More than just a KRAS inhibitor: DCAI abrogates the self-renewal of pancreatic cancer stem cells in vitro
-
Yuan Han Teh



Abstract
Objectives
Growing evidence indicates that pancreatic cancer stem cells (CSCs) contribute to cancer recurrence via chemoresistance, and their growth is sustained by self-renewal. Targeting the self-renewal of pancreatic CSCs is a crucial strategy to eradicate them. Here, we are the first to describe a known KRAS inhibitor, 4,6-dichloro-2-methyl-3-aminoethyl-indole (DCAI), as a novel anti-pancreatic CSC agent that abrogates the self-renewal of pancreatic CSCs.
Methods
Cell viability assay was used to determine the cytotoxicity of KRAS binders in pancreatic cancer cell lines with either wild-type KRAS (BxPC-3) or clinically relevant KRAS mutations (PANC-1, Capan-2, and MIA PaCa-2). The tumoursphere assay was utilised to investigate the effect of DCAI on the self-renewal of pancreatic CSCs, and its mechanism of action was examined by Western blotting.
Results
The growth of pancreatic cancer cells remains unaffected by the binding of Benzimidazole (BZIM) to both wild-type and oncogenic KRAS. DCAI and Kobe0065 were equally potent in pancreatic cancer cell lines, except for Capan-2, in which DCAI (GI50=25.8 ± 0.8 µM) was more potent than Kobe0065 (GI50=54.0 ± 1.0 µM). Capan-2 tumourspheres were markedly irresponsive to gemcitabine (IC50>100 µM), while DCAI abrogated the formation of Capan-2 tumourspheres profoundly (IC50=30 µM). Upon treatment with DCAI, CRAF, ERK1, ERK2, and AKT activations were significantly inhibited, and SOX2 expression was greatly reduced in Capan-2 tumourspheres.
Conclusions
Our present study revealed that DCAI depletes pancreatic CSCs by inhibiting self-renewal via KRAS–CRAF–ERK1/2–SOX2 and KRAS–AKT–SOX2 axes. Our findings suggested that KRAS is a valid therapeutic target in pancreatic CSCs for eradicating cancer recurrence.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer [1]. Current treatment for PDAC is limited to curative resection and adjuvant chemotherapy with gemcitabine, but the post-treatment prognosis is poor, with 80 % of patients experiencing recurrence [2]. Growing evidence has proven the presence of a residual fraction of resilient cancer cells despite a remarkable shrinkage of tumour masses under the influence of chemotherapy. These unique cancer cells, generally termed cancer stem cells (CSCs), remain dormant under the stress of chemotherapy yet revive and regenerate secondary tumours once the chemotherapy is not prescribed [3]. Therefore, it is of utmost importance to eliminate chemoresistant CSCs, which are the root cause of tumourigenesis and cancer recurrence.
Following the isolation and characterisation of pancreatic CSCs based on cell surface markers and an association of these cells with chemoresistanc [4], a few anti-pancreatic CSC agents were discovered. Vismodegib is one of the earliest known anti-pancreatic CSC agents that target Hedgehog signalling [5]. Its activity against pancreatic CSCs was proven to be promising in preclinical studies. Nevertheless, the most recent clinical study revealed that combining vismodegib and gemcitabine did not improve the prognosis of patients with metastatic PDAC [6], suggesting that the anti-pancreatic CSC activity of vismodegib is not translatable from bench to clinical settings. Therefore, it is crucial to seek an alternative to vismodegib.
Oncogenic KRAS mutations have been reported in more than 90 % of PDAC cases [7], indicating its indispensable role in pancreatic carcinogenesis and development. Mutations in the KRAS gene occur in the initial stages of PDAC, and its sustained activity plays a pivotal role in propelling the progression of the disease. The pivotal role of KRAS in promoting stem cell-like characteristics, including self-renewal, has been confirmed in pancreatic cancer cells [8]. It was reported that the KRAS-integrin β3 complex activates nuclear factor-kappa B (NF-κB) signalling, which in turn contributes to the acquisition of stem-cell characteristics, including self-renewal, by a subset of integrin β3-expressing cells in pancreatic cancer cell lines. The findings of this study warranted targeting oncogenic KRAS as one of the critical strategies to eradicate pancreatic CSCs.
KRAS demonstrates slow intrinsic nucleotide exchange and guanosine triphosphate (GTP) hydrolysis, which can be accelerated by guanine nucleotide exchange factor (GEF) and GTPase-activating protein (GAP), respectively [7]. Decades of effort in drugging KRAS have yielded numerous KRAS inhibitors [9]. Benzimidazole (BZIM; Figure 1) binds to KRAS, neither inducing an alteration in its structure nor affecting its function [10]. Although sharing a common backbone with BZIM, additional substituents in 4,6-dichloro-2-methyl-3-aminoethyl-indole (DCAI; Figure 1) molecule induce a conformational change in KRAS upon binding, which has been implicated in its inhibitory effect on KRAS function [10]. They reported that DCAI-induced conformational change in KRAS interferes with the physical interaction between KRAS and GEF without affecting the intrinsic GTPase activity of KRAS and KRAS-effector interaction [10].

Chemical structures of KRAS binders. (A) BZIM binds to KRAS without exerting an effect on KRAS function. (B) DCAI inhibits KRAS activation by interfering KRAS–GEF interaction. (C) Kobe0065 blocks the interaction between KRAS and its multiple effectors, including RAF, PI3K, and RalGDS, which in turn attenuating KRAS signalling. Chemical structures were generated by using PubChem Sketcher V2.4 (https://pubchem.ncbi.nlm.nih.gov/edit3/index.html).
Disruption of KRAS-effector interaction has been reported as the underlying mechanism of action in the inhibition of KRAS signalling by Kobe0065 (Figure 1) [11]. The protein–protein interaction between KRAS and its multiple effectors, including RAF and phosphoinositide 3-kinase (PI3K), was previously found to be effectively blocked by Kobe0065 [11]. The structural alteration of KRAS induced upon the binding of Kobe0065 is distinguishable from that induced by DCAI, which justifies the difference between DCAI and Kobe0065 in their modes of KRAS inhibition. Kobe0065 showed a notable antiproliferative effect on oncogenic KRAS-driven cancer cells in vitro and in vivo [11]. Despite demonstrating profound activity against oncogenic KRAS, none of these inhibitors have been explored for their potential as anti-pancreatic CSC agents.
