinical significance of the FBXO25/SEPT14 fusion gene in CML patients.
Mutations in the ABL1 kinase region (KD) are the main cause of poor TKI efficacy or drug resistance. In recent years, with the application of NGS technology in clinical practice, it has been found that CML patients are often accompanied by mutations in the non-ABL1 kinase region, but it is unclear whether these concomitant mutations are associated with TKI efficacy and disease progression in CML. The heterogeneity of disease duration and TKI efficacy between patients has been a clinical problem. Non-ABL1 KD mutations are frequently detected in CML patients, with epi-genetically related genes such as ASXL1, DNMT3A, and TET2 being the most common types of the mutated gene [18]. Further NGS assays have revealed that non-ABL1 KD mutations are a common molecular event in TKI resistance and disease progression, adding to the significance of these mutations in CML. While ABL1 KD mutations are a major cause of TKI resistance, the relationship between non-ABL1 KD mutations and ABL1 KD secondary mutations in CML patients remains unclear. Studies by Schmidt et al. [19] demonstrated that mutations in genes such as DNMT3A, EZH2, RUNX1, TET2, TP53, U2AF1, and ZRSR2 were detected in Ph-negative clonal hematopoietic cells in 43 % of CML patients even after remission with TKI therapy. Kim et al. [20] also found that 89 % of patients with ABL1 KD mutations carried non-ABL1 KD mutations, and most of them were earlier than ABL1 KD mutations. The above suggests that non-ABL1 KD mutations may not only be present in CML cells to affect the efficacy of TKIs, but also may act as co-stimulatory factors to induce tumorigenesis and promote disease progression in the early stage of the disease. The FBXO25–SEPT14 fusion gene we identified in this patient may be a novel non-ABL1 KD mutation.
The FBXO25–SEPT14 fusion gene present in this case represents the status of this individual patient only. Due to technical limitations, there may be some false positives and false negatives in the sequencing itself, and the corresponding results need to be judged by the clinic in conjunction with the patient’s specific clinical manifestations and other examination results. In addition, low tumor cell content or low expression levels of fusion and mutation genes may affect the accuracy and sensitivity of the sample results. In order to further explore the mechanism of the effect of this fusion gene on patients, CML cell line and mouse models are needed to further investigate the FBXO25–SEPT14 fusion and its impact on progression of CML.
The identification of the FBXO25–SEPT14 fusion gene in this case of CML could be a valuable contribution to the ongoing research on drug resistance mechanisms and treatment options. By studying the functional significance of this fusion gene and its potential association with TKI resistance and disease progression, researchers could gain a deeper understanding of the molecular mechanisms underlying CML pathogenesis and develop more effective therapeutic strategies to overcome resistance and improve patient outcomes. Moreover, this discovery highlights the importance of utilizing advanced genomic technologies and personalized medicine approaches to tailor treatment plans to individual patients based on their unique genetic profiles.
Conclusions
In conclusion, our study utilized a comprehensive NGS assay to identify a rare FBXO25–SEPT14 fusion in a patient with CML. This represents the first documented instance of an FBXO25–SEPT14 fusion in CML, and our findings suggest a potential association between this mutation and resistance to TKIs. We expect that more well-designed cell-animal experiments as well as more studies of patients with similar clinical characteristics will be reported in the future to further enrich our research results.
Acknowledgments
The authors are grateful to the patient.
-
Research ethics: This study was approved by the Clinical Trial Ethics Committee of the Affiliated Hospital of Southwest Medical University (Consent no KY2023220).
-
Informed consent: Written informed consent was obtained from patients to publish their anonymous information in this article.
-
Author contributions: All authors were involved in designing the project. Yun Liao and Haoshu Zhong performed data extraction and analysis. Quality assessment was conducted by Hao Xiong, Yang Liu and Xiaomin Chen. Yun Liao, Jiayue Liu and Rongrong Chen wrote the paper, Chunlan Huang and Mengyu Wei made critical changes to the manuscript and oversaw the project. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Competing interests: Authors state no conflict of interest.
-
Research funding: This study was supported by Sichuan Department of Science and Technology (grant no. 2020YJ0339).
