Osteogenesis imperfecta: shifting paradigms in pathophysiology and care in children
-
Stefanie Stasek
 ,
Frank Zaucke
,
Frank Zaucke


Abstract
The formation of functional bone requires a delicate interplay between osteogenesis and osteolysis. Disturbances in this subtle balance result in an increased risk for fractures. Besides its mechanical function, bone tissue represents a key player in the regulation of calcium homeostasis. Impaired bone formation results in bone fragility, which is especially pronounced in osteogenesis imperfecta (OI). This rare genetic disorder is characterized by frequent fractures as well as extraskeletal manifestations. The current classification of OI includes 23 distinct types. In recent years, several new mutations in different genes have been identified, although the exact pathomechanisms leading to the clinical presentation of OI often remain unclear. While bisphosphonates are still the standard of care, novel therapeutic approaches are emerging. Especially, targeted antibody therapies, originally developed for osteoporosis, are increasingly being investigated in children with OI and represent a promising approach to alleviate the consequences of impaired osteogenesis and improve quality of life in OI patients. This review aims to provide insight into the pathophysiology of OI and the consequences of distinct disease-causing mutations affecting the regulation of bone homeostasis. In this context, we describe the four most recently identified OI-causing genes and provide an update on current approaches for diagnosis and treatment.
Introduction
Although typically regarded as a rare condition, the prevalence of osteogenesis imperfecta (OI) is estimated to be 1:10.000, making it one of the most common skeletal diseases in children [1]. The clinical picture was already documented in 1788 by the Swedish physician Olaus Jakob Ekman [2]. However, significant advances in understanding the pathophysiology of OI were first made in the 1980s, when mutations in COL1A1 and COL1A2, coding for the distinct chains of the triple helical collagen type I, were identified as the primary disease-causing genes [3]. Quantitative and structural defects of collagen type I, the critical component of the bone matrix, result in increased bone fragility and a predisposition to fractures after minor trauma. Initially thought to be a monogenic disorder, it is now known that OI involves a broad spectrum of genes, most of them involved in the synthesis of collagen I. The original classification of OI, introduced by David Sillence in 1979, categorized the disease into four types based on clinical severity: Type I (mild), Type II (perinatal lethal), Type III (severe with significant skeletal deformities), and Type IV (moderate severity) [4]. An alternative, genetic classification was implemented in the last years that defined each OI type based on the affected gene. This classification includes to date 23 different types, which reflects the complexity of this continuously growing field [5]. This review will give a brief overview of the underlying pathophysiology including the four most recently identified OI-causing genes. The focus, however, will be on the clinical aspects of OI, including the clinical presentation, diagnostic approaches, and management strategies.
Pathophysiology of OI
The human skeleton is a dynamic organ, constantly undergoing remodeling through the coordinated actions of bone formation and resorption. This remodeling process allows bones to repair microdamage, respond to changes in mechanical load, and ensure that old or damaged bone is replaced by new, stronger bone tissue [6]. Additionally, dynamic remodeling is essential for maintaining stable extracellular calcium levels needed for various physiological functions such as muscle contraction, enzymatic activity, and neuronal signaling.
Bone metabolism is regulated by three cell types: osteoblasts, osteoclasts, and osteocytes. Osteoblasts, derived from mesenchymal stem cells, are responsible for synthesizing new bone matrix and facilitating mineralization. Once osteoblasts become embedded in the matrix, they differentiate into osteocytes, which act as mechanosensors and regulate bone remodeling. Osteoclasts, on the other hand, are derived from hematopoietic stem cells and resorb bone [7].
The bone forming activity of osteoblasts and bone resorption by osteoclasts is tightly regulated. Mechanical loading, hormones (e.g., growth hormone, PTH, and estrogen), nutritional factors, along with local cytokines and growth factors, regulate this complex balance. Muscle activity plays a critical role in bone remodeling by generating mechanical forces that stimulate bone formation. Osteocytes detect mechanical strain through their dendritic processes within the bone matrix and transduce these signals into biochemical responses [8].
The signaling pathways regulating bone metabolism are highly complex, and a comprehensive discussion is beyond the scope of this review. However, some are essential to understand, as they have already been targeted by pharmacological therapies currently under investigation for the use in OI. These pathways include the Wnt/β-catenin, RANK/RANKL/OPG, and TGF-β pathway.
The Wnt/β-catenin signaling pathway is crucial for promoting osteoblast differentiation and activity. Wnt ligands bind to the transmembrane receptor “Frizzled” and its coreceptor, the low-density lipoprotein receptor–related protein −5 or −6 (LRP5/6) receptors on osteoblasts, leading to β-catenin stabilization and translocation to the nucleus, stimulating the transcription of genes involved in bone formation [9]. Sclerostin, a protein produced by osteocytes, binds to the LRP5/6 receptor and disrupts its interaction with Wnt proteins, thereby acting as a negative regulator of osteoblast activity and bone formation [10]. The RANK/RANKL/OPG pathway plays a key role in osteoclast regulation. RANKL (Receptor Activator of Nuclear Factor κB Ligand), produced by osteoblasts and osteocytes, binds to its receptor RANK on osteoclast precursors, promoting their maturation and activation [11], 12]. Osteoprotegerin (OPG), a secreted decoy receptor for RANKL, is produced by osteoblasts and can inhibit this interaction. TGF-β has a context-dependent effect on bone formation and absorption and interacts with the RANKL pathway. Upon binding to its receptors on osteoblast precursors, TGF-β triggers SMAD proteins, which translocate to the nucleus to upregulate expression of collagen and other components of the extracellular matrix. However, in certain conditions where increased bone resorption is required, TGF-β can enhance the expression of RANK on the surface of osteoclasts and bone marrow stromal cells, thereby promoting osteoclast differentiation and activity [13], 14].
A disruption of this complexly balanced system of bone formation and resorption can cause various bone disorders characterized by impaired bone stability. In osteoporosis, a condition which is commonly seen in elderly woman, a decline in estrogen levels after menopause causes an increased osteoclast activity and loss of bone mass [15]. Conversely, disturbances of the structural composition of bone can also impair its stability. OI is a genetic disorder, primarily caused by quantitative or qualitative defects of collagen I, the main structural protein of most connective tissue in the human body, making OI a multisystemic disease.
OI as a collagen I–related disorder
Collagen I is the primary organic component of bone matrix. It provides a scaffold for mineral deposition and imparts tensile strength to the bone. Collagen I is primarily synthesized by osteoblasts and fibroblasts and undergoes extensive post-translational modifications within the endoplasmic reticulum (ER). Assembled into a stable triple-helical structure, the procollagen chains are secreted into the extracellular matrix. Intra- and intermolecular crosslinks provide mechanical strength of collagen fibers [16].
Mutations in COL1A1 and COL1A2 can lead to OI with a broad phenotypic variety, ranging from mild to perinatal lethal, depending on the specific type of mutation. Mutations such as stop mutations or frameshift mutations can introduce a premature stop codon into the mRNA. In many cases, this leads to nonsense-mediated decay, and no collagen is produced from the mutated allele, resulting in a quantitative collagen deficiency. However, in heterozygous patients, the presence of one normal allele allows for partial compensation, typically leading to a milder phenotype [4].
In contrast, missense mutations that affect glycine residues essential for the proper folding of the triple-helix cause a more severe phenotype. Misfolded collagen is incorporated into the extracellular matrix, where it forms defective collagen fibrils that stimulate bone resorption by osteoclasts. Furthermore, intracellular accumulation of structurally abnormal collagen causes cellular stress, chronic low-grade inflammation, and osteoblast apoptosis [17].
While mutations in COL1A1 and COL1A2 are the most common causes of OI, there are several other genes involved in various processes related to collagen production, post-translational modification, bone formation, and mineralization that can lead to different forms and severities of OI when mutated. In addition to genes causing classical OI, some mutations result in phenotypes that exhibit bone fragility similar to OI, but these conditions may historically be categorized under other syndromes with overlapping features. These are often referred to as “OI-like phenotypes.” A detailed overview of all genes associated with OI and OI-like phenotypes is provided in Table 1.