In the present study, we seized the reliance of pancreatic CSCs on oncogenic KRAS for self-renewal and investigated the potential of DCAI as an anti-pancreatic CSC agent. Tumourspheres are enriched with self-renewing CSCs, and tumoursphere formation assay is an established platform for assessing the anti-CSC activity of chemical compounds in vitro [12, 13]. Using tumoursphere formation assay, we reported for the very first time that DCAI abrogates the self-renewal of pancreatic CSCs with its mechanism of action being elucidated.
Materials and methods
Cell culture
PDAC cell lines were purchased from the American Type Culture Collection (ATCC, USA). BxPC-3 (KRASwild-type) cells were propagated in RPMI-1640 Medium (Gibco, USA), while PANC-1 (KRASG12D) and MIA PaCa-2 (KRASG12C) cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco, USA) with high glucose (4500 mg/L). Capan-2 (KRASG12V) cells were cultured in McCoy’s 5A Medium (Sigma-Aldrich, USA). All basal media were supplemented with 10 % foetal bovine serum (FBS; Sigma-Aldrich, USA). Cells were incubated at 37 °C under humidified 5 % CO2.
Tumoursphere culture
Capan-2 cells were detached and dissociated with Accutase (Nacalai Tesque, Japan). Single cells were seeded in ultra-low attachment culture vessels (Corning, USA) at a density of 5 cells/µL and cultured in CSC culture medium: Dulbecco’s Modified Eagle’s Medium: Nutrient Mixture F-12 (Gibco, USA), 1 × B27 Supplement without vitamin A (Gibco, USA), 20 ng/mL epidermal growth factor (EGF; Nacalai Tesque, Japan), and 20 ng/mL essential fibroblast growth factor (bFGF; Nacalai Tesque, Japan) for seven days. On the third day, 1/10 volume of fresh CSC culture medium was added to the tumoursphere culture.
Preparation of test compounds
Gemcitabine (Gemita; Fresenius Kabi Oncology, India), vismodegib (GDC-0449; APExBIO Technology, USA), BZIM (Sigma-Aldrich, USA), DCAI (Sigma-Aldrich, USA), or Kobe0065 (Glixx Laboratories Inc, USA) was dissolved in sterile, biologically graded dimethyl sulfoxide (DMSO; Sigma-Aldrich, USA).
Cell viability assay
PDAC cells were seeded onto a 96-well plate (TPP, Switzerland) at a cell density of 2 × 103 cells per well. Cells were treated with test compounds for 96 h. Upon this, the fraction of viable cells was determined by measuring their ability in metabolising 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Gibco, USA). MTT (0.4 g/L) was added to wells, and the plates were incubated at 37 °C (humidified 5 % CO2) for 4 h. The insoluble formazan was solubilised with analytical DMSO (Fisher Scientific, USA). The absorbance (Abs) of formazan solution at the wavelength of 550 nm was measured with a microplate reader (Versa Max Microplate Reader, USA). Cell growth (in percent) was calculated by using the following formulae (1) and (2) in respect of the given conditions [14]:
When Absday0<Abstreatment,
When Absday0>Abstreatment,
Dose–response curves were plotted using the percentage of cell growth calculated from formulae (1) and (2). Growth inhibitory parameters (GI50, TGI, and LC50) were determined from the dose–response curves. Growth inhibition of 50 % (GI50) is the compound concentration resulting in a 50 % reduction in the activity of mitochondrial succinate dehydrogenase when it is compared with the control cells during the compound treatment. Referring to the activity of mitochondrial succinate dehydrogenase measured at the beginning of the compound treatment, total growth inhibition (TGI) is the compound concentration resulting in no net change. In contrast, a lethal concentration of 50 % (LC50) is the compound concentration that causes a net loss of 50 % in cells.
Tumoursphere formation assay
Seven-day-old tumoursphere culture was treated with test compounds (gemcitabine, vismodegib, and DCAI) at 30 µM for either 24 h or seven days. At the end of 24 h treatment, tumourspheres were harvested and lysed for immunoblotting analysis. In the seven-day treatment, 1/10 volume of fresh CSC culture medium containing 30 µM of test compound was added to tumuorsphere culture on the third day of treatment. At the end of treatment, multiple images were taken across the entire 96-well using the built-in camera of ZEISS microscope (ZEISS, Germany). The number of tumourspheres with a diameter of >60 µm was counted using ZEISS software (ZEISS, Germany) [15].
Protein analysis
Cells were lysed with immunoprecipitation lysis buffer: 25 mM tris–HCl (pH 7.4) (1st Base, Singapore), 150 mM sodium chloride (HmbG Chemicals, Malaysia), 1 % (v/v) Triton X-100 (Sigma, USA), 1 mM EDTA, 50 mM magnesium chloride (Billerica, USA), 2 % (v/v) glycerol (Nacalai Tesque, Japan), 1 × protease inhibitor cocktail (Nacalai Tesque, Japan), and 1 × phosphatase inhibitor cocktail (Nacalai Tesque, Japan). Cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting. The primary antibodies (Cell Signalling Technology, USA) used include p-CRAF (#9427), CRAF (#12552), p-ERK1/2 (#9106), ERK1/2 (#4696), p-AKT (#4060), AKT (#4685), p-NF-κB P65 (#3033), NF-κB P65 (#8242), p-GSK3β (#5558), GSK3β (#12456), SOX2 (#3579), and GAPDH (#5174). The HRP-linked secondary antibodies (Cell Signalling Technology, USA) used include mouse IgG (#7076) and rabbit IgG (#7074).
Statistical analysis
The results of continuous variables are expressed as mean ± standard deviation (SD). Pairwise multiple comparisons were achieved by performing One-Way ANOVA with post-hoc Tukey’s HSD test on two continuous variables with homogenous variances or the Games-Howell test otherwise. The Shapiro–Wilk test was performed to assess the normality of the continuous variables. Levene’s test evaluated the homogeneity of variances among the continuous variables. All analyses were performed with IBM SPSS Statistics 20 (IBM-SPSS Inc., USA). p values≤0.05 denote statistically significant.
