The CMT1A duplication
-
James R. Lupski

Contents
From the United States
From Europe
References
From the United States
I came to Houston, Texas in 1986 with one goal being to identify “the gene” for Charcot-Marie-Tooth (CMT) disease. I was peripherally aware of the paper by Botstein and colleagues [1] proposing the genetic mapping of human “disease genes” using linked restriction fragment length polymorphisms (RFLPs) to position the gene within the human genome and indeed became very excited as a graduate student when Gusella’s paper [2] appeared in Nature linking the Huntington disease locus to markers on chromosome 4. It was a natural extension to think this “positional cloning” approach might be applied to a host of other human traits. There was a personal, one might say egocentric, reason to choose CMT because I have the disease [3] and, in fact, the first blood samples collected for DNA linkage studies were from my own family wherein CMT segregated as an apparent autosomal recessive trait.
The year 1986 was also somewhat historic for the opportunity to attend the Cold Spring Harbor Symposium on Quantitative Biology, which that year was on “The Molecular Biology of Homo sapiens” [4]. It was there that Kary Mullis first announced publicly the polymerase chain reaction (PCR) technique, and also some of the first “scientific public” debates surrounding the initiation of the Human Genome Project took place. I distinctly remember Kary Mullis arguing during these discussions that if there was going to be a huge amount of DNA sequence determined (like the three billion basepair human genome) then the “G” symbol for the base guanine should be changed to “W” to distinguish it from “C,” which was difficult to do because of the typewriters and printers available at the time. He argued that Crick already had one of the symbols (“C” for cytocine) named after him and Watson should have one. I vaguely remember Jim Watson smiling on the sidelines of the audience. I was married that week in Huntington, New York.
From: Genomic Disorders: The Genomic Basis of Disease Edited by: J. R. Lupski and P. Stankiewicz © Humana Press, Totowa, NJ
The move to Houston was also because of the decision to continue my clinical training and begin internship and residency in pediatrics at Texas Children’s Hospital. This occupied my time immensely and, thus, I was fortunate to be able to join Pragna Patel, a junior faculty member in the Institute for Molecular Genetics, to bank CMT family samples and initiate our genetic linkage studies.
Family collections began in earnest towards the end of my residency (1988–1989). Pragna had known of a physician, Carlos Garcia (New Orleans), who followed a number of families with CMT in Louisiana and we also contacted Jim Killian (Houston), at the time co-chairman of Neurology at Baylor College of Medicine, who published a huge French Acadian pedigree segregating CMT a decade earlier [5]. He had also made the intriguing observation that apparent homozygosity for the dominant CMT gene, a child of two affected parents, resulted in a significantly more severe phenotype [5]. Thus, like many other human traits, CMT is probably better characterized as a semi-dominant disorder.
Carlos Garcia directed the Muscular Dystrophy Association clinics in New Orleans, Baton Rouge, and Lafayette, LA. Once a month, Carlos’ wife Mona would always remark “It’s that time of the month again,” I would fly to New Orleans and stay overnight Monday at the Garcia’s. Carlos and I would awaken and drive a couple of hours to Baton Rouge and see patients from morning until just after lunch, drive to Lafayette (where Carlos followed several hundred CMT patients) and see patients until dinner time. We would have a wonderful Cajun dinner, stay overnight in Lafayette, and the next day start seeing patients early in the morning until late afternoon, then he would drive me back to the New Orleans airport with a suitcase of blood samples in hand. Carlos would clinically examine and oversee nerve conduction velocity (NCV) testing (NCVs are an objective laboratory test for type 1 CMT [CMT1]) while I would draw pedigrees and obtain blood for DNA samples and to make permanent transformed lymphoblastoid cell lines on my return to Houston.
One particular blood collection sticks out in my mind. It took place in a hospital clinic adjacent to the emergency room of a local hospital in Lafayette. We first collected blood from a teenage man distinguished by an unusual haircut and tattoos dressed in an outfit becoming of a punk rocker. When we next began collecting blood from his younger sister, she passed out and started to have myoclonic jerks. Her older brother started to shout “she is throwing a fit.” He then proceeded to stand, look at both Dr. Garcia and I, and stated, “I am going to go get a REAL doctor” and proceeded to the emergency room next door. Needless to say both he and she were just fine and Dr. Garcia and I recovered from our ego bruising.
These monthly trips continued for a few years, but for the collection of very large families we would sometimes arrange a family reunion. It was remarkable how there would be one family member, often an unaffected individual, who could mobilize the entire family because of their belief in the research efforts. Importantly, we had to perform the electrical studies (NCVs) and collect blood samples from all family members. This included unaffected individuals, who were sometimes hesitant, or required further explanation of the need for their samples. I often thought of the irony of the situation. At these reunion parties, Dr. Garcia would oversee the administration of the electrical shocks accompanying nerve conduction studies, I would draw blood from each family member, and they would feed us wonderful Cajun barbeque. We similarly collected the large family reported by Dr. Killian using the family reunion approach. In this case, Jim Killian rented the town hall of a small town in the French Acadian countryside of Louisiana. I remember Dr. Killian asking other family members about one particular family member, expressing some concern during the inquiry. Apparently, during the examinations and home visits that led to the 1979 paper of Killian and Kloepfer [5], this family member drew a gun on Dr. Killian thinking that he was either “the law,” or a tax collector.
Meanwhile in Houston, Pragna had collected several polymorphic DNA markers from the laboratory of Dr. Ray White in Utah and we analyzed systematically the family material that was available. We began with the smaller chromosomes and essentially had ruled out several, including initially chromosome 17, using sparse markers when Jeff Vance (Durham, NC) [6] reported linkage of CMT1A to chromosome 17 using the same marker that revealed linkage to NF1 on chromosome 17. We and others confirmed this chromosome 17 linkage [7].
Much effort was now focused on identifying, and/or making more informative, DNA markers for the pericentromeric region of chromosome 17. Yusuke Nakamura (Tokyo, Japan) had provided some chromosome 17 cosmid clones, which were used to identify chromosome 17 polymorphic markers. Also, Pragna developed a novel method to obtain region specific chromosome 17 markers using differential Alu-PCR [8]. At the time Alu-PCR had been recently developed in our Institute for Molecular Genetics by David Nelson in Tom Caskey’s lab [9]. To identify region specific markers, Alu-PCR was performed on somatic cell hybrids that retain either intact human chromosome 17 or a deleted chromosome from a patient with Smith-Magenis syndrome (SMS) [del(17)(p11.2p11.2)] [10]. Amplification products were compared and if a band was present in the amplification from the hybrid retaining intact chromosome 17, but not from the amplification of the hybrid with the deletion chromosome, then this was surmised to physically come from the specific deleted region. Of course, one also identifies Alu polymorphisms this way. We found that the procedure could be remarkably simplified by first reducing the genome complexities using restriction endonuclease digestion before the Alu-PCR. This, in turn, lead to the development of “restricted-Alu PCR” [11]. By 1989 we had accumulated extensive mapping data to show that the CMT1A locus was on the short arm of chromosome 17 and most tightly linked to markers that were physically located within the common SMS deletion interval in 17p11.2.