Summary of genes and phenotypic characteristics of OI and OI-like phenotypes.
| Category | Affected pathway | Affected gene (mode of inheritance) | Protein | Characteristics and phenotype | Genetic OI type | OMIM |
|---|---|---|---|---|---|---|
| Collagen synthesis and modifications | Collagen synthesis | COL1A1 COL1A2 (AD) |
Pro-alpha 1(I)/pro-alpha 2 (I) chain of type I collagen | I | 166200 | |
| II | 166210 | |||||
| III | 259420 | |||||
| IV | 166220 | |||||
| mRNA stabilization | TENT5A (AR) | Terminal nucleotidyl-transferase 5A (TENT5A) alternative: family with sequence similarity 46, member A (FAM46A) |
|
XVIII | 617952 | |
| Hydroxylation of proline and lysine residues | P4HB (AR) | Protein disulfide isomerase (PDI) alternative: prolyl 4-hydroxylase subunit beta (P4HB) |
|
Not classified | 112240 | |
| CRTAP (AR) | Cartilage-associated protein (CRTAP) | VII | 610682 | |||
| P3H1 Alternative: LEPRE1 (AR) |
Prolyl-3-hydroxylase 1 (P3H1) |
|
VIII | 610915 | ||
| PPIB (AR) | Peptidyl-prolyl-cis-trans-isomerase B (PPIB) alternative: cyclophilin B |
|
IX | 259440 | ||
| Chaperone-assisted formation of procollagen triple helix and quality control | SERPINH1 (AR) | Serpin peptidase inhibitor, clade H, member 1 alternative: heat shock protein 47 (HSP47) |
|
X | 613848 | |
| FKBP10 (AR) | Peptidyl-prolyl cis-trans isomerase FKBP10 (PPIase FKBP10) alternative: 65 kDa FK506-binding protein (FKBP65) |
|
XI | 610968 | ||
| KDELR2 (AR) | (Lys-Asp-Glu-Leu) endoplasmic reticulum protein receptors 2 (KDELR2) |
|
XXI | 619131 | ||
| ER homeostasis | CREB3L1 (AR) | Cyclic AMP-responsive element-binding protein 3-like protein 1 (CR3L1) |
|
XVI | 616229 | |
| MBTPS2 (XLR) | Membrane-bound transcription factor site-2 protease alternative: endopeptidase S2P |
|
XIX | 301014 | ||
| TMEM38B (AR) | Transmembrane protein 38B (TMEM38B) alternative: trimeric intracellular cation channel type B (TRIC-B) |
|
XIV | 615066 | ||
| Vesicular transport to Golgi apparatus and extracellular space | SEC24D (AR) | Selected protein acidic and rich in cysteine (SPARC) |
|
Not classified | 616294 | |
| Extracellular proteolytic processing and conversion into mature collagen | BMP1 (AR) | Bone morphogenic protein 1 (BMP1) |
|
XIII | 614856 | |
| Collagen crosslinking | PLOD2 (AR) | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 (PLOD2) alternative: lysyl hydroxylase 2 (LH2) |
|
Not classified | 609220 | |
| Bone homeostasis | ECM mineralization | SPARC c | Selected protein acidic and rich in cysteine (SPARC) |
|
XVII | 616507 |
| Transduction of mechanical stimuli on osteocytes | PLS3 (XLD) | Plastin-3 (PLS3) |
|
Not classified | 300910 | |
| Regulation of osteoblast differentiation or function and collagen synthesis | WNT1 (AR) | Proto-oncogene Wnt1 (wingless-type MMTV integration site family, member 1, WNT1) |
|
XV | 615220 | |
| MESD (AR) | Mesoderm development LRP chaperone (MESD) |
|
XX | 618644 | ||
| TAPT1 (AR) | Transmembrane anterior posterior transformation 1 (TAPT1) | Not classified | 616897 | |||
| SP7 (AR, AD) | Transcription factor Sp7 alternative: zinc finger protein osterix |
XII | 613849 | |||
| Regulation of osteoclast differentiation or function | SERPINF1 (AR) | Pigment epithelium-derived factor (PEDF) |
|
VI | 613982 | |
| IFITM5 (AR) | Interferon-induced transmembrane protein 5 (IFITM5) alternative: bone-restricted interferon-induced transmembrane protein-like protein (BRIL) |
|
V | 610967 | ||
| CCDC134 (AR) | Secreted coiled-coil domain containing protein 134 |
|
XXII | 619795 | ||
| Unknown | Unknown | PHLDB1 (AR) | Pleckstrin homology-like domain, family B member 1 |
|
XXIII | 620639 |
-
AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive; XLD, X-linked dominant; BMD, bone mineral density. a IFAP: ichthyosis follicularis, alopecia, photophobia. BRESEK: bronchiectasis, restrictive pulmonary disease, encephalopathy, skeletal contractures, and ichthyosis. BRESHECK: brain anomalies, ectodermal dysplasia, skeletal malformations, and cryptorchidism. bCole–Carpenter syndrome is an OI-like syndrome with bone fragility, craniosynostosis, ocular proptosis, hydrocephalus, and distinctive facial features (frontal bossing, midface hypoplasia, micrognathia) [20]. Type I and II exist, differentiation based on genetic findings. cSPARC has a function as a chaperone for collagen folding and is also involved in ECM mineralization. dOlmsted syndrome is a rare keratinization disorder characterized by palmoplantar keratoderma and perioral keratotic plaques [62], 63]. eBruck syndrome is characterized by bone fragility, osteoporosis, and congenital joint contractures. Two types exist classified based on genetic finding.
Novel genetic insights from inherited bone fragility syndromes
Over the last years, genetic analyses of patients with the clinical phenotype of OI led to the discovery of novel genes. The list of OI types has been updated accordingly and currently ends with type XXIII [5]. However, not all newly identified genes have been officially classified as distinct OI types. In the following section, the four latest described genes and the function of the encoded proteins are briefly summarized.
KDELR2 (OI type XXI)
In 2020, three different mutations in KDELR2 (KDEL receptor 2) were first identified in six affected individuals from four families [32]. Later, another two missense variants were reported. All affected individuals had been diagnosed with progressively deforming OI or OI type II; in the latter cases, additional neurodevelopmental features were reported [33]. KDELR2 encodes for a seven transmembrane domain receptor that localizes mainly to the ER, the intermediate ER–Golgi compartment, and the cis-Golgi complex indicating a crucial function in the secretion of proteins. KDELR2 binds proteins harboring a KDEL-like motif and regulates their trafficking between compartments. Interestingly, the collagen chaperone heat shock protein 47 (Hsp47) is one of the substrates and KDELR2 mutations lead to a reduction of intracellular levels and an increased secretion of Hsp 47. Finally, this results in impaired extracellular collagen fibril formation [32].
CCDC134 (OI type XXII)
Whole exome sequencing of three Moroccan patients from two families with severe OI identified a homozygous missense mutation in the first codon of CCDC134 [58]. Later, a male infant [60] and a Brazilian boy with severe OI harboring exactly the same mutation were described [59]. The gene encodes for the secreted coiled-coil domain containing protein 134 that is involved in the transcriptional regulation and signal transduction of MAP kinases. The mutation causes a loss of protein function and the pathomechanisms might involve a dysregulated MAPK/ERK signaling in osteoprogenitors. However, the exact mechanism as well as the overlap with other bone dysplasias with dysregulated MAPK/ERK signaling is not yet fully understood.
PHLDB1 (OI type XXIII)
In 2023, a new mild OI phenotype with regressive spondylometaphyseal alterations was reported in five infants from two unrelated families. In both families, the parents of the affected individuals were first cousins [61]. Two biallelic frameshift variants were identified in the candidate gene PHLDB1 that encodes the protein Pleckstrin homology-like domain, family B, member 1. The frameshift mutations lead to the loss of the C-terminal PH domain. The function of PHLDB1 is largely unknown. It interacts with and is a modulator of the Akt protein kinase [64], and it was suggested that this interaction might affect collagen biosynthesis or osteogenic differentiation [61].
TAPT1
Mutations in the gene TAPT1 were first reported to result in a complex and early osteochondrodysplasia with features of lethal OI in two consanguineous families from Morocco and Syria [51]. In 2023, two groups independently described novel homozygous mutations in TAPT1 in seven patients from consanguineous families that survived and were clinically diagnosed with OI [65], 66]. The function of the encoded protein TAPT1 (transmembrane anterior posterior transformation 1 protein) is not yet completely understood, but mutations led to a disrupted ciliogenesis and an altered morphology of the Golgi apparatus that might affect collagen secretion [51]. Molecular analysis also revealed an impaired collagen fibril formation that might explain brittle bones [66]. Currently TAPT1 is not yet classified as an OI-causing gene.