Results
DCAI demonstrated greater anticancer activity against the Capan-2 cell line than Kobe0065
BZIM exerted neither cytostatic nor cytocidal effect on PDAC cell lines, as evidenced by plateauing dose–response curves and remarkably high growth inhibitory parameters, of which values fall out of the test concentration range (Figure 2). DCAI and Kobe0065 resemble one another in their anticancer activities against PDAC cell lines, except for Capan-2 cell line (Figure 2). ANOVA revealed that significant differences in GI50 values exist between groups (F7,16=178.95, p=5.53 × 10−14). Capan-2 cells were more sensitive to the growth inhibition of DCAI (GI50=25.8 ± 0.8 µM) than Kobe0065 (GI50=54.0 ± 1.0 µM), as suggested by Tukey’s HSD test (p=8.94 × 10−12). Statistically significant between-groups differences in TGI values were verified (F7,16=140.29, p=3.74 × 10−13). Noteworthy, Kobe0065 (TGI>100 µM) treatment did not pose a cytostatic effect on the Capan-2 cell line (Figure 2). The TGI value of Kobe0065 was significantly greater than DCAI (TGI=58.5 ± 7.4 µM), which induced a cytostatic effect in Capan-2 cell line (p=0.045). The replicates of cell viability assay results are shown in Supplementary Figure 2.

Growth inhibitory effect of KRAS binders on PDAC cell lines. PDAC cell lines were treated with test compounds for 96 h. The cytotoxicity of the test compounds was assessed by MTT cell viability assay. (A) Representative dose–response growth inhibitory effect of KRAS binders on PDAC cell lines. (B) Growth inhibitory parameters (GI50, TGI, and LC50) of KRAS binders in PDAC cell lines. Data are expressed in mean ± SD (n=3 independent experiments). BZIM treatment did not affect the growth and survival of PDAC cells. The responses of PDAC cell lines to DCAI and Kobe0065 treatments were found to be comparable, except for Capan-2 cell line, which showed greater sensitivity to DCAI. DCAI demonstrated equal anticancer activity against BxPC-3, PANC-1, and MIA PaCa-2 cells. Statistical significance of the differences relative to BxPC-3 cell line was analysed by Tukey’s HSD or Games-Howell test of SPSS One-Way ANOVA (ns, not significant; *p≤0.05; **p≤0.01; ***p≤0.001).
DCAI inhibited the self-renewal of Capan-2 tumourspheres
Capan-2 tumourspheres remained intact with defined spherical morphology and smooth surfaces when they were challenged with gemcitabine treatment (Figure 3A). Capan-2 tumourspheres showed noticeable shrinkage when they were treated with 30 µM vismodegib (Figure 3A). Even though appearing smaller in sizes, the morphology of the vismodegib-treated tumourspheres resembled that of DMSO-treated tumourspheres (Figure 3A). DCAI treatment resulted in phenotypic changes, including the presentation of bleb-like structures around the surfaces of the tumourspheres, which seemingly signifies their deformation (Figure 3A). Similar to vismodegib-treated tumourspheres, DCAI-treated tumourspheres appeared markedly smaller (Figure 3A).

Effect of test compounds on the self-renewal of Capan-2 tumourspheres. Capan-2 tumourspheres were cultured in serum-free and non-attached conditions for seven days, followed by seven-day treatments with DMSO vehicle control (0.03 %), gemcitabine, vismodegib, and DCAI. All test compounds were tested at 30 µM. (A) Representative microscopic images show post-treatment morphologies of Capan-2 tumourspheres. (Inset) enlarged images of selected tumourspheres (white boxes). Scale bar, 100 µm. The size and shape of Capan-2 tumourspheres were not affected by gemcitabine treatment. Although the integrity of sphere-like colonies was unaffected, the size of Capan-2 tumourspheres was greatly reduced by vismodegib. DCAI induced bleb-like structures around the tumourspheres, which seemingly signifies their deformation, in addition to inhibiting their growth. (B) Representative dose–response inhibitory effect of test compounds on Capan-2 tumourspheres. Sphere-like multicellular colonies with a diameter≥60 µm were counted as tumourspheres. Data are expressed in mean ± SD (n=3 replicates). Capan-2 tumourspheres were irresponsive to gemcitabine treatment, whereas vismodegib and DCAI inhibited the self-renewal of tumourspheres. Statistical significance of the differences relative to DMSO vehicle control was analysed by Tukey’s HSD test of SPSS One-Way ANOVA (ns, not significant; *p≤0.05; **p≤0.01; ***p≤0.001).
The self-renewal efficiency of gemcitabine-treated Capan-2 tumourspheres was consistent with their DMSO-treated counterparts, despite a 10-fold increment in concentration from 0.01 to 100 µM (Figure 3B). This observation was confirmed by ANOVA (F5,12=1.76, p=0.195). Vismodegib, a known anti-pancreatic CSC agent, attenuated the self-renewal efficiency of Capan-2 tumourspheres at IC50=35 µM, while DCAI exerted comparable strength of inhibition at IC50=30 µM (Figure 3B). ANOVA reported significant differences between various concentrations of vismodegib or DCAI and their respective DMSO counterparts (F7,16=12.43, p=2.10 × 10−5), which is indicative of an inhibition of self-renewal by these compounds. Post-hoc comparison using Tukey’s HSD test revealed no significant difference between the anti-self-renewal activities of 10 µM vismodegib and 15 µM DCAI (p=0.943). Similarly, the anti-self-renewal activities of 50 µM vismodegib and 45 µM DCAI were not significantly different (p=0.999). The replicates of tumoursphere formation assay results are shown in Supplementary Figure 3.
DCAI deactivated MAPK and PI3K-AKT pathways and reduced SOX2 expression in self-renewing Capan-2 tumourspheres
Treatments with test compounds resulted in significant alterations in the activation levels of CRAF (F3,8=5.32, p=0.03), ERK1 (F3,8=18.89, p=5.47 × 10−4), and ERK2 (F3,8=10.19, p=0.004). Specifically, DCAI deactivated CRAF (p=0.02), ERK1 (p=0.03), and ERK2 (p=0.03), indicating that MAPK signalling was diminished in Capan-2 tumourspheres upon treatment with DCAI (Figure 4).

Molecular mechanism underlying the inhibition of self-renewing Capan-2 tumourspheres by test compounds. Capan-2 tumourspheres were cultured in serum-free and non-attached conditions for seven days, followed by 24-h treatments with DMSO vehicle control (0.03 %), gemcitabine, vismodegib, and DCAI. All test compounds were tested at 30 µM. (A) Representative immunoblot shows the molecular alterations occurred in tumourspheres in response to treatments, including the activations of CRAF, ERK1, ERK2, AKT, and P65, the inhibition of GSK3β, and the expression of SOX2. The activation levels of CRAF, ERK1, ERK2, AKT, and P65 were quantified by normalising phosphoproteins to their respective total proteins, whereas normalising inhibitory phosphoprotein of GSK3β to its total protein evaluates the inhibition level. SOX2 expression level was quantified by normalising total SOX2 protein to GAPDH. GAPDH is a housekeeping protein serving as loading control. (B) Bar chart shows the quantitative analysis of the immunoblot. Data are expressed in mean ± SD (n=3 independent experiments). No significant molecular alteration was observed in gemcitabine-treated and vismodegib-treated tumourspheres. DCAI remarkably inhibited CRAF, ERK1, ERK2, and AKT activations and reduced SOX2 expression. Statistical significance of the differences relative to DMSO vehicle control was analysed by Tukey’s HSD or Games-Howell test of SPSS One-Way ANOVA (ns, not significant; *p≤0.05; **p≤0.01).