References
1. Ben-Neriah, Y, Daley, GQ, Mes-Masson, AM, Witte, ON, Baltimore, D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986;233:212–4. https://doi.org/10.1126/science.3460176.Suche in Google Scholar PubMed
2. Hochhaus, A, Larson, RA, Guilhot, F, Radich, JP, Branford, S, Hughes, TP, et al.. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med 2017;376:917–27. https://doi.org/10.1056/NEJMoa1609324.Suche in Google Scholar PubMed PubMed Central
3. Hochhaus, A, Baccarani, M, Silver, RT, Schiffer, C, Apperley, JF, Cervantes, F, et al.. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020;34:966–84. https://doi.org/10.1038/s41375-020-0776-2.Suche in Google Scholar PubMed PubMed Central
4. Druker, BJ, Guilhot, F, O’Brien, SG, Gathmann, I, Kantarjian, H, Gattermann, N, et al.. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006;355:2408–17. https://doi.org/10.1056/NEJMoa062867.Suche in Google Scholar PubMed
5. Melo, JV, Barnes, DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 2007;7:441–53. https://doi.org/10.1038/nrc2147.Suche in Google Scholar PubMed
6. Chomel, JC, Bonnet, ML, Sorel, N, Bertrand, A, Meunier, MC, Fichelson, S, et al.. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 2011;118:3657–60. https://doi.org/10.1182/blood-2011-02-335497.Suche in Google Scholar PubMed PubMed Central
7. Wang, W, Chen, Z, Hu, Z, Yin, CC, Li, S, Bai, S, et al.. Clinical significance of trisomy 8 that emerges during therapy in chronic myeloid leukemia. Blood Cancer J 2016;6:e490. https://doi.org/10.1038/bcj.2016.96.Suche in Google Scholar PubMed PubMed Central
8. Johansson, B, Fioretos, T, Mitelman, F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol 2002;107:76–94. https://doi.org/10.1159/000046636.Suche in Google Scholar PubMed
9. Arber, DA, Orazi, A, Hasserjian, R, Thiele, J, Borowitz, MJ, Le Beau, MM, et al.. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405. https://doi.org/10.1182/blood-2016-03-643544.Suche in Google Scholar PubMed
10. Zhang, Y, Yan, G, Zhai, L, Xu, S, Shen, H, Yao, J, et al.. Proteome atlas of human chromosome 8 and its multiple 8p deficiencies in tumorigenesis of the stomach, colon, and liver. J Proteome Res 2013;12:81–8. https://doi.org/10.1021/pr300834r.Suche in Google Scholar PubMed
11. La Rosa, S, Bernasconi, B, Vanoli, A, Sciarra, A, Notohara, K, Albarello, L, et al.. c-MYC amplification and c-myc protein expression in pancreatic acinar cell carcinomas. New insights into the molecular signature of these rare cancers. Virchows Arch 2018;473:435–41. https://doi.org/10.1007/s00428-018-2366-5.Suche in Google Scholar PubMed
12. Yamamoto, K, Kawamoto, S, Kurata, K, Kitao, A, Mizutani, Y, Ichikawa, H, et al.. MYC amplification in the form of ring chromosomes 8 in acute myeloid leukemia with t(11;16)(q13;p11.2). Cytogenet Genome Res 2017;153:131–7. https://doi.org/10.1159/000486328.Suche in Google Scholar PubMed
13. Kuzmanov, A, Johansen, P, Hofbauer, G. FBXO25 promotes cutaneous squamous cell carcinoma growth and metastasis through cyclin D1. J Invest Dermatol 2020;140:2496–504. https://doi.org/10.1016/j.jid.2020.04.003.Suche in Google Scholar PubMed
14. Jiang, GY, Zhang, XP, Wang, L, Lin, XY, Yu, JH, Wang, EH, et al.. FBXO25 promotes cell proliferation, invasion, and migration of NSCLC. Tumour Biol 2016;37:14311–9. https://doi.org/10.1007/s13277-016-5298-1.Suche in Google Scholar PubMed
15. Baumann, U, Fernández-Sáiz, V, Rudelius, M, Lemeer, S, Rad, R, Knorn, AM, et al.. Disruption of the PRKCD-FBXO25-HAX-1 axis attenuates the apoptotic response and drives lymphomagenesis. Nat Med 2014;20:1401–9. https://doi.org/10.1038/nm.3740.Suche in Google Scholar PubMed
16. Frattini, V, Trifonov, V, Chan, JM, Castano, A, Lia, M, Abate, F, et al.. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 2013;45:1141–9. https://doi.org/10.1038/ng.2734.Suche in Google Scholar PubMed PubMed Central
17. Li, Y, Zhang, HB, Chen, X, Yang, X, Ye, Y, Bekaii-Saab, T, et al.. A rare EGFR-SEPT14 fusion in a patient with colorectal adenocarcinoma responding to erlotinib. Oncologist 2020;25:203–7. https://doi.org/10.1634/theoncologist.2019-0405.Suche in Google Scholar PubMed PubMed Central
18. Kim, T, Tyndel, MS, Zhang, Z, Ahn, J, Choi, S, Szardenings, M, et al.. Exome sequencing reveals DNMT3A and ASXL1 variants associate with progression of chronic myeloid leukemia after tyrosine kinase inhibitor therapy. Leuk Res 2017;59:142–8. https://doi.org/10.1016/j.leukres.2017.06.009.Suche in Google Scholar PubMed
19. Schmidt, M, Rinke, J, Schäfer, V, Schnittger, S, Kohlmann, A, Obstfelder, E, et al.. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia 2014;28:2292–9. https://doi.org/10.1038/leu.2014.272.Suche in Google Scholar PubMed
20. Kim, T, Tyndel, MS, Kim, HJ, Ahn, JS, Choi, SH, Park, HJ, et al.. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood 2017;129:38–47. https://doi.org/10.1182/blood-2016-04-708560.Suche in Google Scholar PubMed
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/oncologie-2023-0217).