Clinical presentation
Osteogenesis imperfecta presents with a wide spectrum of clinical manifestations, ranging from mild to severe, depending on the specific type of OI and the underlying genetic mutations. While some infants present with multiple fractures already at birth, others remain asymptomatic until fractures occur later in childhood, often after they start walking. While early medical and physiotherapeutic interventions can help those with mild to moderate forms to maintain mobility, more severely affected individuals are typically wheelchair dependent throughout life.
Fractures occur typically in the long bones of the upper and lower extremities as well as in vertebrae and can lead to severe skeletal deformities, such as bowed limbs and scoliosis (Figure 1). Thorax deformities can restrict lung expansion and impair respiratory function [67]. In severe cases, scoliosis may cause neurological complications due to compression of the spinal cord [68], 69]. Basilar invagination is a condition more commonly observed in severe forms of OI, where recurrent microfractures cause the skull base to flatten, leading to spine migration into the skull [70]. This can compress the brainstem and cerebellum, causing hydrocephalus due to disrupted cerebrospinal fluid circulation and cranial nerve damage. Early screening for new neurological symptoms is crucial. Growth is often affected due to disruptions in the growth plates, reflecting the disease’s profound impact on bone development [71]. Repeated immobilization following fractures contributes to muscle atrophy and secondary bone loss, further aggravating bone fragility. Chronic pain is often underrecognized by parents and caregivers and consequently insufficiently managed [72]. This significantly impacts quality of life in children and adolescents with OI, as highlighted by the recently published IMPACT survey, which documented experiences of individuals with OI and their caregivers [73], 74].
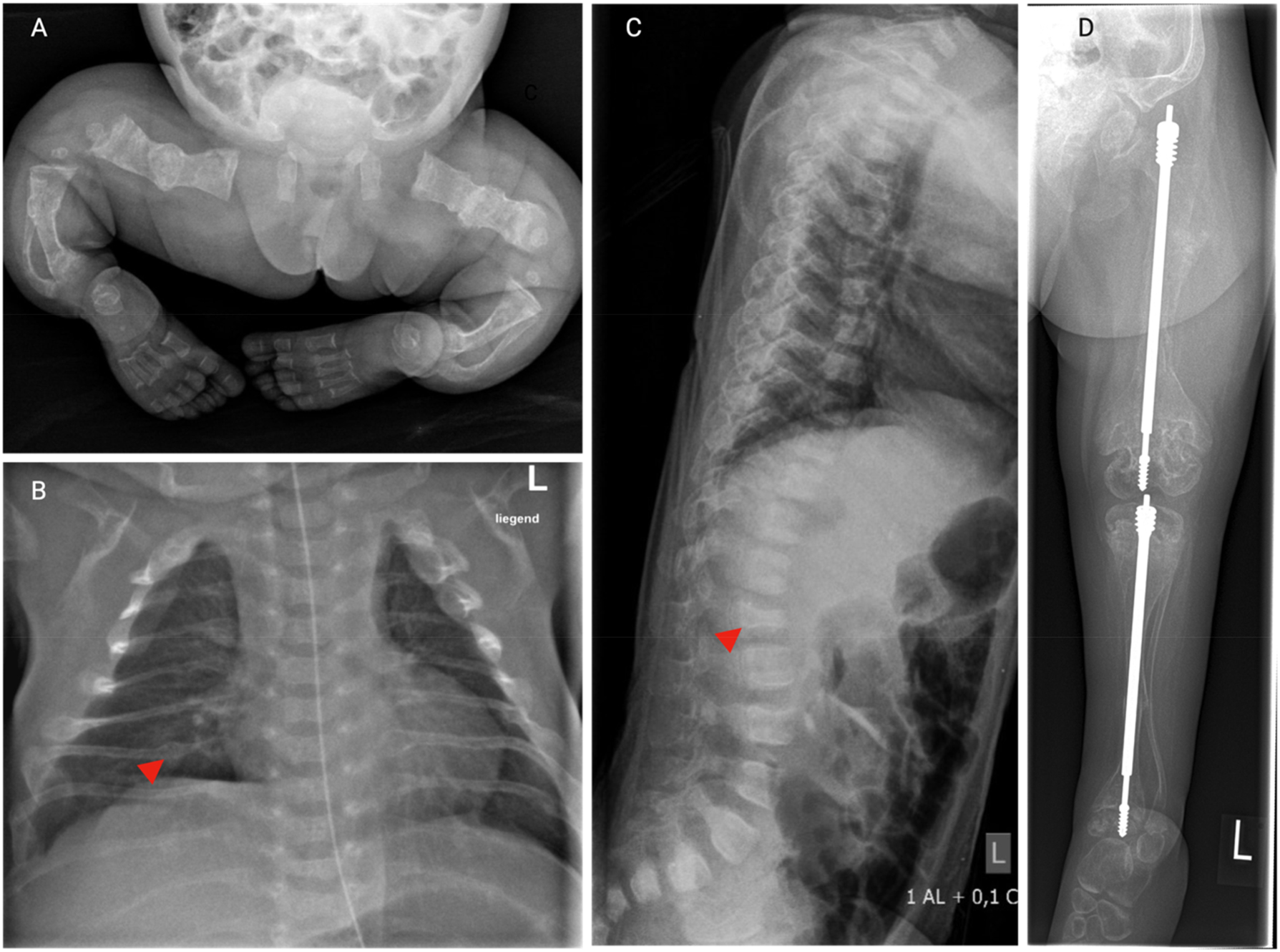
Skeletal manifestations of OI. Series of X-rays from a young girl with severe OI type VIII. (A) X-ray of the lower body 3 days after birth showing multiple healed fractures, generalized osteopenia, bowing and shortening of the long bones. (B) Chest X-ray 3 days after birth. Notice the thin rips with irregularly shaped, thickened areas (arrow head) consistent with healed fractures. (C) Lateral X-ray of the spine in the age of 1.5 years reveals flattened, irregularly shaped vertebral bodies (arrow head), indicative of compression fractures, along with pronounced kyphosis. (D) X-ray of the lower extremity in the age of 5 years after surgical insertion of telescopic nails in the tibia and femur. Notice the generalized osteopenia, thin cortical bones, multiple fractures in the tibia and femur, and bowing of the thin fibula. The distal metaphyses of the femur appear wide and irregular.
Extraskeletal manifestations
Due to the widespread role of collagen I in various tissues, OI also presents with different extraskeletal manifestations (Figure 2). Characteristic facial features like a triangular face shape, blue sclerae, and brittle and discolored teeth (dentinogenesis imperfecta) can provide diagnostic indicators [75], 76]. Neurodevelopmental delay indicate the possibility of OI type XV, a form associated with cognitive impairment caused by mutations in WNT1 [77].