ANOVA suggested that the AKT activation level of Capan-2 tumourspheres, as indicated by its phosphorylation at Serine-473, was significantly altered by treatments with test compounds (F3,8=6.80, p=0.01). Compared to DMSO vehicle control (0.03 %), 30 µM DCAI remarkably inhibited AKT activation in Capan-2 tumourspheres (0.14 ± 0.05 fold, p=0.003; Figure 4). SOX2 expression of Capan-2 tumourspheres was considerably affected by treatments with test compounds, as revealed by ANOVA (F3,8=7.71, p=0.01). The post-hoc comparison revealed that DCAI significantly reduced SOX2 expression level relative to DMSO vehicle control (0.16 ± 0.13 fold, p=0.02; Figure 4).
The inhibitions of the P65 subunit of NF-κB (F3,8=0.77, p=0.54) and glycogen synthase kinase-3β (GSK3β) of WNT signalling (F3,8=3.33, p=0.08) by test compounds were not statistically significant as compared with DMSO vehicle control. The replicates of protein analyses are shown in Supplementary Figure 4.
Discussion
CSCs can increase in the absence of serum and an anchorage to a surface with supplementation of EGF and bFGF. In these unique culture conditions, self-renewing CSCs propagate as spherical multicellular colonies known as tumourspheres in vitro [16]. This hallmark characteristic of CSCs has been observed in a vast spectrum of cancers, including PDAC [4, 17], [18], [19], [20], [21]. Tumoursphere formation assay is renowned for reliably assessing the responses of CSCs to chemical compounds, which are claimed to possess anti-self-renewal properties [12, 13].
In the present study, we were intrigued to find that TGI was attained in DCAI-treated Capan-2 cells at 58 µM. In contrast, TGI was not achievable even when these exceptionally chemoresistant cells were challenged with gemcitabine at a concentration as high as 100 µM (Supplementary Figure 1). Gemcitabine is the gold-standard chemotherapeutic drug used in the treatment of PDAC. Patients usually respond well to gemcitabine treatment [22]. Yet, they often lose the battle with recurrent PDAC when secondary tumours arise from residual CSCs, which have been proven resistant to gemcitabine [2, 3]. Our findings suggested that DCAI effectively eliminates the chemoresistant fraction of the Capan-2 cell line, which is highly likely to be constituted by CSCs.
Profound chemoresistance of the Capan-2 cell line coincides with its tumoursphere-forming ability, suggesting the presence of CSCs in the cell line. In our study, the self-renewal ability of Capan-2 tumourspheres was not significantly suppressed by gemcitabine treatment, even at a concentration as high as 100 µM. Upon gemcitabine treatment, the integrity of Capan-2 tumourspheres was well preserved. Once again, our observations aligned with growing evidence that chemoresistance is an associative feature of CSCs [23]. Most importantly, it is proven that Capan-2 tumourspheres served as a valid test subject for the present study. By employing Capan-2 tumourspheres as the test subject, we discovered a novel role of DCAI as an inhibitor of CSC self-renewal and its existing role as a KRAS inhibitor [10, 24].
Vismodegib was reported to inhibit self-renewal and induce apoptosis in pancreatic CSCs by suppressing Hedgehog signalling [5]. We observed that vismodegib treatment reduced the sizes of Capan-2 tumourspheres, which indicates impaired self-renewal in the tumourspheres. Our observation was consistent with the anti-pancreatic CSC property of vismodegib reported by Singh et al. [5] DCAI abrogated the self-renewal of Capan-2 tumourspheres to a degree similar to vismodegib. Our novel finding strongly suggested that DCAI possesses anti-self-renewal properties and has great potential to further develop into an anti-pancreatic CSC agent.
It is not surprising to find anti-self-renewal properties in DCAI, as KRAS signalling is associated with regulating the self-renewal of pancreatic CSCs [25]. This groundbreaking study by Seguin et al. [25] provided us with insight into exploring the possibility of repurposing KRAS inhibitors to develop potent anti-pancreatic CSC drugs. The two most well-studied KRAS-driven pathways, mitogen-activated protein kinase (MAPK) and PI3K-AKT are implicated in maintaining the pluripotency of human embryonic stem cells (hESCs) [26], and the ability of self-renewing in pancreatic CSCs [27, 28]. In the present study, we elucidated the mechanism of action underlying the anti-self-renewal activity of DCAI. As expected, DCAI markedly attenuated MAPK and AKT pathways in Capan-2 tumourspheres.
Ectopic expressions of four embryonic transcription factors, namely SRY-box Transcription Factor 2 (SOX2), Kruppel-like Factor 4 (KLF4), Cellular Myelocytomatosis Oncogene (CMYC), and Octamer-binding Protein 4 (OCT4), collectively known as Yamanaka factors, in mammalian somatic cells were evidenced to have driven the dedifferentiation of these cells into pluripotent stem cells [29]. Ever since the groundbreaking introduction of Yamanaka factors as the key inducers of stem-cell pluripotency, the expressions of these pluripotency factors become one of the hallmark features of pancreatic CSCs [3, 30]. Among these pluripotency factors, SOX2 is of greater interest because its expression alone was found to induce and sustain stem-cell characteristics, including self-renewal, in pancreatic cancer cells [31]. Consistent with this finding, our study shows that SOX2 expression was significantly upregulated in Capan-2 tumourspheres, presumably of pancreatic CSCs (Supplementary Figure 2). Interestingly, the expression levels of the other dedifferentiation markers, namely KLF4, CMYC, and OCT4, were not found to be upregulated in Capan-2 tumourspheres (Supplementary Figure 2), suggesting a passive role of these markers in the self-renewal of pancreatic CSCs as reported by precedent [32], [33], [34], [35]. In the present study, a reduction of SOX2 expression level was observed in association with DCAI-induced inactivations of MAPK and AKT pathways. Based on this observation, DCAI could potentially solve pancreatic cancer recurrence by targeting the self-renewal of pancreatic CSCs.
