Advancing Charcot-Marie-Tooth disease diagnostics, through the UK 100,000 Genomes Project
-
Menelaos Pipis


Abstract
Whole genome sequencing (WGS) is regarded by many as the pinnacle of contemporary molecular genetic testing, and has only been possible because of the rapid development and roll-out of next-generation sequencing technologies. It provides a phenotype-agnostic analysis of the genome and has important advantages compared to other techniques including a consistent coverage across the coding and non-coding genome, the application of high resolution homozygosity mapping and the ability to detect and highlight structural variation.
Realising this potential and with a bid to sequence 100,000 genomes, the UK rolled out the 100,000 Genomes Project as a proof of concept of integrating genomics in the national health service. Participants with cancer and rare diseases enrolled in the project whose infrastructure comprises of a central national biorepository and 13 regional genomic medicine centres where clinicians, geneticists and other scientists work as part of a multidisciplinary team. Amongst participants are also patients with genetically unclassified Charcot-Marie-Tooth disease who have benefited substantially from improved diagnostic rates and many more stand to benefit as the analysis of genomic data is ongoing.
WGS is an important tool as we head towards more personalised medicine and in our quest to improve public health and treat and where possible prevent disease.
The completion of the Human Genome Project at the start of the century was a major breakthrough in our efforts to better understand human health and disease, but it was not until the advent and general roll-out of next-generation sequencing (NGS) a few years later that the genomic era truly began. Genomics is pivotal in the personalisation of healthcare as well as prediction and prevention of disease with applications ranging from diagnostics and improving patient management in inherited diseases and molecular oncology, to pharmacogenomics and microbe sequencing and resistance identification.
Unlocking the potential of genomic sequencing carries important benefits for patient care and in 2012, the UK Government announced funding for 100,000 genomes to undergo whole genome sequencing (WGS). Cancer (with enrolled cases having paired sequencing of germline and somatic samples) and rare diseases (germline sample sequencing) were selected as the disciplines that would most benefit initially from WGS. Approximately 8,000 individuals, both patients and relatives, with neurological and neurodegenerative diseases were expected to enrol in the 100,000 Genomes Project (100KGP), via the Neurology Genomics Clinical Interpretation Partnership (GeCIP).

Workflow of the 100KGP and its integration into routine clinical care. The patient is initially consented and enrolled in the 100KGP through the local GMC, which is also responsible for the DNA extraction and QC. The quality-passed DNA is sent to the central national biorepository for WGS and storage. The NGS-derived data are fed through the bioinformatics pipeline for processing, calling and annotating variants before being returned to the local GMC. An MDT meeting consisting of clinicians and geneticists with expertise in CMT and related disorders review each individual case’s phenotype, apply the relevant WGS virtual gene panels to the returned variants and review the candidate variants. Any positive or negative findings are then returned to the patient during a clinical visit. Abbreviations: GMC, genomic medicine centre; QC, quality control; WGS, whole genome sequencing; NGS, next-generation sequencing; MDT, multidisciplinary team; CMT, Charcot-Marie-Tooth disease.
The workflow of the 100KGP from the point of patient consent and DNA collection through to the return of clinically significant results to each patient was designed as a proof of concept for integrating WGS into routine clinical care (Fig. 1). In the UK, this is in the form of a National Health Service (NHS) Genomic Medicine Service which has a centralised provision for WGS and the related bioinformatics analysis, whereas the downstream variant interpretation and clinical reporting occurs locally through 13 regional genomic medicine centres (GMCs) acting as hubs throughout the country [1].