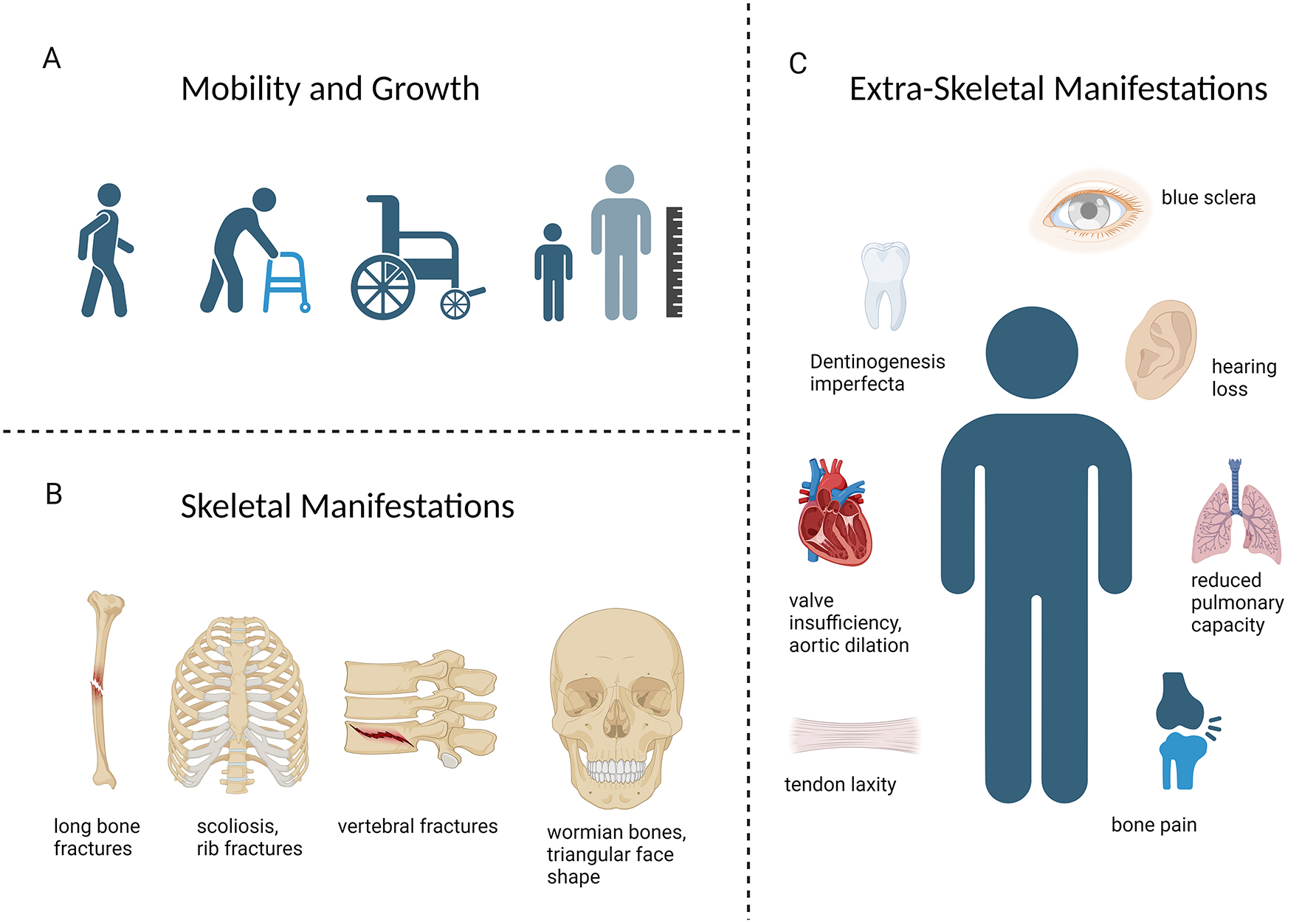
Clinical manifestations of OI. (A) The phenotype of OI ranges from patients with mild forms who maintain full mobility, to those who rely on walking aids or are wheelchair dependent. Short stature is commonly present. (B) Skeletal manifestations of OI include fractures and deformities of the long bones, scoliosis, vertebral fractures, and craniofacial features like wormian bones, and a characteristic triangular face shape. (C) Extraskeletal manifestations occur due to the widespread presence of collagen I in various organ systems. Structural abnormalities in heart valves, blood vessels, and the lung parenchyma can restrict cardiopulmonary function and cause potentially lethal complications. Blue sclera and dentinogenesis imperfecta are characteristic facial features. Early-onset hearing loss and ligamentous laxity can also occur.
Other manifestations usually appear in adulthood and are, therefore, in most cases not relevant for the diagnosis but need to be considered by the medical team and care takers as they have significant impact on the patient’s quality of life. Fragility and deformation of the ossicles in the middle ear, as well as the abnormal remodeling of the otic capsule in the inner ear, can cause early hearing loss [78]. Abnormalities in collagen fibers can affect the structural integrity of heart valves and blood vessels, potentially leading to cardiovascular complications such as valve insufficiency and increased susceptibility to aortic dilation or rupture [79]. Thoracic deformities and intrinsic defects in the lung parenchyma restrict lung expansion and function [67]. In OI type II, the most severe type, prenatal rib fractures and underdeveloped lungs lead to respiratory failure and usually cause death in the perinatal period [80].
Diagnosis
To confirm the diagnosis of OI, a combination of clinical evaluation, imaging, and genetic testing is typically required. Importantly, not all patients exhibit the full spectrum of symptoms and individuals with milder forms may be misdiagnosed with conditions like juvenile osteoporosis or remain undiagnosed until adulthood. However, certain clinical signs should raise suspicion of OI. Children typically present with recurrent fractures without a clear cause or with an injury disproportionate to the reported trauma, a constellation of symptoms that often alerts clinicians and raises suspicion of child abuse.
Like other monogenetic diseases, a detailed family history might pave the way to the correct diagnosis. Recurrent fractures in one parent can guide the physician in the direction of OI, although not all patients have a positive family history, as spontaneous mutations are common [81].
Radiographic imaging is often the first diagnostic tool, revealing generalized osteopenia, multiple fractures at different stages of healing and bone deformities. Wormian bones are extra bone pieces that occur particularly along the lambdoid suture. So-called popcorn calcifications are disorganized hyperdense lines around the metaphyseal growth plates of long bones [82], 83]. The measurement of bone density via dual-energy X-ray absorptiometry (DXA) scans plays a supporting role in the diagnosis of OI. It does not help to differentiate OI from other bone diseases like osteoporosis, but it remains an important tool for monitoring effectiveness of antiresorptive treatments.
Laboratory diagnostics including measurements of calcium, phosphate, alkaline phosphatase (AP), vitamin D, and parathyroid hormone levels are recommended to exclude metabolic bone disorders associated with increased bone fragility, such as nutritional rickets or X-linked hypophosphatemia [81].
Genetic testing is currently gaining more and more relevance and the number of identified genes contributing to the OI phenotype is continuously growing. Genetic and phenotypical overlap with other connective tissue disorders, such as Ehlers–Danlos syndrome and Bruck syndrome, make the classification of patients into distinct diagnostic categories increasingly challenging [84]. For diagnosing OI, genetic testing is not mandatory. However, the deeper understanding of the genetic background and pathophysiology of OI resulted in a paradigm shift in the approach to manage this condition, as patients are no longer treated as one homogenous group. Instead, with the increasing knowledge of the disease’s pathophysiology, more targeted therapeutic strategies are currently investigated.
Management and therapy
The management of OI is primarily focused on symptomatic and supportive care, as there is currently no curative treatment available. A multidisciplinary approach (Figure 3) involving pediatricians, orthopedic surgeons, physiotherapists, and many other disciplines is essential to account for the complex needs of OI patients [81], 85]. Patients with OI and their caregivers, including health care professionals, often develop a fear of injuries and fractures [86]. Parents and hospital staff should, therefore, be trained how to handle OI patients safely from birth on. Overprotection due to fear of fractures impairs the social and motor development of the child. Therefore, education of the caregivers is a key component of the multidisciplinary treatment approach [87]. Psychological support and patient support groups can help patients and their families coping with fears and challenges of living with a chronic condition.