Sun et al. [36] reported that the NF-κB signalling pathway is activated in pancreatic CSCs and associated with the acquisition of invasive and tumourigenic properties. In their study, SOX9, a stem cell-specific transcription factor known to be an essential regulator of self-renewal and differentiation of intestinal and neural stem cells, was identified as a novel target gene of NF-κB signalling. SOX9 expression is regulated by NF-κB signalling through direct binding of NF-κB P65 subunit at its promoter region, which leads to recruiting a demethylating protein complex to facilitate SOX9 gene expression [32]. WNT signalling is well known for its pivotal role in positively regulating normal stem cells’ self-renewal, including hematopoieticand neural stem cells [37, 38]. The indispensable role of this pathway in the self-renewal of pancreatic CSCs was also revealed by several studies [39, 40].
Previous studies have confirmed a direct involvement of MAPK and PI3K-AKT pathways in activating NF-κB signalling [41, 42], whereas a direct inhibition of GSK3β activity by MAPK and PI3K-AKT pathways was also reported [43]. GSK3β inactivates WNT signalling by phosphorylating β-catenin at Serine-33 and Serine-37 residues for ubiquitin-mediated protein degradation [44, 45]. Phosphorylation of GSK3β at its Serine-9 residue inhibits its function, following which WNT signalling is activated [43], [44], [45]. Therefore, we hypothesised that P65 activity would be reduced and GSK3β activity would be increased following DCAI-induced deactivations of MAPK and PI3K-AKT pathways.
Nonetheless, our study showed that DCAI treatment did not affect P65 and GSK3β activities. We postulated that DCAI would affect P65 and GSK3β activities beyond 24 h post-treatment, considering that P65 and GSK3β activities were regulated downstream of MAPK and PI3K-AKT pathways. The resultant effects from inhibiting MAPK and PI3K-AKT pathways may require a longer time to effectively modulate downstream pathways. The mechanism of action underlying the depletion of self-renewing Capan-2 tumourspheres by DCAI is illustrated in Figure 5. Even though our study has shown that DCAI may potentially be developed into an inhibitor of CSC self-renewal, which has not been reported elsewhere, the mechanism of action depicted in Figure 5 is speculative at best. Moreover, we have not verified anti-CSC in vivo.
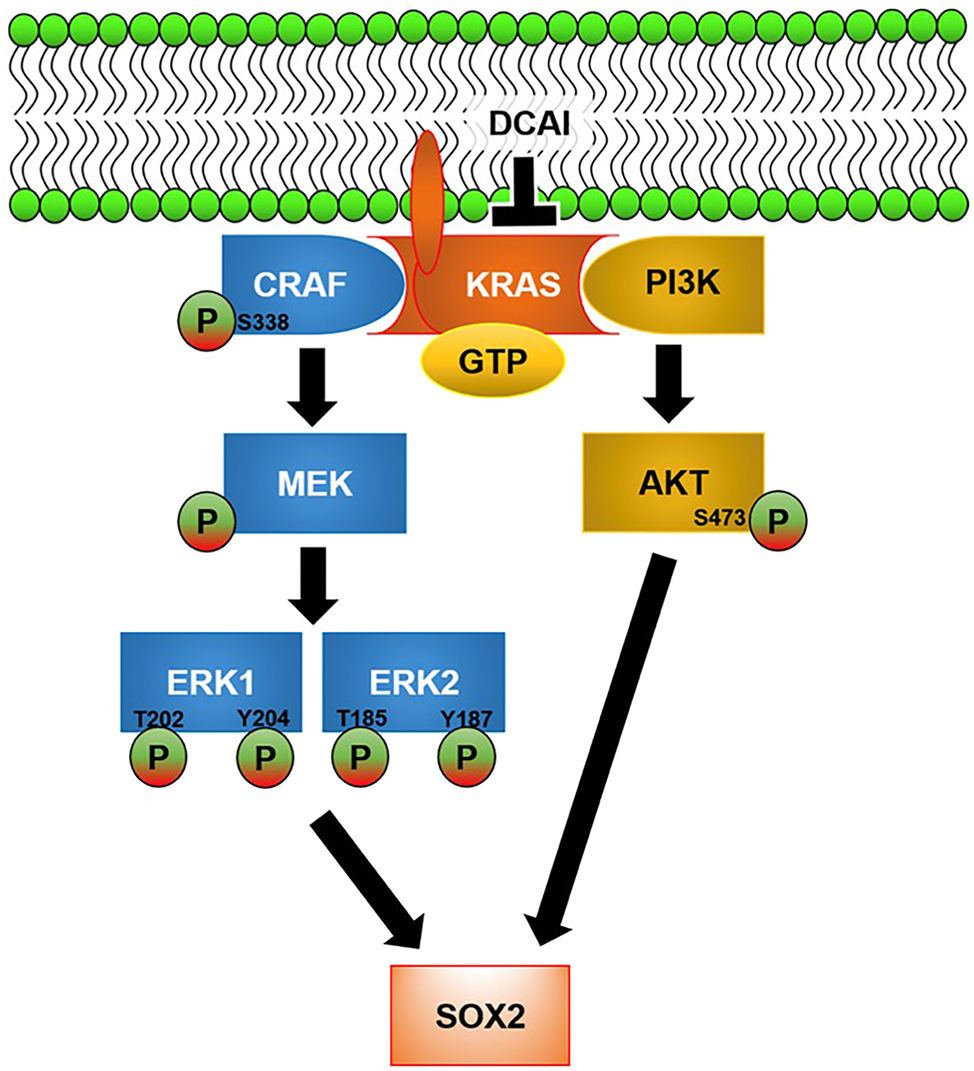
Proposed mechanism of action underlying the depletion of self-renewing Capan-2 tumourspheres by DCAI. DCAI abrogates GTP loading of KRAS and its activation by blocking KRAS–GEF interaction. The inhibitory effect of DCAI on KRAS signalling is transmitted to MAPK and PI3K-AKT pathways, as evidenced by reduced phosphorylations of CRAF (Serine-338), ERK1 (Threonine-202, Tyrosine-204), ERK2 (Threonine-185, Tyrosine-187), and AKT (Serine-473). SOX2 expression is attenuated by DCAI-induced inactivation of MAPK and PI3K-AKT pathways.
Our study revealed that DCAI and vismodegib abrogated the self-renewal of pancreatic CSCs through distinguishable mechanisms of action. DCAI induced the formation of bleb-like structures around the surfaces of tumourspheres, which is an apparent sign of deformation. At the same time, vismodegib reduced the sizes of tumourspheres without noticeable morphological changes. Whereas DCAI targets KRAS–CRAF–ERK1/2–SOX2 and KRAS–AKT–SOX2 signalling axes, vismodegib inhibits Hedgehog signalling. While a recent study showed that vismodegib did not work as promisingly as at the bench in human subjects, we firmly believe that DCAI is an on-time alternative to vismodegib.