Neuromuscular diseases, which include Charcot-Marie-Tooth disease (CMT) and related disorders, is one of most heavily recruited categories in the Neurology GeCIP and the rare disease programme in general. Participants with CMT (probands and relatives) have been recruited in a mixture of singletons, duos, classic trios and a few families with multiple affected family members. A breakdown of the clinical phenotype of 290 CMT kindreds enrolled from our centre reveals that the majority have CMT2 (56 %), followed by complex neuropathy syndromes (17 %), hereditary motor neuropathy (13 %), hereditary sensory neuropathy (10 %) and CMT1 (4 %). Even though the rare disease programme anticipated an initial average diagnostic rate of 22 % [1], the observed diagnostic rate in CMT and related disorders has been 12–15 %. Obvious reasons for this perceived low rate are the extensive pretesting undertaken in many cases that have been reviewed in specialist peripheral neuropathy clinics prior to enrolment in the 100KGP (with genetic tests that often include more than one disease gene panel, mitochondrial DNA sequencing and whole exome sequencing) and the recruitment of sporadic patients with late onset neuropathies felt to be most likely inherited. However, a genomic approach to CMT diagnostics remains superior to other forms of genetic testing, not only because it enhances the diagnostic approaches that can be deployed as discussed below, but furthermore the WGS data can be banked and re-analysed at a later stage and it also provides the opportunity of screening for genetic risks and modifiers in CMT and related disorders.
WGS virtual gene panels for groups of hereditary neurological conditions have been developed and are regularly updated by the clinical scientific community with expertise in that field [2]. For example, the WGS hereditary neuropathy gene panel contains 290 genes that have been reviewed and agreed by a team of 19 clinicians and scientists with expertise in the molecular genetics of inherited neuropathies [3]. During the multidisciplinary team (MDT) meeting, which usually occurs on a monthly basis, clinicians and geneticists review the phenotype with the associated Human Phenotype Ontology terms and apply the relevant disease-specific virtual gene panels to the quality-controlled annotated sequenced data, thus yielding candidate variants (Fig. 1). For the interpretation of candidate variants in CMT and related disorders we take into consideration many factors including the strength of the genotype–phenotype correlation, population allele frequency of the variant and zygosity of the variant in the context of the gene’s known inheritance pattern [4]. At our centre, WGS has also enabled a genetic diagnosis in three patients with a fast-paced progressive non-length dependent neuropathy and no relevant family history (two patients with biallelic variants in MME and 1 patient with a de novo pathogenic variant in TFG), which also meant that further investigations such as imaging and plans for a sural nerve biopsy were abandoned.
Each MDT has the discretion to apply all the necessary virtual gene panels to a particular case and this is particularly important in CMT and related disorders, which exhibit considerable phenotypic and genetic heterogeneity. The gene composition of each virtual gene panel necessitates a trade-off between diagnostic yield and background genetic noise, both of which increase with increasing number of genes on the panel. Therefore, not all genes that have been associated with syndromes of which peripheral neuropathy is one feature are present on the WGS hereditary neuropathy gene panel. An example is the case of a 60-year-old man with childhood onset slowly progressive optic neuropathy who also developed a peripheral sensorimotor neuropathy in his forties. Following the WGS hereditary neuropathy gene panel which returned no variants, the WGS optic neuropathy gene panel was applied which identified two heterozygous variants in the SLC25A46 gene which is strongly associated with an optic-peripheral neuropathy phenotype [5]. This sequential use of WGS virtual gene panels (i. e. applying other gene panels as guided by the phenotype if the primary gene panel returns no candidate variants) has proved very efficient in practice for achieving a genetic diagnosis. Other examples include applying the WGS hereditary spastic paraplegia panel, following the WGS hereditary neuropathy gene panel in cases of peripheral neuropathy that also show a subtle pyramidal syndrome and applying the WGS ataxia panel in cases with subtle cerebellar features. Furthermore, through the Neurology GeCIP research environment, investigators working in any GMC have access to the complete set of whole genome sequence data of all patients from all other GMCs that have been recruited in the Neurology GeCIP. This process facilitates novel gene discovery, and the respective GeCIP ensures that all the relevant parties, including the recruiting GMC and clinical care team, are involved in this process.
WGS carries significant advantages over other genetic tests including a reliable and persistently uniform coverage across coding and non-coding regions of both the nuclear and mitochondrial genomes that contribute to the genetic heterogeneity in CMT and high density homozygosity mapping for the identification of recessive traits [4]. The former property is an important one to consider as it allows the identification of structural variants (SVs) and their accurate breakpoints, which often lie in intronic or intergenic areas, as well as repeat nucleotide expansions. Such variants and repeat expansions are now increasingly recognised as causes of hereditary forms of neuropathy [6], [7] and two examples from our cohort also illustrate the usefulness of WGS in this regard.