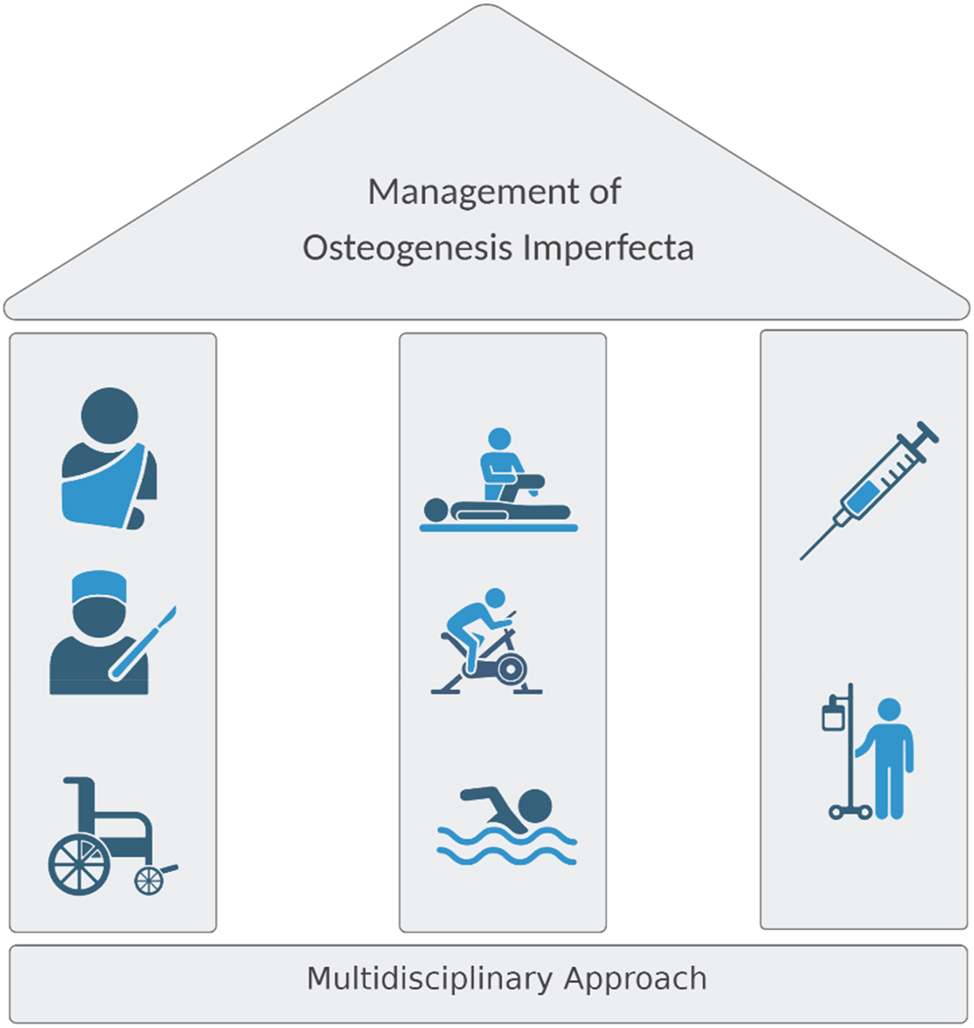
The three pillars of OI therapy. The multidisciplinary approach for OI patients includes physiotherapy, orthopedic management of acute fractures, and surgical correction of deformities and pharmacological therapy. Psychological support should always be provided to help the family managing a chronic, life-long condition.
Orthopedic management involves acute fracture treatment and the correction of chronic bone deformities. Fractures are usually treated by immobilization (casts, splints, bandages) while severe deformities and dislocated fractures require surgical interventions. Telescopic intramedullary rods, which prolong as the child grows, offer long-term stability and help maintain bone alignment [88]. Spinal surgery is indicated for progressive spinal deformities to correct misalignments, improve respiratory function, relieve pain through neural decompression, and enhance spinal stability [68], 69]. Concerns about potential side effects and lack of standardized pain assessment in young children often lead to insufficient pain management in chronically ill children [74], 89]. However, adequate pain management is essential to prevent immobilization due to fear of new fractures; therefore, its effectiveness should be evaluated regularly.
Physiotherapy plays a key role in enhancing mobility, motor function, and independence in daily activities [87]. Rehabilitation should aim to reduce the risk of fractures and deformities, while allowing the child to safely engage in activities that promote development. Especially after fractures, it is important to prevent contractures and joint misalignment and reduce secondary osteopenia from immobilization. Exercises focusing on improving muscle strength, coordination, and endurance are essential. Weight-bearing activities, isometric exercises, and functional training can significantly improve walking ability and bone mineral density (BMD) in OI patients [90]. Specific physiotherapy modalities exist for OI patients that provide a low-impact environment and minimize the risk of injury, such as whole-body vibration training and aquatic physiotherapy [87], 91].
Pharmacological treatment approaches
The pharmacological management of OI focuses on two primary therapeutic strategies: antiresorptive and osteoanabolic therapies. Both approaches have been adapted from treatments commonly used in adults with osteoporosis, although their application in OI and in children remains largely under investigation. An overview of the current medical treatment approaches is provided in Figure 4.
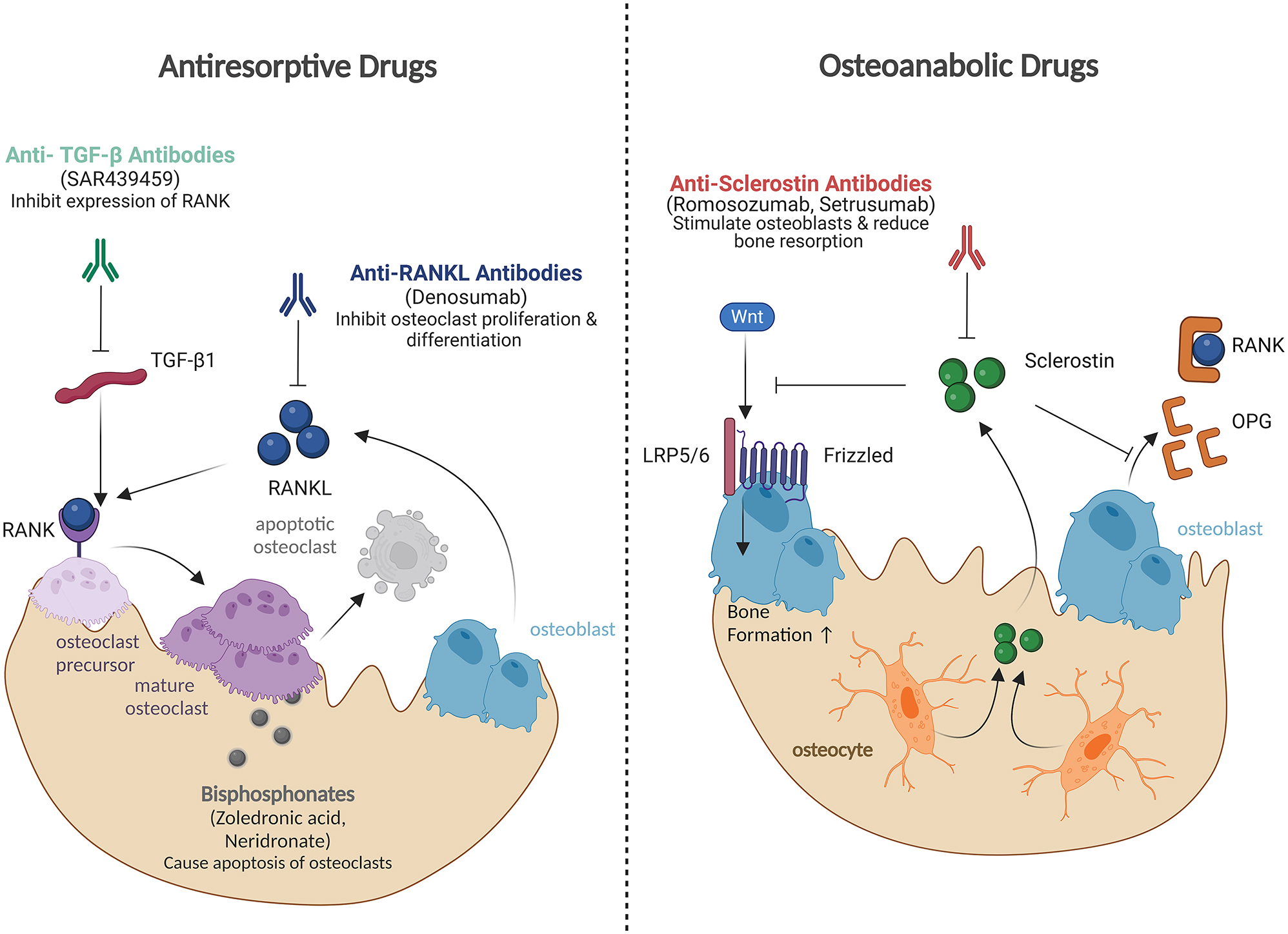
Pharmacological treatment approaches for OI. Most of these approaches are currently being investigated in clinical trials and are not yet approved for OI. Antiresorptive drugs primarily inhibit bone resorption. Denosumab, an anti-RANKL antibody, prevents RANKL-induced osteoclast proliferation and differentiation. Anti-TGF-β antibodies reduce TGF-β-stimulated osteoclastogenesis and expression of RANK on osteoclast precursors. Bisphosphonates (zoledronic acid, neridronate), the current standard of care for OI, induce apoptosis of osteoclasts. Osteoanabolic drugs primarily promote bone formation. Antisclerostin antibodies (romosozumab, setrusumab) inhibit sclerostin, leading to activation of the Wnt signaling pathway and subsequently stimulation of osteoblast activity. Additionally, antisclerostin antibodies exert antiresorptive effects by promoting the secretion of osteoprotegerin (OPG), which inhibits the RANK–RANKL interaction.