Here I must digress to say that much of our daily business was centered around marker genotyping using RFLPs. It was clear that some markers worked better than others, for some the segregating alleles were easier to score than for others, and in general each DNA marker had its own “personality.” There were clearly certain DNA markers that appeared to show an artifact of different hybridization intensities for cross-hybridizing bands on genomic Southern blots. However, individuals from the same families were not always run adjacent to each other on the genomic Southerns. By no means did we initially recognize that the presumed artifact of “dosage differences between cross-hybridizing bands” segregated in a Mendelian fashion. Scoring of alleles was done independent of knowledge of affection status. Linkage analyses were performed in collaboration with Aravinda Chakravarti (Baltimore, MD), and I worked mostly with his student Susan Slaugenhaupt, who would input the data from the scoring sheets for the analyses.
Although RFLP mapping was proceeding, much effort was also expended on screening the proximal 17p linked probes for the presence of simple sequence repeats (SSR; e. g.,

Nucleotide sequence of the simple sequence repeat RM11-GT. Autoradiogram of a DNA sequencing gel showing the repeat, which lies at the basis of the polymorphic DNA marker RM11-GT. Initial evidence for the Charcot-Marie-Tooth type 1A (CMT1A) duplication was revealed by this marker that showed three alleles (i. e., triallelic) in fully informative CMT patients.
Roberto examined some of the Southern blots that utilized an RFLP marker from the same locus and noted that often when there were three alleles revealed by RM11-GT [14], a dosage difference could be observed between the two alleles if the affected individual was heterozygous for that RFLP. These initial observations suggested that there may be three copies of the genomic region that was being assayed, potentially reflecting genomic duplication at the CMT1A locus. The entire laboratory now focused on the “duplication hypothesis” and, to keep our hypothesis quiet, it was referred to as the “D” word within the laboratory because we all focused on gathering data to support or refute the duplication hypothesis using multiple independent molecular approaches. When now correcting for diagnosis (i. e., making sure that all apparent unaffected individuals did not have subclinical disease by performing NCVs and systematically examining CMT1A families), the putative duplication appeared to cosegregate with the CMT1A phenotype as determined by objective NCV measurements [15].
To reconcile dosage differences of heterozygous RFLP alleles with the three RM11-GT alleles observed in CMT1A duplication patients, Pragna performed an important experiment. For one of the RFLP markers revealing dosage differences, and from which the PCR-typeable RM11-GT marker was derived, the MspI alleles were separated on preparative agarose gels and used as templates for PCR amplifications of RM11-GT. As anticipated, from the RFLP allele showing increased dosage she could amplify 2 RM11-GT alleles, whereas only one was found from the PCR of the other RFLP allele that displayed normal dosage [15]. Similar types of experiments, to examine the molecular basis for the dosage differences of alleles, were performed by first physically separating the two chromosome homologs in rodent somatic cell hybrids [15].
The allele dosage differences revealed by RFLP analyses could also be observed, although it was much more difficult to see and less informative, for two other CMT1A-linked markers that by genetic mapping studies were adjacent to the initial marker revealing duplication. This suggested the putative duplication might be large and we, thus, attempted to obtain further, physical evidence for its existence. Pentao Liu applied pulsed-field gel electrophoresis (PFGE) as a means to try to resolve a potentially large genomic change. Indeed, he was able to identify a 500-kb apparent junction fragment in CMT1A patients that was not observed in controls. Furthermore, he showed that this junction fragment cosegregated with CMT1A [15]. Interestingly, this junction fragment was increased in dosage in a patient, whom we presumed was homozygous for the CMT mutation given the severe clinical picture, where both parents had CMT1A. In collaboration with Barbara Trask (Seattle, WA), we attempted to resolve the duplication by fluorescence in situ hybridization of metaphase spreads from lymphoblastoid cell lines constructed from CMT1A patients and controls. The metaphase analysis failed, but on interphase spreads she could identify duplication on one of the two chromosome 17 homologs [15]. Moreover, she identified an apparent duplication on both homologous chromosomes in the patient presumed to be homozygous on clinical grounds. Because these were interphase cells, it was important to distinguish duplication from replication and this was done by comparison to a nearby control probe. To our knowledge, this was the first time that a common autosomal dominant human disease trait was diagnosed using a microscopic technique. Although it was 55 years later, I think we were probably as excited as Calvin Bridges was when he initially applied the then new technique of polytene chromosomes to the study of fruit fly traits and found that the Bar gene was a duplication [16].