The first case is a 48-year-old woman who developed walking difficulties in childhood and had associated hearing loss and mild scoliosis. She had two other similarly affected siblings and healthy parents and her neurophysiological studies revealed a length dependent neuropathy with slowing of the upper limb motor nerve conduction velocities (18 m/s). A WGS hereditary neuropathy gene panel initially identified a heterozygous stop gain variant in SH3TC2 (p.Tyr169*) and due to a high clinical suspicion of CMT4C, we manually interrogated the aligned sequence reads from the WGS data (Fig. 2). This revealed a small 2 kb deletion in SH3TC2 spanning exons 5 to 8 in an apparent heterozygous fashion and the exact breakpoint junctions enabled the development of further long range sequencing required to confirm the SV.

Aligned sequence reads at the SH3TC2 locus visualised in the IGV browser. In the blue circle is evidence of sequence reads showing a germline deletion, depicted by thick black horizontal lines (split reads). The deletion is at the SH3TC2 locus (gene reference shown at the bottom), spans exons 5 to 8 and by crude estimation involves half of the reads at that point. This is also evident by the approximate 50 % reduction in the sequence trace shown at the top (within the blue circle as well).
The second case is a 75-year-old man who presented with a sensory-predominant neuropathy in his thirties which slowly progressed and who had subtle upper motor neuron signs in his legs when examined. His neurophysiological studies confirmed a sensory-predominant neuropathy with attenuated but present tibial and peroneal compound muscle action potentials. A WGS hereditary neuropathy gene panel identified the p.Gly130Val variant in FXN in the heterozygous state, which has been previously associated with atypical Friedreich ataxia [8]. This prompted a targeted review of the aligned whole genome sequence reads at the FXN locus and revealed a cluster of unmapped sequence reads in the first intron (Fig. 3). The sequences of the unmapped reads were complementary to each either on either side of the partial drop in coverage (not shown here), thus indicating a GAA triplet repeat expansion, which was subsequently confirmed. In addition to these examples, we have three further cases where interrogation of the WGS data has identified structural variants at known CMT gene loci and a novel structural variant which is currently being prepared for publication.