DCAI has not been explored for its potential in any disease model since its discovery as a KRAS inhibitor. This compound’s pharmacokinetic and pharmacodynamic aspects have not been investigated in depth. Therefore, the first step to bringing DCAI from the bench to the bedside is to determine how much dosage an individual could tolerate DCAI, beyond which systemic toxicity could happen. It is critical to know the bioavailability of DCAI at a given dose, which measures how much this compound is absorbed bodily and distributed to the target site, where an outgrowth of tumour mass is located and exerts the desired effect. These measures are necessary for effectively dosing the patients while minimising the adverse effects.
Conclusions
The present study has shed some light on the novel therapeutic role of DCAI as an anti-pancreatic CSC agent. Its anti-pancreatic CSC activity warrants further in-depth investigation and development to maximise its potential as a treatment for pancreatic cancer recurrence.
Funding source: Fundamental Research Grant Scheme Grant
Award Identifier / Grant number: 04-02-13-1324FR
-
Research ethics: This is an in vitro study. As such, ethics approval from the local Institutional Review Board is deemed unnecessary.
-
Informed consent: Not applicable.
-
Author contributions: Johnson Stanslas contributed to the funding acquisition, study conception and design, and supervision. Yuan Han Teh performed material preparation, data collection, and analysis. Rajesh Ramasamy, Kok Lian Ho, and Sreenivasa Rao Sagineedu were involved in supervision and mentorship. Yuan Han Teh and Rui Jing wrote the first draft of the manuscript, and all authors commented on previous versions. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Competing interests: The authors state no conflict of interest.
-
Research funding: This work was supported by the Ministry of Education, Malaysia, through the Fundamental Research Grant Scheme Grant (04-02-13-1324FR).
-
Data availability: The authors confirm that the data supporting the findings of this study are available within the article [and/or its supplementary materials].
References
1. Taherian, M, Wang, H, Wang, H. Pancreatic ductal adenocarcinoma: molecular pathology and predictive biomarkers. Cells 2022;11:3068. https://doi.org/10.3390/cells11193068.Suche in Google Scholar PubMed PubMed Central
2. Gbolahan, OB, Tong, Y, Sehdev, A, O’Neil, B, Shahda, S. Overall survival of patients with recurrent pancreatic cancer treated with systemic therapy: a retrospective study. BMC Cancer 2019;19:468. https://doi.org/10.1186/s12885-019-5630-4.Suche in Google Scholar PubMed PubMed Central
3. Perelló-Reus, CM, Rubio-Tomás, T, Cisneros-Barroso, E, Ibargüen-González, L, Segura-Sampedro, JJ, Morales-Soriano, R, et al.. Challenges in precision medicine in pancreatic cancer: a focus in cancer stem cells and microbiota. Front Oncol 2022;12:995357.10.3389/fonc.2022.995357Suche in Google Scholar PubMed PubMed Central
4. Sumbly, V, Landry, I. Understanding pancreatic cancer stem cells and their role in carcinogenesis: a narrative review. Stem Cel Invest 2022;9:1. https://doi.org/10.21037/sci-2021-067.Suche in Google Scholar PubMed PubMed Central
5. Singh, BN, Fu, J, Srivastava, RK, Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS One 2011;6:e27306. https://doi.org/10.1371/journal.pone.0027306.Suche in Google Scholar PubMed PubMed Central
6. De Jesus-Acosta, A, Sugar, EA, O’Dwyer, PJ, Ramanathan, RK, Von Hoff, DD, Rasheed, Z, et al.. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br J Cancer 2020;122:498–505. https://doi.org/10.1038/s41416-019-0683-3.Suche in Google Scholar PubMed PubMed Central
7. Bannoura, SF, Uddin, MH, Nagasaka, M, Fazili, F, Al-Hallak, MN, Philip, PA, et al.. Targeting KRAS in pancreatic cancer: new drugs on the horizon. Cancer Metastasis Rev 2021;40:819–35. https://doi.org/10.1007/s10555-021-09990-2.Suche in Google Scholar PubMed PubMed Central
8. Philip, PA, Azar, I, Xiu, J, Hall, MJ, Hendifar, AE, Lou, E, et al.. Molecular characterization of KRAS wild-type tumors in patients with pancreatic adenocarcinoma. Clin Cancer Res 2022;28:2704–14. https://doi.org/10.1158/1078-0432.ccr-21-3581.Suche in Google Scholar
9. Quah, SY, Tan, MS, Teh, YH, Stanslas, J. Pharmacological modulation of oncogenic Ras by natural products and their derivatives: renewed hope in the discovery of novel anti-Ras drugs. Pharmacol Ther 2016;162:35–57. https://doi.org/10.1016/j.pharmthera.2016.03.010.Suche in Google Scholar PubMed
10. Maurer, T, Garrenton, LS, Oh, A, Pitts, K, Anderson, DJ, Skelton, NJ, et al.. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A 2012;192:5299–304. https://doi.org/10.1073/pnas.1116510109.Suche in Google Scholar PubMed PubMed Central
11. Shima, F, Yoshikawa, Y, Ye, M, Araki, M, Matsumoto, S, Liao, J, et al.. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci U S A 2013;110:8182–7. https://doi.org/10.1073/pnas.1217730110.Suche in Google Scholar PubMed PubMed Central
12. Lee, CH, Yu, CC, Wang, BY, Chang, WW. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016;7:1215–26. https://doi.org/10.18632/oncotarget.6261.Suche in Google Scholar PubMed PubMed Central
13. Nallasamy, P, Nimmakayala, RK, Karmakar, S, Leon, F, Seshacharyulu, P, Lakshmanan, I, et al.. Pancreatic tumor microenvironment factor promotes cancer stemness via SPP1-CD44 Axis. Gastroenterology 2021;161:1998–2013.e7. https://doi.org/10.1053/j.gastro.2021.08.023.Suche in Google Scholar PubMed PubMed Central
14. Jada, SR, Matthews, C, Saad, MS, Hamzah, AS, Lajis, NH, Stevens, MF, et al.. Benzylidene derivatives of andrographolide inhibit growth of breast and colon cancer cells in vitro by inducing G(1) arrest and apoptosis. Br J Pharmacol 2008;155:641–54. https://doi.org/10.1038/bjp.2008.368.Suche in Google Scholar PubMed PubMed Central