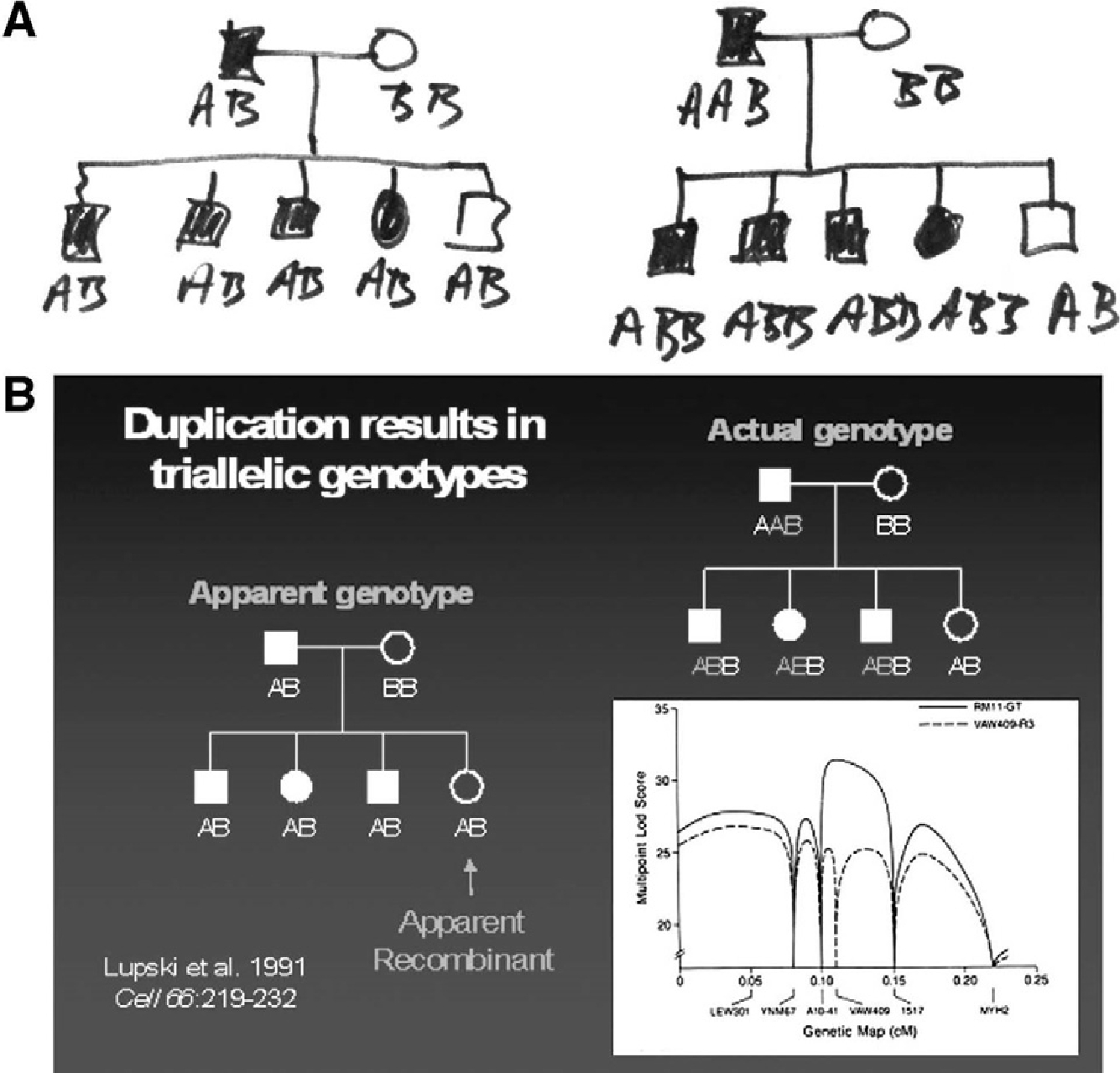
Triallelic marker genotypes and false recombinants. (A) Actual note paper wherein biallelic marker genotype scoring was compared to triallelic marker genotype scoring to reveal the molecular genetic basis of false recombinants. (B) The effects of molecular duplication on the interpretation of marker genotypes and linkage mapping. Standard pedigree symbols are used; females depicted as circles and males by squares. Filled-in symbols denote affected individuals. On the left is a simple pedigree with marker genotypes scored as a usual biallelic system with one of the two alleles inherited from each parent. One unaffected daughter is an apparent recombinant (false recombinant) because she has the same apparent genotype as her three affected siblings. To the right is shown the actual genotypes scored as a triallelic system accounting for the molecular duplication. The lower right shows how the different scoring biallelic (dashed line) vs triallelic (bold line), affects the multipoint LOD-score. Note the differences in peak LOD scores and the fact that the failure to account for three alleles (or dosage differences in heterozygous restriction fragment length polymorphisms) results in an erroneous map position.
We had accumulated very strong physical evidence thus far; 3 GT alleles, dosage differences of heterozygous RFLP alleles, a PFGE junction fragment, and interphase fluorescence in situ hybridization revealing a duplicated signal, for the CMT1A duplication. However, what remained was reconciliation with the genetic data. The marker VAW409, from which RM11-GT was derived and which physically revealed the duplication by virtue of dosage differences of heterozygous RFLP alleles in CMT1A patients, also appeared to reveal recombinants in the genetic analysis. We thought it would be hard to publish our CMT1A duplication findings without reconciling the physical and genetic data. Through conversations I was having with Markus Grompe (Portland, OR), a then clinical genetics fellow with me at Baylor, it became clear that the duplication could have consequences for the interpretation of marker genotypes and thus linkage analyses. The failure to account for the duplication in linkage analyses produces false recombinants (Fig. 2A). Linkage programs score genetic transmission data using a biallelic system—one inherits one allele from each of two parents. However, the duplication produces three alleles. The failure to account for dosage differences at a two allele (biallelic) RFLP in linkage analysis, when it exists, leads to the misinterpretation of the parental origin of alleles (Fig. 2B). Importantly, when we rescored the marker genotypes as a triallelic system, both the genetic data and the physical duplication data converged on the same locus [15].
We now thought that the problem was solved, but convincing one’s colleagues and peer reviewers is another challenge. Pragna and I initially showed all the data to our Chairman Tom Caskey. He said that he was not completely convinced, but we better be absolutely sure if we were going to publish this from his department. Art Beaudet, a senior colleague, seemed to be convinced and made some helpful comments on both of the manuscripts. I say two manuscripts because the amount of accumulated data was extensive; there was one entire paper that covered the genetic analyses and the second manuscript described the physical evidence for duplication.
We sent both manuscripts to Science and they were both rejected. Interestingly, I subsequently learned from Christine Van Broeckhoven (Antwerp, Belgium), whose laboratory independently identified the CMT1A duplication in Europe, that she had submitted their paper to Science the same month that we submitted our papers. There were referees’ and editors’ comments to both of us from a couple of journals that we “had not identified the gene.” This pretty clearly showed that the reviewers completely misunderstood the novelty of our findings, as did the editor handling the manuscript, thus, the burden was on us to make it clearer. At the time, I certainly do not think that I understood the implications of the CMT1A duplication for other human diseases that result from genome rearrangements; a class of conditions subsequently referred to as genomic disorders that represent recombination-based disease resulting form DNA rearrangements owing to genome architecture [17], [18]. Nor did I anticipate that the requirement for three alleles to manifest a trait, triallelic inheritance, might apply to the genetic transmission of other conditions [19]. A revision and resubmission of both manuscripts to Cell was met with more favorable reviews. They each insisted on condensation to one large paper, because of the interdependence of the genetic and physical data, and suggested the deletion of some material. Although heated discussions concerning authorships and positions on the paper ensued, Pragna, Aravinda, and I agreed with the reviewers’ ideas that because of interdependence of the data, it would be best presented as a single paper. Whether Pragna or I would be first or last author was mainly settled by which person would now condense these two papers into one and address each of the reviewer’s thoughtful comments.
We first presented the data for the CMT1A duplication at a small CMT meeting in Tucson, AZ hosted by the Muscular Dystrophy Association (MDA). Christine Van Broeckhoven spoke first about a duplication they identified in CMT patients from Europe [20]. I felt immediate relief and excitement—our hypothesis and supporting data were already reproduced in another part of the world. I spoke after her and described the multiple methods we used to obtain evidence in support of the duplication and how this genomic rearrangement affected the interpretation of marker genotypes. During the lunch break that followed our talks, multiple audience members called their respective laboratories and, indeed, review of their Southern blots revealed RFLP dosage differences for the appropriate markers. The existence of the CMT1A duplication had now almost instantaneously been confirmed around the world.