Antiresorptive drugs
Bisphosphonates
Bisphosphonates, first introduced as a pharmacological treatment for children with OI in the 1980s, still remain the standard of care [81], 92]. These compounds bind to hydroxyapatite crystals in bone and are internalized by osteoclasts upon their activation and attachment to the bone surface. Once internalized, bisphosphonates disrupt key metabolic pathways within the osteoclasts, leading to functional inhibition or apoptosis, thereby reducing bone resorption. This results in increases of bone mass, reduced incidence of vertebral compression fractures and bone pain, and vertebral remodeling [93]. While generally well tolerated, short-term side effects include flu-like symptoms, such as fever, bone pain, and fatigue [94]. Oral administration of bisphosphonates may cause gastrointestinal irritation; therefore, intravenous application is usually preferred [95]. Since mild hypocalcemia can occur due to decreased bone resorption 24–48 h after administration of bisphosphonates, calcium and vitamin D status should be assessed before initiating therapy [96]. Osteonecrosis of the jaw is a serious side effect documented in adult patients treated with bisphosphonates. However, there is no evidence of an increased risk of bisphosphonate-related osteonecrosis in children with OI [97], 98]. The response to bisphosphonates in children with OI is variable and seems to be related to the underlying genetic mutation: Children with structural collagen defects or nonautosomal-dominant inheritance showed less increase of BMD with zoledronic acid therapy [99] and also type VI OI is characterized by a poor response to bisphosphonates [55]. This highlights the limitations of bisphosphonate therapy and the need for tailored therapeutic approaches for certain genetic subtypes.
Anti-RANKL antibodies
Denosumab is a monoclonal antibody administered via subcutaneous injection every 3 to 6 months. It specifically targets and inhibits the receptor activator of nuclear factor kappa-B ligand (RANKL), thereby inhibiting RANKL-induced differentiation of osteoclast precursors to mature bone resorbing cells. Furthermore, denosumab reduces activity and survival time of mature osteoclasts [100], [101], [102], [103]. In contrast to bisphosphonates, which remain in the organism for decades [104], denosumab is fully metabolized after a few months [105].
Denosumab effectively increased bone mass and areal BMD measured by DXA and reduced fracture incidence in osteoporosis patients [106], 107]. A single-center, 1-year study by Liu et al. (2024) showed previously denosumab to be as effective as the bisphosphonate zoledronic acid in increasing aBMD in children with OI [108]. However, large-scale studies investigating the effects of denosumab in OI are currently lacking due to safety concerns about calcium-related side effects. A pronounced rebound effect 3–7 months after denosumab injections was reported in children and adults [109], 110]. This rebound is associated with excessive bone resorption, leading to a loss of bone mass to pretreatment levels, increased calcium mobilization to the circulation, and an increased risk of fractures [111]. Case reports have described severe hypercalcemia requiring acute hospital admission with hydration and antiresorptive therapy to prevent acute kidney failure [108], 112], 113]. These safety concerns led to the early termination of a large multicenter study that aimed to investigate denosumab in children with OI [114]. Denosumab is, therefore, currently not recommended for the treatment of pediatric OI patients.
Anti-TGF-β antibodies
Excessive TGF-β signaling has been identified in both dominant and recessive forms of OI [115], 116]. Anti-TGF-β treatment using neutralizing antibodies increased bone mass and improved bone architecture in OI mouse models [115] and in adults receiving fresolimumab [117]. A phase 1 clinical study is currently investigating the safety and tolerability of a single dose of SAR439459, another human anti-TGF-β monoclonal antibody, in adult participants with OI [118].
Osteoanabolic drugs
Recombinant human PTH
Parathyroid hormone (PTH) regulates bone metabolism by stimulating osteoclast activity to increase calcium release into the bloodstream. However, when administered intermittently, PTH stimulates bone formation [119]. In adults with OI, particularly patients with OI type I, teriparatide, a recombinant human PTH, has shown significant increases in BMD compared to placebo and bisphosphonates [120], 121]. However, preclinical studies in rodents receiving high-dose PTH treatment raised concerns about an increased risk of osteosarcoma with long-term use [122]. Therefore, this therapy is not used in children. The TOPaZ trial is currently investigating whether a 2-year treatment with teriparatide followed by a single infusion of zoledronic acid can reduce fracture risk in adult patients with OI compared to standard therapy with zoledronic acid alone [123].
Antisclerostin antibodies
Sclerostin, a glycoprotein predominantly expressed by osteocytes, regulates bone formation by binding to the LRP5/6 receptors on osteoblasts. It inhibits the Wnt/β-catenin signaling pathway, which is essential for osteoblast proliferation, differentiation, and activity. Inhibiting sclerostin reduces β-catenin degradation, thereby enhancing osteoblast activity and bone formation [124], 125]. In murine OI models, treatment with an antisclerostin antibody decreased fracture rates and increased cortical bone thickness and BMD [126], 127]. The effectiveness of the antisclerostin antibody setrusumab was already shown in adult osteoporosis and OI patients [128]. Currently, two antisclerostin antibodies are investigated in phase 3 multicenter studies in pediatric patients with OI to evaluate their effectiveness compared to bisphosphonates: Romosozumab, a monoclonal antibody, which is injected subcutaneously monthly [129], and setrusumab, which is administered monthly via intravenous infusion [130].
Future therapy approaches
Emerging therapies such as mesenchymal stem cell (MSC) therapy and gene therapy offer promising new avenues for treating OI. These approaches aim to address the underlying genetic and cellular defects of the disease, potentially providing long-term or even curative effects.
MSC therapy involves the use of multipotent stromal cells that can differentiate into osteoblasts and chondrocytes. Preclinical studies with animal models have shown that human fetal MSCs can target bone lesions, reduce brittleness, and increase bone thickness and collagen content in OI mice [131]. In humans, bone marrow transplants from siblings have increased growth rates and reduced fractures in children with OI type III [132]. Prenatal MSC transplantation followed by additional postnatal treatments improved growth and fracture reduction in severe OI types III and IV [133]. Challenges include ensuring the effective integration and long-term viability of transplanted MSCs, along with risks such as immune rejection and potential malignant transformation. The BOOSTB4 clinical trial is currently evaluating the long-term safety and efficacy of postnatal vs. prenatal and postnatal administration of fetal MSCs in severe OI [134].
Gene therapy aims to treat OI by directly addressing its genetic defect. Techniques like CRISPR/Cas9 are being explored for precise gene editing in other genetic diseases [135]. However, challenges include developing effective delivery systems and avoiding potential off-target effects.
Summary
OI is one of the most common inherited skeletal disorders. Over the past decades, there has been substantial progress in understanding its genetic background and underlying molecular mechanisms, as well as in improving treatment strategies. Nevertheless, current treatment for OI remains primarily supportive. Future approaches may focus increasingly on personalized, gene-targeted therapies to offer long-term solutions for even the most severe cases. As our understanding of the molecular mechanisms in this disease deepens, we are moving closer to curative therapies that could transform patient care in the future.
Funding source: Deutsche Forschungsgemeinschaft
Award Identifier / Grant number: FOR2722
Acknowledgments
Figures were created in BioRender. Metz, M. (2024) https://BioRender.com/q44t448.
-
Research ethics: Not applicable.
-
Informed consent: Not applicable.
-
Author contributions: All authors were involved in structuring and preparation of the manuscript. SS wrote the manuscript and prepared the figures. FZ contributed by describing the four new genes. JE, FZ, and IA proofread the biochemical section. OS, SR, and HH provided critical revisions and contributed to the clinical section. All authors reviewed and approved the final manuscript. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: All other authors state no conflict of interest.
-
Research funding: Parts of this work were supported by DFG funding FOR 2722 (SE2373/1–2384170921; ZA 561/3–2407168728; ET 144/3–2384170921). SS and HH work as Clinician Scientists within the project.