15. Singh, JK, Farnie, G, Bundred, NJ, Simões, BM, Shergill, A, Landberg, G, et al.. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin Cancer Res 2013;19:643–56. https://doi.org/10.1158/1078-0432.ccr-12-1063.Suche in Google Scholar
16. Pastrana, E, Silva-Vargas, V, Doetsch, F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011;8:486–98. https://doi.org/10.1016/j.stem.2011.04.007.Suche in Google Scholar PubMed PubMed Central
17. Li, N, Li, Y, Zheng, P, Zhan, X. Cancer stemness-based prognostic immune-related gene signatures in lung adenocarcinoma and lung squamous cell carcinoma. Front Endocrinol 2021;12:755805. https://doi.org/10.3389/fendo.2021.755805.Suche in Google Scholar PubMed PubMed Central
18. Li, WJ, Liu, X, Dougherty, EM, Tang, DG. MicroRNA-34a, prostate cancer stem cells, and therapeutic development. Cancers 2022;14:4538. https://doi.org/10.3390/cancers14184538.Suche in Google Scholar PubMed PubMed Central
19. Chen, W, Wei, W, Yu, L, Ye, Z, Huang, F, Zhang, L, et al.. Mammary development and breast cancer: a notch perspective. J Mammary Gland Biol Neoplasia 2021;26:309–20. https://doi.org/10.1007/s10911-021-09496-1.Suche in Google Scholar PubMed PubMed Central
20. Sarabia-Sánchez, MA, Moreno-Londoño, AP, Castañeda-Patlán, MC, Alvarado-Ortiz, E, Martínez-Morales, JC, Robles-Flores, M. Non-canonical Wnt/Ca2+ signaling is essential to promote self-renewal and proliferation in colon cancer stem cells. Front Oncol 2023;13:1121787.10.3389/fonc.2023.1121787Suche in Google Scholar PubMed PubMed Central
21. Lah, TT, Novak, M, Breznik, B. Brain malignancies: glioblastoma and brain metastases. Semin Cancer Biol 2020;60:262–73. https://doi.org/10.1016/j.semcancer.2019.10.010.Suche in Google Scholar PubMed
22. Zeng, S, Pöttler, M, Lan, B, Grützmann, R, Pilarsky, C, Yang, H. Chemoresistance in pancreatic cancer. Int J Mol Sci 2019;20:4504. https://doi.org/10.3390/ijms20184504.Suche in Google Scholar PubMed PubMed Central
23. Parejo-Alonso, B, Royo-García, A, Espiau-Romera, P, Courtois, S, Curiel-García Á, Zagorac, S, et al.. Pharmacological targeting of the receptor ALK inhibits tumorigenicity and overcomes chemoresistance in pancreatic ductal adenocarcinoma. Biomed Pharmacother 2023;158:114162. https://doi.org/10.1016/j.biopha.2022.114162.Suche in Google Scholar PubMed
24. Donohue, E, Khorsand, S, Mercado, G, Varney, KM, Wilder, PT, Yu, W, et al.. Second harmonic generation detection of Ras conformational changes and discovery of a small molecule binder. Proc Natl Acad Sci U S A 2019;116:17290–7. https://doi.org/10.1073/pnas.1905516116.Suche in Google Scholar PubMed PubMed Central
25. Seguin, L, Kato, S, Franovic, A, Camargo, MF, Lesperance, J, Elliott, KC, et al.. An Integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol 2014;16:457–68. https://doi.org/10.1038/ncb2953.Suche in Google Scholar PubMed PubMed Central
26. Mossahebi-Mohammadi, M, Quan, M, Zhang, JS, Li, X. FGF signaling pathway: a key regulator of stem cell pluripotency. Front Cell Dev Biol 2020;8:79. https://doi.org/10.3389/fcell.2020.00079.Suche in Google Scholar PubMed PubMed Central
27. Ischenko, I, Petrenko, O, Hayman, MJ. Analysis of the tumor-initiating and metastatic capacity of PDX1-positive cells from the adult pancreas. Proc Natl Acad Sci U S A 2014;111:3466–71. https://doi.org/10.1073/pnas.1319911111.Suche in Google Scholar PubMed PubMed Central
28. Matsubara, S, Ding, Q, Miyazaki, Y, Kuwahata, T, Tsukasa, K, Takao, S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep 2013;3:3230. https://doi.org/10.1038/srep03230.Suche in Google Scholar PubMed PubMed Central
29. Patel, I, Parchem, RJ. Regulation of Oct4 in stem cells and neural crest cells. Birth Defects Res 2022;114:983–1002. https://doi.org/10.1002/bdr2.2007.Suche in Google Scholar PubMed PubMed Central
30. Müller, M, Hermann, PC, Liebau, S, Weidgang, C, Seufferlein, T, Kleger, A, et al.. The role of pluripotency factors to drive stemness in gastrointestinal cancer. Stem Cell Res 2016;16:349–57. https://doi.org/10.1016/j.scr.2016.02.005.Suche in Google Scholar PubMed
31. Sharma, NS, Gupta, VK, Dauer, P, Kesh, K, Hadad, R, Giri, B, et al.. O-GlcNAc modification of Sox2 regulates self-renewal in pancreatic cancer by promoting its stability. Theranostics 2019;9:3410–24. https://doi.org/10.7150/thno.32615.Suche in Google Scholar PubMed PubMed Central
32. Yan, Y, Li, Z, Kong, X, Jia, Z, Zuo, X, Gagea, M, et al.. KLF4-Mediated suppression of CD44 signaling negatively impacts pancreatic cancer stemness and metastasis. Cancer Res 2016;76:2419–31. https://doi.org/10.1158/0008-5472.can-15-1691.Suche in Google Scholar PubMed PubMed Central
33. Lin, WC, Rajbhandari, N, Liu, C, Sakamoto, K, Zhang, Q, Triplett, AA, et al.. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res 2013;73:1821–30. https://doi.org/10.1158/0008-5472.can-12-2067.Suche in Google Scholar
34. Sancho, P, Burgos-Ramos, E, Tavera, A, Bou Kheir, T, Jagust, P, Schoenhals, M, et al.. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab 2015;22:590–605. https://doi.org/10.1016/j.cmet.2015.08.015.Suche in Google Scholar PubMed
35. Herreros-Villanueva, M, Zhang, JS, Koenig, A, Abel, EV, Smyrk, TC, Bamlet, WR, et al.. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2013;2:e61. https://doi.org/10.1038/oncsis.2013.23.Suche in Google Scholar PubMed PubMed Central
36. Sun, L, Mathews, LA, Cabarcas, SM, Zhang, X, Yang, A, Zhang, Y, et al.. Epigenetic regulation of SOX9 by the NF-κB signaling pathway in pancreatic cancer stem cells. Stem Cell 2013;31:1454–66. https://doi.org/10.1002/stem.1394.Suche in Google Scholar PubMed PubMed Central