From Europe
I started my PhD in October 1988 at the University of Antwerp, Belgium in the laboratory of Christine Van Broeckhoven, currently the scientific director of the Molecular Genetics Department affiliated to the Flanders Interuniversity Institute for Biotechnology. I became interested in her molecular genetic research of neurological disorders, after reading a paper in Nature on Alzheimer’s disease [21]. I selected this paper as a topic for a course in the frame of my master studies in biotechnology (applied in agriculture) at the University of Leuven, Belgium. When I joined the Antwerp team, Peter Raeymaekers was the only other PHD student, performing molecular genetics on a multi-generation Belgian CMT family with autosomal dominant transmission. In fact, Peter initially started his PHD on genetics of Alzheimer disease, but because the families were still being sampled, he initially spent a lot of effort in developing protocols for isolating human DNA, and in cloning probes that recognized RFLPs. When Peter De Jonghe and Jan Gheuens, clinical neurologists at the Neurology Department of the University Hospital Antwerp, presented to him the large pedigree of a CMT family, he decided to switch to research into genetics of CMT. It became apparent that the pedigree of this CMT family was huge, with more than 350 family members in five generations. In total we sampled 51 affected and 60 healthy relatives for linkage studies. Because we were convinced that CMT was a very rare disorder at that time, we used alphabetical letters in the acronyms of the CMT families we ascertained in Belgium. Still, we had not changed our opinion of the disease frequency when we reached the letter Z, and considered starting again with A-A, in retrospect it is fortunate that we decided to switch to numbers. At this moment we have nearly 2000 CMT families under investigation, either sampled in Belgium or obtained through international collaboration, particularly within the European CMT consortium founded in 1991. However, looking back it seems like the alphabetical letters had some magic value: the CMTA family turned out to belong to the CMT1A subtype [22], family CMT-B belongs to the CMT1B subtype [23], and CMT-M has a pure motor phenotype [24].
Fortunately, Belgium is a small country (you can hardly drive 2 hours by car without ending up in a neighboring country), in which people tend to continue living in the village where they were born. Every week Peter De Jonghe and his wife Gisèle Smeyers, at that time the research nurse on the project, made many trips visiting family members of family CMT-A at their homes to collect blood samples. Gisèle made the first contact with the patients and relatives to explain the aims of the study and to ask whether she could visit again, but now with the neurologist Peter De Jonghe, who was leading the project. What she did not tell was that the neurologist was in fact her husband, because she wanted the family members to feel free to criticize doctors because of lack of attention for the problems of a CMT patient. However, there soon came a moment when she had to disclose the husband–wife relationship. When visiting a CMT patient whose husband was a forester, she was offered a rabbit to take home. The next visit, the man offered her again a rabbit but said “and here is one for the doctor too.” Not to look greedy, she disclosed that the doctor was her husband Peter De Jonghe, but still received the two rabbits.
In the Belgian CMT-A family, Peter Raeymaekers used RFLPs to exclude the first CMT locus on chromosome 1-designated CMT1B in 1982 [25], and initiated a genome search using some of his in-house developed RFLPs. However, shortly before Peter Raeymaekers’ PhD thesis defense June 1989, Jeffery Vance (Durham, NC) reported linkage with two chromosome 17p markers (D17S58 and D17S71) in CMT1A families [6]. We confirmed the linkage with the two 17p DNA markers in the Belgian CMT-A family, and obtained a log of the odd (LOD) score of 10.67 (significant linkage is obtained when the LOD > 3) [22]. We proceeded with the genetic analysis of eight additional chromosome 17 markers, and showed that the CMT1A mutation was mapped in the 17p11.2-p12 region between the marker D17S71 and the gene coding for myosin heavy polypeptide 2 (MYH2) [26].
At that time only partial genetic maps were available for linkage studies [27], [28]. To finemap the CMT1A locus, we genotyped additional RFLPs and detected informative recombinants in family CMT-A. However, the genotypes obtained for two DNA markers (pVAW409R1 and pVAW409R3), representing the same locus D17S122, were hard to interpret on RFLP analysis. For one marker we obtained a significant LOD score of 16.20, but it recombined with the second marker at D17S122. These results were hard to believe, and our first reaction was that we had misinterpreted the genotypes. The autoradiograms of the Southern blots were messy, with high backgrounds owing to the presence of repetitive sequences that made it difficult to “read” the MspI alleles. To avoid these “dirty blots,” we decided to “clean up” the pVAW409R1 and pVAW409R3 clones, by recloning the non-repetitive restriction fragments into derivative probes designated pVAW409R1b and pVAW409R3a. The hybridization results were squeaky clean; however, much to my amazement, in each genotype one allele had the double density of the other. There was also no consistency because in one patient it was the upper band and in another patient the lower band revealing the increased dosage intensity. After checking and rechecking, I realized that the data could only be explained if one of the alleles was duplicated. I reinterpreted the genotypes, and yes, the recombinants had disappeared.