The 100KGP is not immune to the challenges that accompany WGS, of which the biggest is the prioritisation and interpretation of NGS-derived variants. Aside from interpreting the wealth of these genomic variants, another challenge is overcoming what has been labelled as their ‘narrative potential’. The swathes of candidate variants in complete sequence data simply increase the likelihood of encountering compelling causal mutations and the potential of assigning a disease-causing narrative to some of them [9], thus increasing the likelihood of variant over-interpretation and over-diagnosis. Furthermore, NGS technologies, and WGS in particular, magnify pre-existing ethical, social and legal issues encountered in genomic medicine, such as non-paternity, consent of individuals for research and sharing and storage of sequence data [10], [11]. The wealth of NGS data and the challenges in interpreting the data necessitate interdisciplinary collaborations between various clinicians and scientists.
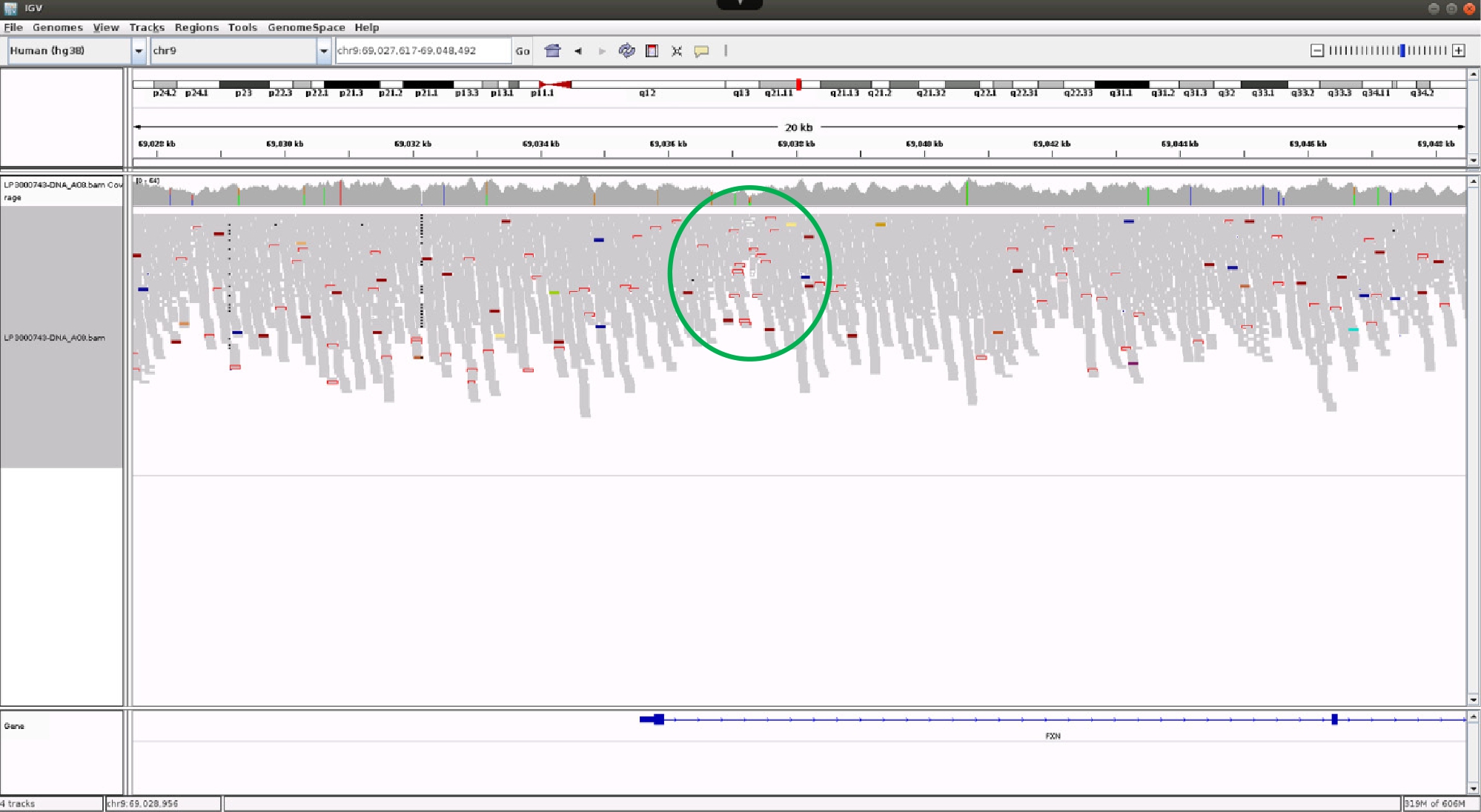
Aligned sequence reads at the FXN locus visualised in the IGV browser. There is a cluster of sequence reads (green circle), shown as rectangles with open red borders, which have unmapped paired reads. These account for approximately 50 % of all sequence reads and the unmapped reads show continuous traces of GAAGAA(n) and CTTCTT(n) (not shown here) on either side of the midline of this cluster, which is the presumed site of the repeat expansion.
In the UK, the 100KGP has revolutionised genomic medicine, is promoting important research collaborations, and is allowing an ever greater number of patients with CMT and related disorders to receive a genetic diagnosis. The 100KGP has provided a platform to recruit patients and centrally sequence genomes and the best mechanism (through MDTs) to interpret genome sequencing results in rare disorders. On this basis, WGS is moving into mainstream NHS genetic testing, based on our experience and diagnostic rate from the 100KGP, to analyse certain neurological disorders such as neuromuscular diseases, hereditary ataxias and movement disorders. With pre-clinical studies revealing the efficacy of potential genetic therapies and human clinical trials on the horizon, this could not have come at a better time for patients with CMT and their families.
Funding statement: MP is funded by the NIH and the Inherited Neuropathy Consortium (INC; grant U54NS065712) as well as the BMA (Vera Down grant). MMR is grateful to the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712) and to the Muscular Dystrophy Association (MDA510281) for their support. This research was also supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Acknowledgment
This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support.