-
Data availability: Not applicable.
References
1. Orioli, IM, Castilla, EE, Barbosa-Neto, JG. The birth prevalence rates for the skeletal dysplasias. J Med Genet 1986;23:328–32. https://doi.org/10.1136/jmg.23.4.328.Search in Google Scholar PubMed PubMed Central
2. Olaus, JE, Acrel, JG. Dissertatio medica descriptionem et casus aliquot osteomalaciæ sistens. Uppsala: Disciplinary Domain of Medicine and Pharmacy, Faculty of Medicine; 1788.Search in Google Scholar
3. Marini, JC, Forlino, A, Bächinger, HP, Bishop, NJ, Byers, PH, De Paepe, A, et al.. Osteogenesis imperfecta. Nat Rev Dis Prim 2017;3:1–19. https://doi.org/10.1038/nrdp.2017.52.Search in Google Scholar PubMed
4. Shapiro, JR. Clinical and genetic classification of osteogenesis Imperfecta and epidemiology. London: Academic Press; 2014:15–22 pp.10.1016/B978-0-12-397165-4.00002-2Search in Google Scholar
5. Jovanovic, M, Marini, JC. Update on the genetics of osteogenesis imperfecta. Calcif Tissue Int 2024;115:891–914. https://doi.org/10.1007/s00223-024-01266-5.Search in Google Scholar PubMed PubMed Central
6. Kenkre, JS, Bassett, JHD. The bone remodelling cycle. Ann Clin Biochem 2018;55:308–27. https://doi.org/10.1177/0004563218759371.Search in Google Scholar PubMed
7. Mohamed, AMFS. An overview of bone cells and their regulating factors of differentiation. Malays J Med Sci 2008;15:4.Search in Google Scholar
8. Qin, L, Liu, W, Cao, H, Xiao, G. Molecular mechanosensors in osteocytes. Bone Res 2020;8:1–24. https://doi.org/10.1038/s41413-020-0099-y.Search in Google Scholar PubMed PubMed Central
9. Ozhan, G, Iscan, E, Schambony, A, Liu, Y, Ren, Q, Chen, J. LRP5 and LRP6 in Wnt signaling: similarity and divergence. Front Cell Dev Biol 2021;9. https://doi.org/10.3389/FCELL.2021.670960.Search in Google Scholar
10. Marini, F, Giusti, F, Palmini, G, Brandi, ML. Role of Wnt signaling and sclerostin in bone and as therapeutic targets in skeletal disorders. Osteoporos Int 2023;34:213–38. https://doi.org/10.1007/s00198-022-06523-7.Search in Google Scholar PubMed
11. Boyce, BF, Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 2008;473:139–46. https://doi.org/10.1016/j.abb.2008.03.018.Search in Google Scholar PubMed PubMed Central
12. Anandarajah, AP. Role of RANKL in bone diseases. Trends Endocrinol Metab 2009;20:88–94. https://doi.org/10.1016/j.tem.2008.10.007.Search in Google Scholar PubMed
13. Rahman, MS, Akhtar, N, Jamil, HM, Banik, RS, Asaduzzaman, SM. TGF-β/BMP signaling and other molecular events: regulation of osteoblastogenesis and bone formation. Bone Res 2015;3:15005. https://doi.org/10.1038/boneres.2015.5.Search in Google Scholar PubMed PubMed Central
14. Yan, T, Riggs, BL, Boyle, WJ, Khosla, S. Regulation of osteoclastogenesis and RANK expression by TGF‐β1. J Cell Biochem 2001;83:320–5. https://doi.org/10.1002/jcb.1200.Search in Google Scholar PubMed
15. Buckley, L, Humphrey, MB. Glucocorticoid-induced osteoporosis. N Engl J Med 2018;379:2547–56. https://doi.org/10.1056/NEJMcp1800214.Search in Google Scholar PubMed
16. Forlino, A, Cabral, WA, Barnes, AM, Marini, JC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 2011;7:540–57. https://doi.org/10.1038/nrendo.2011.81.Search in Google Scholar PubMed PubMed Central
17. Lisse, TS, Thiele, F, Fuchs, H, Hans, W, Przemeck, GKH, Abe, K, et al.. ER stress-mediated apoptosis in a new mouse model of osteogenesis imperfecta. PLoS Genet 2008;4:e7. https://doi.org/10.1371/journal.pgen.0040007.Search in Google Scholar PubMed PubMed Central
18. Doyard, M, Bacrot, S, Huber, C, Di Rocco, M, Goldenberg, A, Aglan, MS, et al.. FAM46A mutations are responsible for autosomal recessive osteogenesis imperfecta. J Med Genet 2018;55:278–84. https://doi.org/10.1136/JMEDGENET-2017-104999.Search in Google Scholar
19. Rauch, F, Fahiminiya, S, Majewski, J, Carrot-Zhang, J, Boudko, S, Glorieux, F, et al.. Cole-Carpenter syndrome is caused by a heterozygous missense mutation in P4HB. Am J Hum Genet 2015;96:425–31. https://doi.org/10.1016/J.AJHG.2014.12.027.Search in Google Scholar PubMed PubMed Central
20. Balasubramanian, M, Pollitt, RC, Chandler, KE, Mughal, MZ, Parker, MJ, Dalton, A, et al.. CRTAP mutation in a patient with Cole-Carpenter syndrome. Am J Med Genet A 2015;167:587–91. https://doi.org/10.1002/AJMG.A.36916.Search in Google Scholar PubMed
21. Valli, M, Barnes, AM, Gallanti, A, Cabral, WA, Viglio, S, Weis, MA, et al.. Deficiency of CRTAP in non-lethal recessive osteogenesis imperfecta reduces collagen deposition into matrix. Clin Genet 2012;82:453–9. https://doi.org/10.1111/J.1399-0004.2011.01794.X.Search in Google Scholar
22. Cabral, WA, Chang, W, Barnes, AM, Weis, M, Scott, MA, Leikin, S, et al.. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 2007;39:359–65. https://doi.org/10.1038/ng1968.Search in Google Scholar PubMed PubMed Central
23. Willaert, A, Malfait, F, Symoens, S, Gevaert, K, Kayserili, H, Megarbane, A, et al.. Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J Med Genet 2009;46:233–41. https://doi.org/10.1136/JMG.2008.062729.Search in Google Scholar PubMed
24. Barnes, AM, Carter, EM, Cabral, WA, Weis, M, Chang, W, Makareeva, E, et al.. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N Engl J Med 2010;362:521–8. https://doi.org/10.1056/nejmoa0907705.Search in Google Scholar PubMed PubMed Central
25. van Dijk, FS, Nesbitt, IM, Zwikstra, EH, Nikkels, PGJ, Piersma, SR, Fratantoni, SA, et al.. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet 2009;85:521–7. https://doi.org/10.1016/j.ajhg.2009.09.001.Search in Google Scholar PubMed PubMed Central
26. Pyott, SM, Schwarze, U, Christiansen, HE, Pepin, MG, Leistritz, DF, Dineen, R, et al.. Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum Mol Genet 2011;20:1595–609. https://doi.org/10.1093/HMG/DDR037.Search in Google Scholar
27. Marshall, C, Lopez, J, Crookes, L, Pollitt, RC, Balasubramanian, M. A novel homozygous variant in SERPINH1 associated with a severe, lethal presentation of osteogenesis imperfecta with hydranencephaly. Gene 2016;595:49–52. https://doi.org/10.1016/J.GENE.2016.09.035.Search in Google Scholar
28. Christiansen, HE, Schwarze, U, Pyott, SM, AlSwaid, A, Al Balwi, M, Alrasheed, S, et al.. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet 2010;86:389–98. https://doi.org/10.1016/j.ajhg.2010.01.034.Search in Google Scholar PubMed PubMed Central
29. Duran, I, Nevarez, L, Sarukhanov, A, Wu, S, Lee, K, Krejci, P, et al.. HSP47 and FKBP65 cooperate in the synthesis of type I procollagen. Hum Mol Genet 2015;24:1918–28. https://doi.org/10.1093/HMG/DDU608.Search in Google Scholar