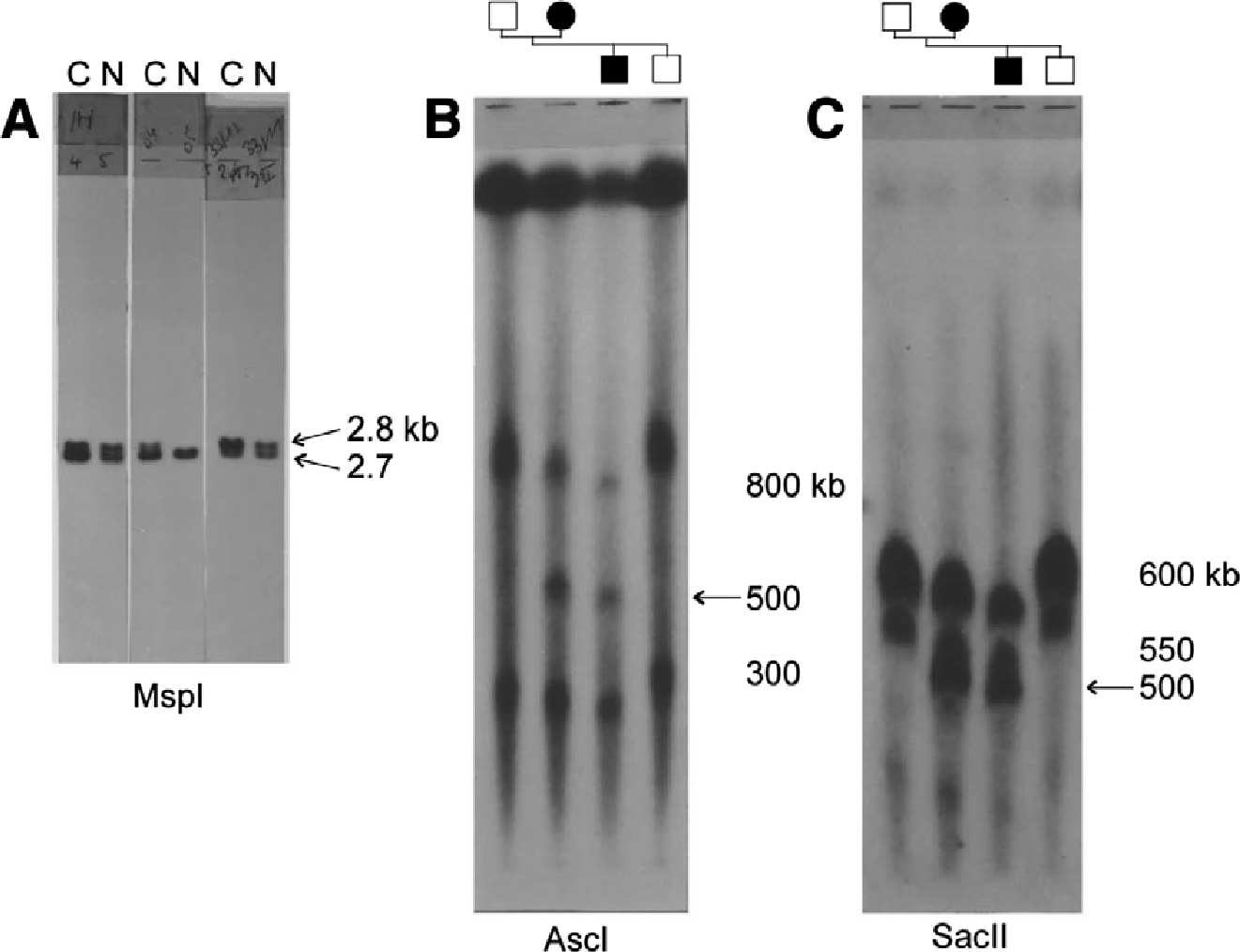
Hybridization signals obtained with probe pVAW409R3a (D17S122). (A) Southern blot of genomic DNA digested with MspI of Charcot-Marie-Tooth neuropathy type 1A (CMT1A) duplication patients (C) and healthy relatives (N) of three different CMT families. Dosage differences between the alleles are seen in each patient, either in the upper allele (2.8 kb) or lower allele (2.7 kb). (B,C) Southern patterns of genomic DNA digested with rare cutter restriction enzymesAscI and SacII. DNA fragments were separated by pulsed-field gel electrophoresis. The 500-kb junction fragment (arrow) is only present in the CMT1A duplication patients belonging to a small branch of the Belgian family CMT-A.
I can still feel the excitement that went through my body that summer in 1990. Although I was convinced that the data were true, I refrained from telling Peter Raeymaekers and my supervisor Christine. Could it still be that I mixed up samples? I redid the entire experiment, but no, the same results appeared (Fig. 3A). Now it was time to share my findings! Suddenly, the project became the hottest one in the group, we worked hard and step-by-step discovered that the duplication had to be >1 Mb in size based on other duplicated markers in the region (pVAW412R3 [D17S125] and pEW401 [D17S61]) and PFGE data. We wrote the paper and submitted it to Nature. While under editorial review, we continued the work and found the real genetic proof that it was the duplication that caused the disease, namely in one family we observed the duplication appearing de novo together with the disease. In this family (CMT-G), the grandparents were unaffected, and among their six children there was only one patient, who transmitted CMT to his son.
Peter Raeymaekers, Peter De Jonghe and I attended the seventh International Congress on Neuromuscular Diseases in Münich, September 1990. During this meeting our excitement about the finding of the CMT1A duplication gradually turned into sheer paranoia. Jeffery Vance was giving the plenary lecture on CMT; did he not know about the duplication, or did he and would not tell? Who was chatting to whom during the poster session and what about? Could it be that the CMT1A duplication would be revealed in the “surprise box,” the last presentation of the meeting? To us shareholders of the CMT1A duplication an extraordinary event took place at this meeting. During the poster session, authors had to present their poster in three slides in a session chaired by Peter James Dyck (Rochester, NY) one of the forefathers of the entire field of peripheral neuropathies. At some point, there was a heated discussion regarding controversial data presented by Victor Ionascescu (Iowa City, IA). To change the subject and calm the audience, P. J. Dyck suddenly asked, “in all these fancy molecular genetic studies, has someone of you ever seen something special like a duplication?” Peter Raeymaekers turned pale and almost fainted. However, nobody noticed the remark, and later it became apparent that P. J. Dyck had absolutely no knowledge of the CMT1A duplication at that time.
Nature did not send our paper for review. Christine, my supervisor, had many hours of discussion with the associate editor Kevin Davies handling the paper; she added new data to the paper (e. g., the de novo duplication data, the size of the duplication estimated at around 1 Mb, additional CMT1A duplication families), but nothing helped. The final verdict was “since we were so close to the gene we might consider coming back when we found it.” This illustrated the disbelief in the scientific community that an autosomal dominant disease could result from a gene dosage defect, a genetic mechanism that now is commonly accepted! Also, in 1990 we did not have available the technology we have today, and cloning a gene from a larger than 1 Mb region was still a major challenge. Later, Kevin Davies became editor of Nature Genetics, and invited Christine to tell the story of the CMTA duplication at the first International Conference of Nature Genetics on “Human Genetics: Mapping the Future,” Washington, April 1993.
We sent the paper to Science and Lancet and neither were prepared to send it for review and comments ranged from “not interesting for the larger public” to “the duplication does not provide insight in the identity of the genetic defect causing CMT1A.” By now it was spring 1991, we were desperate and terribly disappointed, while running into another problem.
Christine was organizing and chairing the eighth workshop on “The Genetics of Hereditary Motor and Sensory Neuropathies” sponsored by the European NeuroMuscular Center (ENMC), in May 1991, in The Netherlands. The paper was not yet resubmitted, and thus we were confronted with a major dilemma: should we be quiet while the workshop aimed at defining criteria for sampling CMT families using strict diagnostic criteria of CMT1 for mapping and cloning of the CMT1A defect, although we knew about the duplication? We decided not, what could we lose at this point. Also, if it became later known to the participants that we as organizers had this information at the time of the workshop, all European CMT researchers would feel deceived. We decided to share the exciting, although still unpublished data, having it presented by Peter Raeymaekers.