References
[1] Turnbull C et al. The 100 000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ. 2018;361:k1687.10.1136/bmj.k1687Suche in Google Scholar PubMed
[2] Martin AR et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. 2019;51:1560–1565.10.1038/s41588-019-0528-2Suche in Google Scholar PubMed
[3] England G. Genomics England PanelApp. Available at: https://panelapp.genomicsengland.co.uk/ (Accessed: 21st April 2020).Suche in Google Scholar
[4] Pipis M, Rossor AM, Laura M, Reilly MM. Next-generation sequencing in Charcot-Marie-Tooth disease: opportunities and challenges. Nat Rev Neurol. 2019;15:644–656.10.1038/s41582-019-0254-5Suche in Google Scholar PubMed
[5] Abrams AJ et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet. 2015;47:926–32.10.1038/ng.3354Suche in Google Scholar PubMed PubMed Central
[6] Cutrupi AN, Brewer MH, Nicholson GA, Kennerson ML. Structural variations causing inherited peripheral neuropathies: A paradigm for understanding genomic organization, chromatin interactions, and gene dysregulation. Mol Genet Genomic Med. 2018;6:422–33.10.1002/mgg3.390Suche in Google Scholar PubMed PubMed Central
[7] Cortese A et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019;51:649–58.10.1038/s41588-019-0372-4Suche in Google Scholar PubMed PubMed Central
[8] Bidichandani SI, Ashizawa T, Patel PI. Atypical Friedreich ataxia caused by compound heterozygosity for a novel missense mutation and the GAA triplet-repeat expansion. Am J Hum Genet. 1997;60:1251–6.Suche in Google Scholar
[9] Goldstein DB et al. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 2013;14:460–70.10.1038/nrg3455Suche in Google Scholar PubMed PubMed Central
[10] Burke W et al. The translational potential of research on the ethical, legal and social implications of genomics. Genet Med. 2015;17:12–20.10.1038/gim.2014.74Suche in Google Scholar PubMed PubMed Central
[11] Hercher L, Jamal L. An old problem in a new age: Revisiting the clinical dilemma of misattributed paternity. Appl Transl Genomics. 2016;8:36–9.10.1016/j.atg.2016.01.004Suche in Google Scholar PubMed PubMed Central
© 2020 Pipis et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- MAIN TOPIC: GENETICS OF PERIPHERAL NEUROPATHIES
- Peripheral Neuropathies
- The CMT1A duplication
- Charcot-Marie-Tooth disease and hereditary motor neuropathies – Update 2020
- Charcot-Marie-Tooth neuropathy and pregnancy: general and specific issues
- Advancing Charcot-Marie-Tooth disease diagnostics, through the UK 100,000 Genomes Project
- Peripheral sensory neuropathies – pain loss vs. pain gain
- On the differential diagnosis of neuropathy in neurogenetic disorders
- Spinal muscular atrophy (5qSMA): best practice of diagnostics, newborn screening and therapy
- BERICHTE AUS DER HUMANGENETIK
- Zur Geschichte der Humangenetik
- Der Weg in die Einheit: Das Fach Humangenetik 1990–1991. Ein Erfahrungsbericht
- Personalia
- Personalia
- BVDH-Verbandsmitteilungen
- BVDH-Verbandsmitteilungen
- Aktuelle Nachrichten
- Aktuelle Nachrichten
Artikel in diesem Heft
- Frontmatter
- MAIN TOPIC: GENETICS OF PERIPHERAL NEUROPATHIES
- Peripheral Neuropathies
- The CMT1A duplication
- Charcot-Marie-Tooth disease and hereditary motor neuropathies – Update 2020
- Charcot-Marie-Tooth neuropathy and pregnancy: general and specific issues
- Advancing Charcot-Marie-Tooth disease diagnostics, through the UK 100,000 Genomes Project
- Peripheral sensory neuropathies – pain loss vs. pain gain
- On the differential diagnosis of neuropathy in neurogenetic disorders
- Spinal muscular atrophy (5qSMA): best practice of diagnostics, newborn screening and therapy
- BERICHTE AUS DER HUMANGENETIK
- Zur Geschichte der Humangenetik
- Der Weg in die Einheit: Das Fach Humangenetik 1990–1991. Ein Erfahrungsbericht
- Personalia
- Personalia
- BVDH-Verbandsmitteilungen
- BVDH-Verbandsmitteilungen
- Aktuelle Nachrichten
- Aktuelle Nachrichten