© 2023 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
{kind=link}
Artikel in diesem Heft
- Frontmatter
- Review Articles
- Mitochondrial thermogenesis in cancer cells
- Application of indocyanine green in the management of oral cancer: a literature review
- Long non-coding RNA, FOXP4-AS1, acts as a novel biomarker of cancers
- The role of synthetic peptides derived from bovine lactoferricin against breast cancer cell lines: a mini-review
- Single cell RNA sequencing – a valuable tool for cancer immunotherapy: a mini review
- Research Articles
- Global patterns and temporal trends in ovarian cancer morbidity, mortality, and burden from 1990 to 2019
- The association between NRF2 transcriptional gene dysregulation and IDH mutation in Grade 4 astrocytoma
- More than just a KRAS inhibitor: DCAI abrogates the self-renewal of pancreatic cancer stem cells in vitro
- DUSP1 promotes pancreatic cancer cell proliferation and invasion by upregulating nephronectin expression
- IMMT promotes hepatocellular carcinoma formation via PI3K/AKT/mTOR pathway
- MiR-100-5p transfected MSCs-derived exosomes can suppress NSCLC progression via PI3K-AKT-mTOR
- Inhibitory function of CDK12i combined with WEE1i on castration-resistant prostate cancer cells in vitro and in vivo
- Prognostic potential of m7G-associated lncRNA signature in predicting bladder cancer response to immunotherapy and chemotherapy
- Case Reports
- A rare FBXO25–SEPT14 fusion in a patient with chronic myeloid leukemia treatment to tyrosine kinase inhibitors: a case report
- Stage I duodenal adenocarcinoma cured by a short treatment cycle of pembrolizumab: a case report
- Rapid Communication
- ROMO1 – a potential immunohistochemical prognostic marker for cancer development
- Article Commentary
- A commentary: Role of MTA1: a novel modulator reprogramming mitochondrial glucose metabolism
Artikel in diesem Heft
- Frontmatter
- Review Articles
- Mitochondrial thermogenesis in cancer cells
- Application of indocyanine green in the management of oral cancer: a literature review
- Long non-coding RNA, FOXP4-AS1, acts as a novel biomarker of cancers
- The role of synthetic peptides derived from bovine lactoferricin against breast cancer cell lines: a mini-review
- Single cell RNA sequencing – a valuable tool for cancer immunotherapy: a mini review
- Research Articles
- Global patterns and temporal trends in ovarian cancer morbidity, mortality, and burden from 1990 to 2019
- The association between NRF2 transcriptional gene dysregulation and IDH mutation in Grade 4 astrocytoma
- More than just a KRAS inhibitor: DCAI abrogates the self-renewal of pancreatic cancer stem cells in vitro
- DUSP1 promotes pancreatic cancer cell proliferation and invasion by upregulating nephronectin expression
- IMMT promotes hepatocellular carcinoma formation via PI3K/AKT/mTOR pathway
- MiR-100-5p transfected MSCs-derived exosomes can suppress NSCLC progression via PI3K-AKT-mTOR
- Inhibitory function of CDK12i combined with WEE1i on castration-resistant prostate cancer cells in vitro and in vivo
- Prognostic potential of m7G-associated lncRNA signature in predicting bladder cancer response to immunotherapy and chemotherapy
- Case Reports
- A rare FBXO25–SEPT14 fusion in a patient with chronic myeloid leukemia treatment to tyrosine kinase inhibitors: a case report
- Stage I duodenal adenocarcinoma cured by a short treatment cycle of pembrolizumab: a case report
- Rapid Communication
- ROMO1 – a potential immunohistochemical prognostic marker for cancer development
- Article Commentary
- A commentary: Role of MTA1: a novel modulator reprogramming mitochondrial glucose metabolism