While Peter was gradually building the story towards the discovery of the CMT1A duplication, one could notice the increasing excitement among the participants who became silent, stopped taking notes and started whispering “they found it.” After the applause, Alan Emery, former research director of the ENMC stood up, congratulated the Antwerp researchers with their important and fascinating finding that they were sharing prepublication. He asked all participants to keep all the presented data confidential, to remember they heard about this unpublished data at the ENMC workshop, and refrain from publishing their data on the duplication obtained without crediting the Antwerp group. To us, he suggested to publish our manuscript in Neuromuscular Disorders, a new journal of his good friend Victor Dubowitz [20]. This explains why our most cited paper was published in the second issue of a journal that had not yet had an impact factor in 1991. Later on in June 1991, my supervisor Christine Van Broeckhoven attended the MDA-organized workshop on CMT in Tucson, AZ, chaired by Kurt Fischbeck (Bethesda, MD). Here, again there was the dilemma, but now all European researchers that were present at the workshop had brought data from their families confirming that the duplication was the major CMT1A mutation. The evening before the workshop Christine informed Kurt Fischbeck. Also, present in the bar was Garth Nicholson (Sidney, Australia) who bought a bottle of champagne, and made a phone call to his co-workers who, while he was asleep, collected all the data on the Australian families. The next day, Christine presented the Antwerp data and referenced the data of the European groups. Next there was a presentation by Jim Lupski (Houston, TX) who had similar data that was in press in Cell [15]. A more extreme difference in journal impact factors, is hard to imagine. Though, we had not published this major finding in a major journal, we did receive substantial recognition thanks to the many European and American colleagues who always cited, and still do, our paper in Neuromuscular Disorders [20]. Also, since the MDA workshop in 1991, the Antwerp and Houston labs had a special bond based on mutual respect and friendship, and have been collaborating on the genetics of different inherited peripheral neuropathies ever since.
After the discovery of the CMT1A duplication many labs requested our “clean” RFLP probes for research and DNA-diagnosis of CMT neuropathies. I remember the many tubes we had to prepare to distribute the clones around the world. In the same year we reported our findings to the patients in Belgium. Since then, we organize yearly meetings for the Belgian CMT organization. In some families, such as CMT-G, the disease appeared simultaneously with a de novo duplication originating from an unequal crossover event between two homologous chromosomes [20]. These findings indicated that the CMT1A duplication in 17p11.2 was the disease-causing mutation. At that time it was thought that isolated cases of hereditary motor and sensory neuropathies represented autosomal recessive traits. We and others demonstrated that the CMT1A duplication was responsible for most cases of autosomal dominant CMT1, but that de novo mutations occurred in 9 out of 10 sporadic patients. This finding became important for genetic counseling of isolated CMT patients [29].
Because the duplicated markers in CMT1A spanned a minimal distance of approx 10 cM on the genetic map of chromosome 17p11.2-p12, we constructed a physical map of the CMT1A region using rare cutter restriction enzymes in combination with PFGE. This was a very laborious undertaking that resulted in determining the size of the CMT1A duplication to about 1.5 Mb. The discrepancy between the genetic and physical map distances suggested that the 17p11.2 region was extremely prone to recombination events, and that the high recombination rate could be a contributing factor to the genetic instability of this chromosomal region. We also determined by PFGE mapping the position of the duplication breakpoints. The discovery of extra restriction fragments or “duplication junction fragments” with the markers in 1.5-Mb region, provided a more accurate DNA-diagnostic tool for the screening of CMT1A patients (Fig. 3B, C). In addition to the unequal crossover resulting into the CMT1A duplication, we also observed in some of our CMT1 families recombination between DNA markers located on the chromosome transmitting the CMT1A duplication, making our research a puzzling event [30].
After the proposed genetic mechanism causing the CMT1A duplication was determined to be owing to unequal crossover during meiosis, we studied the parental origin of the duplication in genetically sporadic CMT1A patients. We demonstrated that in all cases the mutation was the product of an unequal nonsister chromatid exchange during spermatogenesis. The fact that only paternal de novo duplications were observed in the sporadic CMT1A patients, suggested that male-specific factors may be operating during spermatogenesis that either aid in the formation of the duplication and/or stabilize the duplicated chromosome [31]. Later, de novo duplications were also described on the maternal chromosome.
The next step was to identify the gene interrupted by the duplication, or to find a dosagesensitive gene (three copies instead of two copies), or one in which a position effect on one or more genes is involved. One year after the discovery of the CMT1A duplication, Ueli Suter (Zürich, Switzerland) reported two independent mutations in the transmembrane domain of the mouse peripheral myelin protein 22 (PMP22) gene. These missense mutations occurred spontaneously in the trembler (Tr) and trembler-j (
Finally, the work was published in the first issue of Nature Genetics [34], [35], [36], [37]. The proof that PMP22 was the disease-causing gene for CMT1A was made after the identification of point mutations in some rare patients [38], [39], [40].
After my PhD defense in 1993, Phillip Chance (Seattle, WA) demonstrated that the condition known as hereditary neuropathy with liability to pressure palsies (HNPP) was associated with an interstitial deletion of the same 1.5-Mb region that is duplicated in CMT1A patients [41]. The mechanism for unequal crossover was explained by the misalignment at flanking repeat sequences (CMT1A-REPs) leading to a tandem duplication in CMT1A and the reciprocal deletion in HNPP [42], [43], and subsequently confirmed by many labs. As a result of another paper by Lupski’s team [44], Jim invited me in 1995 as a visiting scientist at the Baylor College of Medicine in Houston, to screen markers located within the CMT1A-REP. Our joint effort allowed analyzing a large group of unrelated CMT1A duplication and HNPP deletion patients from different European countries for the presence of a recombination hotspot in the CMT1A-REP sequences. We confirmed the hotspot for unequal crossover between the misaligned flanking CMT1A-REP elements, and detected novel junction fragments in more than 70 % of the unrelated patients. This recombination hotspot was also present in de novo CMT1A duplication and HNPP deletion patients. Our data also indicated that the hotspot of unequal crossover occurred in several populations independent of ethnic background. We concluded that the detection of junction fragments from the CMT1A-REP element on Southern blots could be used as a novel and reliable DNA-diagnostic tool in most patients [45]. Nowadays, the Southern blot method (Fig. 3A) has been replaced by PCR methods making use of highly informative short tandem repeat markers in the CMT1A region or specific primers located within the CMT1A-REP region.