37. Katoh, M, Katoh, M. WNT signaling and cancer stemness. Essays Biochem 2022;66:319–31. https://doi.org/10.1042/ebc20220016.Suche in Google Scholar PubMed PubMed Central
38. Wen, X, Wu, Y, Awadasseid, A, Tanaka, Y, Zhang, W. New advances in canonical Wnt/β-catenin signaling in cancer. Cancer Manag Res 2020;12:6987–98. https://doi.org/10.2147/cmar.s258645.Suche in Google Scholar PubMed PubMed Central
39. Zhang, Z, Xu, Y, Zhao, C. Fzd7/Wnt7b signaling contributes to stemness and chemoresistance in pancreatic cancer. Cancer Med 2021;10:3332–45. https://doi.org/10.1002/cam4.3819.Suche in Google Scholar PubMed PubMed Central
40. Zhang, Z, Xu, Y. FZD7 accelerates hepatic metastases in pancreatic cancer by strengthening EMT and stemness associated with TGF-β/SMAD3 signaling. Mol Med 2022;28:82. https://doi.org/10.1186/s10020-022-00509-1.Suche in Google Scholar PubMed PubMed Central
41. Liu, R, Chen, Y, Liu, G, Li, C, Song, Y, Cao, Z, et al.. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis 2020;11:797. https://doi.org/10.1038/s41419-020-02998-6.Suche in Google Scholar PubMed PubMed Central
42. Yu, H, Lin, L, Zhang, Z, Zhang, H, Hu, H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Targeted Ther 2020;5:209. https://doi.org/10.1038/s41392-020-00312-6.Suche in Google Scholar PubMed PubMed Central
43. Sutherland, C, Leighton, IA, Cohen, P Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 1993;296:15–9. https://doi.org/10.1042/bj2960015.Suche in Google Scholar PubMed PubMed Central
44. Wang, B, Wang, T, Zhu, H, Yan, R, Li, X, Zhang, C, et al.. Neddylation is essential for β-catenin degradation in Wnt signaling pathway. Cell Rep 2022;38:110538. https://doi.org/10.1016/j.celrep.2022.110538.Suche in Google Scholar PubMed
45. Lorzadeh, S, Kohan, L, Ghavami, S, Azarpira, N. Autophagy and the Wnt signaling pathway: a focus on Wnt/β-catenin signaling. Biochim Biophys Acta, Mol Cell Res 2021;1868:118926. https://doi.org/10.1016/j.bbamcr.2020.118926.Suche in Google Scholar PubMed
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/oncologie-2023-0214).
© 2023 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- Review Articles
- Mitochondrial thermogenesis in cancer cells
- Application of indocyanine green in the management of oral cancer: a literature review
- Long non-coding RNA, FOXP4-AS1, acts as a novel biomarker of cancers
- The role of synthetic peptides derived from bovine lactoferricin against breast cancer cell lines: a mini-review
- Single cell RNA sequencing – a valuable tool for cancer immunotherapy: a mini review
- Research Articles
- Global patterns and temporal trends in ovarian cancer morbidity, mortality, and burden from 1990 to 2019
- The association between NRF2 transcriptional gene dysregulation and IDH mutation in Grade 4 astrocytoma
- More than just a KRAS inhibitor: DCAI abrogates the self-renewal of pancreatic cancer stem cells in vitro
- DUSP1 promotes pancreatic cancer cell proliferation and invasion by upregulating nephronectin expression
- IMMT promotes hepatocellular carcinoma formation via PI3K/AKT/mTOR pathway
- MiR-100-5p transfected MSCs-derived exosomes can suppress NSCLC progression via PI3K-AKT-mTOR
- Inhibitory function of CDK12i combined with WEE1i on castration-resistant prostate cancer cells in vitro and in vivo
- Prognostic potential of m7G-associated lncRNA signature in predicting bladder cancer response to immunotherapy and chemotherapy
- Case Reports
- A rare FBXO25–SEPT14 fusion in a patient with chronic myeloid leukemia treatment to tyrosine kinase inhibitors: a case report
- Stage I duodenal adenocarcinoma cured by a short treatment cycle of pembrolizumab: a case report
- Rapid Communication
- ROMO1 – a potential immunohistochemical prognostic marker for cancer development
- Article Commentary
- A commentary: Role of MTA1: a novel modulator reprogramming mitochondrial glucose metabolism
Artikel in diesem Heft
- Frontmatter
- Review Articles
- Mitochondrial thermogenesis in cancer cells
- Application of indocyanine green in the management of oral cancer: a literature review
- Long non-coding RNA, FOXP4-AS1, acts as a novel biomarker of cancers
- The role of synthetic peptides derived from bovine lactoferricin against breast cancer cell lines: a mini-review
- Single cell RNA sequencing – a valuable tool for cancer immunotherapy: a mini review
- Research Articles
- Global patterns and temporal trends in ovarian cancer morbidity, mortality, and burden from 1990 to 2019
- The association between NRF2 transcriptional gene dysregulation and IDH mutation in Grade 4 astrocytoma
- More than just a KRAS inhibitor: DCAI abrogates the self-renewal of pancreatic cancer stem cells in vitro
- DUSP1 promotes pancreatic cancer cell proliferation and invasion by upregulating nephronectin expression
- IMMT promotes hepatocellular carcinoma formation via PI3K/AKT/mTOR pathway
- MiR-100-5p transfected MSCs-derived exosomes can suppress NSCLC progression via PI3K-AKT-mTOR
- Inhibitory function of CDK12i combined with WEE1i on castration-resistant prostate cancer cells in vitro and in vivo
- Prognostic potential of m7G-associated lncRNA signature in predicting bladder cancer response to immunotherapy and chemotherapy
- Case Reports
- A rare FBXO25–SEPT14 fusion in a patient with chronic myeloid leukemia treatment to tyrosine kinase inhibitors: a case report
- Stage I duodenal adenocarcinoma cured by a short treatment cycle of pembrolizumab: a case report
- Rapid Communication
- ROMO1 – a potential immunohistochemical prognostic marker for cancer development
- Article Commentary
- A commentary: Role of MTA1: a novel modulator reprogramming mitochondrial glucose metabolism