At the second CMT workshop, sponsored by the ENMC in The Netherlands, researchers from several European countries agreed to contribute to a large study with the aim to estimate the frequency of the CMT1A duplication and HNPP deletion, and to make the first inventory of mutations in the myelin genes causing CMT. I remember the many phone calls (e-mail was not yet available in all 28 centers involved in the study) Eva Nelis made to find out that the CMT1A duplication was present in more than 70 % of 800 unrelated CMT1 patients, and the deletion in 84 % of more than 150 unrelated HNPP patients. In CMT1 patients negative for the duplication, mutations were identified in PMP22, myelin protein zero (MPZ), and connexin 32 (GJB1/Cx32) [46]. These data resulted in the Inherited Peripheral Neuropathy Mutation Database developed and maintained by Eva Nelis (http://www.molgen.ua.ac.be/CMTMutations/).
Without the excellent contacts between our lab and the many CMT patients and their families involved in this research, we could have never detected the CMT1A duplication and the many disease-causing genes currently involved in distinct types of inherited peripheral neuropathies. Professor Alan Emery said at the first European CMT workshop: “This is another step towards discovery of the causes of all these disorders, which will open doors to possible treatments in the future.” The CMT1A duplication mechanism is now referred to in many textbooks on Human Molecular Genetics.
Article note
Reprinted by permission from Springer Nature/ Springer eBook: Lupski J. R., Timmerman V. (2006) The CMT1A Duplication. In: Lupski J. R., Stankiewicz P. (eds) Genomic Disorders. Humana Press. pp 3–17, https://doi.org/10.1007/978-1-59745-039-3_1 (Under License Number: 4891960762450; License date: Aug 18, 2020)
Acknowledgment
I appreciate the help of both Christine Van Broeckhoven and Peter De Jonghe for their input on this historical perspective.
References
[1] Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 1980;32:314–331.Suche in Google Scholar
[2] Gusella JF, Wexler NS, Conneally PM et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature. 1983;306:234–238.10.1038/306234a0Suche in Google Scholar
[3] Breo DL. Researchers Lupski and Chance study a baffling genetic disease—their own. J Am Med Assoc. 1993;270:2374–2375.10.1001/jama.270.19.2374Suche in Google Scholar
[4] Molecular biology of Homo sapiens. Cold Spring Harb Symp Quant Biol. 1986;51:1–702.Suche in Google Scholar
[5] Killian JM, Kloepfer HW. Homozygous expression of a dominant gene for Charcot-Marie-Tooth neuropathy. Ann Neurol. 1979;5:515–522.10.1002/ana.410050604Suche in Google Scholar
[6] Vance JM, Nicholson GA, Yamaoka LH et al. Linkage of Charcot-Marie-Tooth neuropathy type 1a to chromosome 17. Exp Neurol. 1989;104:186–189.10.1016/S0014-4886(89)80013-5Suche in Google Scholar
[7] Patel PI, Franco B, Garcia C et al. Genetic mapping of autosomal dominant Charcot-Marie-Tooth disease in a large French-Acadian kindred: identification of new linked markers on chromosome 17. Am J Hum Genet. 1990;46:801–809.Suche in Google Scholar
[8] Patel PI, Garcia C, Montes de Oca-Luna R et al. Isolation of a marker linked to the Charcot-Marie-Tooth disease type IA gene by differential Alu-PCR of human chromosome 17-retaining hybrids. Am J Hum Genet. 1990;47:926–934.Suche in Google Scholar
[9] Nelson DL, Ballabio A, Victoria MF et al. Alu-primed polymerase chain reaction for regional assignment of 110 yeast artificial chromosome clones from the human X chromosome: identification of clones associated with a disease locus. Proc Natl Acad Sci USA. 1991;88:6157–6161.10.1073/pnas.88.14.6157Suche in Google Scholar
[10] Greenberg F, Guzzetta V, Montes de Oca-Luna R et al. Molecular analysis of the Smith-Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2). Am J Hum Genet. 1991;49:1207–1218.Suche in Google Scholar
[11] Guzzetta V, Montes de Oca-Luna R, Lupski JR, Patel PI. Isolation of region-specific and polymorphic markers from chromosome 17 by restricted Alu polymerase chain reaction. Genomics. 1991;9:31–36.10.1016/0888-7543(91)90217-3Suche in Google Scholar
[12] Litt M, Luty JA. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am J Hum Genet. 1989;44:397–401.Suche in Google Scholar
[13] Weber J, May P. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989;44:388–396.Suche in Google Scholar
[14] Montes de Oca-Luna R. Localization of the Charcot-Marie-Tooth Type 1A Disease Locus: A DNA Duplication Associated With Disease. PHD thesis. Monterrey, Mexico: Universidad Antóma de Neuvo León; 1991.Suche in Google Scholar
[15] Lupski JR, de Oca-Luna RM Slaugenhaupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232.10.1016/0092-8674(91)90613-4Suche in Google Scholar
[16] Bridges C. The Bar “gene” duplication. Science. 1936;83:210–211.10.1126/science.83.2148.210Suche in Google Scholar
[17] Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422.10.1016/S0168-9525(98)01555-8Suche in Google Scholar
[18] Lupski JR. Curt Stern Award Address. Genomic disorders recombination-based disease resulting from genomic architecture. Am J Hum Genet. 2002;2003(72):246–252.10.1086/346217Suche in Google Scholar
[19] Katsanis N, Ansley SJ, Badano JL et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259.10.1126/science.1063525Suche in Google Scholar PubMed
[20] Raeymaekers P, Timmerman V, Nelis E et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group Neuromuscul Disord. 1991;1:93–97.10.1016/0960-8966(91)90055-WSuche in Google Scholar
[21] Van Broeckhoven C, Genthe AM, Vandenberghe A et al. Failure of familial Alzheimer’s disease to segregate with the A4-amyloid gene in several European families. Nature. 1987;329:153–155.10.1038/329153a0Suche in Google Scholar
[22] Raeymaekers P, Timmerman V, De Jonghe P et al. Localization of the mutation in an extended family with Charcot-Marie-Tooth neuropathy (HMSN I). Am J Hum Genet. 1989;45:953–958.Suche in Google Scholar
[23] Nelis E, Timmerman V, De Jonghe P, Muylle L, Martin JJ. Van Broeckhoven C. Linkage and mutation analysis in an extended family with Charcot-Marie-Tooth disease type 1B. J Med Genet. 1994;31:811–815.10.1136/jmg.31.10.811Suche in Google Scholar
[24] Timmerman V, De Jonghe P, Simokovic S et al. Distal hereditary motor neuropathy type II (distal HMN II): mapping of a locus to chromosome 12q24. Hum Mol Genet. 1996;5:1065–1069.10.1093/hmg/5.7.1065Suche in Google Scholar
[25] Stebbins NB, Conneally PM. Linkage of dominantley inherited Charcot-Marie-Tooth neuropathy to the Duffy locus in an Indiana family. Am J Hum Genet. 1982;34:A195.Suche in Google Scholar
[26] Timmerman V, Raeymaekers P, De Jonghe P et al. Assignment of the Charcot-Marie-Tooth neuropathy type 1 (CMT1A) gene to 17p11.2-p12. Am J Hum Genet. 1990;47:680–685.Suche in Google Scholar
[27] Nakamura Y, Lathrop M, O’Connell P et al. A mapped set of DNA markers for human chromosome 17. Genomics. 1988;2:302–309.10.1016/0888-7543(88)90018-3Suche in Google Scholar
[28] Wright EC, Goldgar DE, Fain PR, Barker DF, Skolnick MH. A genetic map of human chromosome 17p. Genomics. 1990;7:103–109.10.1016/0888-7543(90)90524-XSuche in Google Scholar
[29] Hoogendijk JE, Hensels GW, Gabreels-Festen AA et al. De-novo mutation in hereditary motor and sensory neuropathy type I. Lancet. 1992;339:1081–1082.10.1016/0140-6736(92)90668-SSuche in Google Scholar
[30] Raeymaekers P, Timmerman V, Nelis E et al. Estimation of the size of the chromosome 17p11.2 duplication in Charcot-Marie-Tooth neuropathy type 1a (CMT1a). HMSN Collaborative Research Group J Med Genet. 1992;29:5–11.10.1136/jmg.29.1.5Suche in Google Scholar
[31] Palau F, Lofgren A, De Jonghe P et al. Origin of the de novo duplication in Charcot-Marie-Tooth disease type 1A: unequal nonsister chromatid exchange during spermatogenesis. Hum Mol Genet. 1993;2:2031–2035.10.1093/hmg/2.12.2031Suche in Google Scholar PubMed
[32] Suter U, Welcher AA, Ozcelik T et al. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992;356:241–244.10.1038/356241a0Suche in Google Scholar PubMed
[33] Suter U, Moskow JJ, Welcher AA et al. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci USA. 1992;89:4382–4386.10.1073/pnas.89.10.4382Suche in Google Scholar PubMed PubMed Central
[34] Patel PI, Roa BB, Welcher AA et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:159–165.10.1038/ng0692-159Suche in Google Scholar
[35] Valentijn LJ, Bolhuis PA, Zorn I et al. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:166–170.10.1038/ng0692-166Suche in Google Scholar
[36] Timmerman V, Nelis E, Van Hul W et al. The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat Genet. 1992;1:171–175.10.1038/ng0692-171Suche in Google Scholar
[37] Matsunami N, Smith B, Ballard L et al. Peripheral myelin protein-22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot-Marie-Tooth 1A. Nat Genet. 1992;1:176–179.10.1038/ng0692-176Suche in Google Scholar
[38] Valentijn LJ, Baas F, Wolterman RA et al. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;2:288–291.10.1038/ng1292-288Suche in Google Scholar
[39] Roa BB, Garcia CA, Suter U et al. Charcot-Marie-Tooth disease type 1A. Association with a spontaneous point mutation in the PMP22 gene. N Engl J Med. 1993;329:96–101.10.1056/NEJM199307083290205Suche in Google Scholar
[40] Nelis E, Timmerman V, De Jonghe P, Van Broeckhoven C. Identification of a 5’ splice site mutation in the PMP22 gene in autosomal dominant Charcot-Marie-Tooth disease type 1. Hum Mol Genet. 1994;3:515–516.10.1093/hmg/3.3.515Suche in Google Scholar
[41] Chance PF, Alderson MK, Leppig KA et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151.10.1016/0092-8674(93)90058-XSuche in Google Scholar
[42] Pentao L, Wise CA, Chinault AC, Patel PI, Lupski JR. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat Genet. 1992;2:292–300.10.1038/ng1292-292Suche in Google Scholar PubMed
[43] Chance PF, Abbas N, Lensch MW et al. Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet. 1994;3:223–228.10.1093/hmg/3.2.223Suche in Google Scholar PubMed
[44] Reiter LT, Murakami T, Koeuth T et al. A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat Genet. 1996;12:288–297.10.1038/ng0396-288Suche in Google Scholar PubMed
[45] Timmerman V, Rautenstrauss B, Reiter LT et al. Detection of the CMT1A/HNPP recombination hotspot in unrelated patients of European descent. J Med Genet. 1997;34:43–49.10.1136/jmg.34.1.43Suche in Google Scholar PubMed PubMed Central
[46] Nelis E, Van Broeckhoven C, De Jonghe P et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. 1996;4:25–33.10.1159/000472166Suche in Google Scholar PubMed
© 2020 Lupski and Timmerman, published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- MAIN TOPIC: GENETICS OF PERIPHERAL NEUROPATHIES
- Peripheral Neuropathies
- The CMT1A duplication
- Charcot-Marie-Tooth disease and hereditary motor neuropathies – Update 2020
- Charcot-Marie-Tooth neuropathy and pregnancy: general and specific issues
- Advancing Charcot-Marie-Tooth disease diagnostics, through the UK 100,000 Genomes Project
- Peripheral sensory neuropathies – pain loss vs. pain gain
- On the differential diagnosis of neuropathy in neurogenetic disorders
- Spinal muscular atrophy (5qSMA): best practice of diagnostics, newborn screening and therapy
- BERICHTE AUS DER HUMANGENETIK
- Zur Geschichte der Humangenetik
- Der Weg in die Einheit: Das Fach Humangenetik 1990–1991. Ein Erfahrungsbericht
- Personalia
- Personalia
- BVDH-Verbandsmitteilungen
- BVDH-Verbandsmitteilungen
- Aktuelle Nachrichten
- Aktuelle Nachrichten
Artikel in diesem Heft
- Frontmatter
- MAIN TOPIC: GENETICS OF PERIPHERAL NEUROPATHIES
- Peripheral Neuropathies
- The CMT1A duplication
- Charcot-Marie-Tooth disease and hereditary motor neuropathies – Update 2020
- Charcot-Marie-Tooth neuropathy and pregnancy: general and specific issues
- Advancing Charcot-Marie-Tooth disease diagnostics, through the UK 100,000 Genomes Project
- Peripheral sensory neuropathies – pain loss vs. pain gain
- On the differential diagnosis of neuropathy in neurogenetic disorders
- Spinal muscular atrophy (5qSMA): best practice of diagnostics, newborn screening and therapy
- BERICHTE AUS DER HUMANGENETIK
- Zur Geschichte der Humangenetik
- Der Weg in die Einheit: Das Fach Humangenetik 1990–1991. Ein Erfahrungsbericht
- Personalia
- Personalia
- BVDH-Verbandsmitteilungen
- BVDH-Verbandsmitteilungen
- Aktuelle Nachrichten
- Aktuelle Nachrichten