30. Shaheen, R, Al-Owain, M, Sakati, N, Alzayed, ZS, Alkuraya, FS. FKBP10 and bruck syndrome: phenotypic heterogeneity or call for reclassification? Am J Hum Genet 2010;87:306–7. https://doi.org/10.1016/j.ajhg.2010.05.020.Search in Google Scholar PubMed PubMed Central
31. Alanay, Y, Avaygan, H, Camacho, N, Utine, GE, Boduroglu, K, Aktas, D, et al.. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet 2010;86:551–9. https://doi.org/10.1016/j.ajhg.2010.02.022. 20362275.Search in Google Scholar PubMed PubMed Central
32. Van Dijk, FS, Semler, O, Etich, J, Köhler, A, Jimenez-Estrada, JA, Bravenboer, N, et al.. Interaction between KDELR2 and HSP47 as a key determinant in osteogenesis imperfecta caused by Bi-allelic variants in KDELR2. Am J Hum Genet 2020;107:989–99. https://doi.org/10.1016/j.ajhg.2020.09.009.Search in Google Scholar PubMed PubMed Central
33. Efthymiou, S, Herman, I, Rahman, F, Anwar, N, Maroofian, R, Yip, J, et al.. Two novel bi-allelic KDELR2 missense variants cause osteogenesis imperfecta with neurodevelopmental features. Am J Med Genet A 2021;185:2241–9. https://doi.org/10.1002/AJMG.A.62221.Search in Google Scholar
34. Keller, RB, Tran, TT, Pyott, SM, Pepin, MG, Savarirayan, R, Mcgillivray, G, et al.. Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively. Genet Med 2017;20. https://doi.org/10.1038/gim.2017.115.Search in Google Scholar PubMed PubMed Central
35. Lindert, U, Cabral, WA, Ausavarat, S, Tongkobpetch, S, Ludin, K, Barnes, AM, et al.. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun 2016;7. https://doi.org/10.1038/NCOMMS11920.Search in Google Scholar
36. Webb, EA, Balasubramanian, M, Fratzl-Zelman, N, Cabral, WA, Titheradge, H, Alsaedi, A, et al.. Phenotypic spectrum in osteogenesis imperfecta due to mutations in TMEM38B: unraveling a complex cellular defect. J Clin Endocrinol Metab 2017;102:2019–28. https://doi.org/10.1210/JC.2016-3766.Search in Google Scholar PubMed PubMed Central
37. Garbes, L, Kim, K, Rieß, A, Hoyer-Kuhn, H, Beleggia, F, Bevot, A, et al.. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am J Hum Genet 2015;96:432. https://doi.org/10.1016/J.AJHG.2015.01.002.Search in Google Scholar
38. Valencia, M, Caparrós-Martin, JA, Sirerol-Piquer, MS, García-Verdugo, JM, Martínez-Glez, V, Lapunzina, P, et al.. Report of a newly identified patient with mutations in BMP1 and underlying pathogenetic aspects. AJMG 2014;164A:1143–50. https://doi.org/10.1002/AJMG.A.36427.Search in Google Scholar PubMed
39. Xu, X-J, Lv, F, Song, Y-W, Li, L-J, Asan, Wei, X-X, et al.. Novel mutations in BMP1 induce a rare type of osteogenesis imperfecta. Clin Chim Acta 2019;489:21–8. https://doi.org/10.1016/j.cca.2018.11.004. 30408480.Search in Google Scholar PubMed
40. Puig-Hervás, MT, Temtamy, S, Aglan, M, Valencia, M, Martínez-Glez, V, Ballesta-Martínez, MJ, et al.. Mutations in PLOD2 cause autosomal-recessive connective tissue disorders within the Bruck syndrome—osteogenesis imperfecta phenotypic spectrum. Hum Mutat 2012;33:1444–9. https://doi.org/10.1002/HUMU.22133.Search in Google Scholar
41. Ha-Vinh, R, Alanay, Y, Bank, RA, Campos-Xavier, AB, Zankl, A, Superti-Furga, A, et al.. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet A 2004;131A:115–20. https://doi.org/10.1002/AJMG.A.30231.Search in Google Scholar
42. Durkin, A, DeVile, C, Arundel, P, Bull, M, Walsh, J, Bishop, NJ, et al.. Expanding the phenotype of SPARC-related osteogenesis imperfecta: clinical findings in two patients with pathogenic variants in SPARC and literature review. J Med Genet 2022;59:810–6. https://doi.org/10.1136/JMEDGENET-2021-107942.Search in Google Scholar PubMed
43. Zhong, W, Pathak, JL, Liang, Y, Zhytnik, L, Pals, G, Eekhoff, EMW, et al.. The intricate mechanism of PLS3 in bone homeostasis and disease. Front Endocrinol 2023;14. https://doi.org/10.3389/FENDO.2023.1168306.Search in Google Scholar PubMed PubMed Central
44. Fahiminiya, S, Majewski, J, Mort, J, Moffatt, P, Glorieux, FH, Rauch, F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet 2013;50:345–8. https://doi.org/10.1136/jmedgenet-2013-101567.Search in Google Scholar PubMed
45. Lu, Y, Ren, X, Wang, Y, Bardai, G, Sturm, M, Dai, Y, et al.. Novel WNT1 mutations in children with osteogenesis imperfecta: clinical and functional characterization. Bone 2018;114:144–9. https://doi.org/10.1016/J.BONE.2018.06.018.Search in Google Scholar
46. Kuptanon, C, Srichomthong, C, Sangsin, A, Kovitvanitcha, D, Suphapeetiporn, K, Shotelersuk, V. The most 5’ truncating homozygous mutation of WNT1 in siblings with osteogenesis imperfecta with a variable degree of brain anomalies: a case report. BMC Med Genet 2018;19:117. https://doi.org/10.1186/s12881-018-0639-0. 30012084.Search in Google Scholar PubMed PubMed Central
47. Moosa, S, Yamamoto, GL, Garbes, L, Keupp, K, Beleza-Meireles, A, Moreno, CA, et al.. Autosomal recessive mutations in MESD cause osteogenesis imperfecta. Am J Hum Genet 2019;105:836–43. https://doi.org/10.1016/J.AJHG.2019.08.008.Search in Google Scholar
48. Stürznickel, J, Jähn-Rickert, K, Zustin, J, Hennig, F, Delsmann, MM, Schoner, K, et al.. Compound heterozygous frameshift mutations in MESD cause a lethal syndrome suggestive of osteogenesis imperfecta type XX. J Bone Miner Res 2021;36:1077–87. https://doi.org/10.1002/JBMR.4277.Search in Google Scholar PubMed
49. Uludağ Alkaya, D, Uyguner, ZO, Güneş, N, Tüysüz, B. Long‐term follow‐up findings in a Turkish girl with osteogenesis imperfecta type XX caused by a homozygous MESD variant. Am J Med Genet A 2022;188:1639–46. https://doi.org/10.1002/AJMG.A.62664.Search in Google Scholar PubMed
50. Etich, J, Semler, O, Stevenson, NL, Stephan, A, Besio, R, Garibaldi, N, et al.. TAPT1 —at the crossroads of extracellular matrix and signaling in Osteogenesis imperfecta. EMBO Mol Med 2023;15. https://doi.org/10.15252/emmm.202317528.Search in Google Scholar PubMed PubMed Central
51. Symoens, S, Barnes, AM, Gistelinck, C, Malfait, F, Guillemyn, B, Steyaert, W, et al.. Genetic defects in TAPT1 disrupt ciliogenesis and cause a complex lethal osteochondrodysplasia. Am J Hum Genet 2015;97:521–34. https://doi.org/10.1016/j.ajhg.2015.08.009.Search in Google Scholar PubMed PubMed Central
52. Gauthier, LW, Fontanges, E, Chapurlat, R, Collet, C, Rossi, M. Long-term follow-up of severe autosomal recessive SP7-related bone disorder. Bone 2024;179:116953. https://doi.org/10.1016/J.BONE.2023.116953.Search in Google Scholar PubMed
53. Ludwig, K, Ward, LM, Khan, N, Robinson, ME, Miranda, V, Bardai, G, et al.. Dominant osteogenesis imperfecta with low bone turnover caused by a heterozygous SP7 variant. Bone 2022;160:116400. https://doi.org/10.1016/J.BONE.2022.116400.Search in Google Scholar
54. Al-Mutairi, DA, Jarragh, AA, Alsabah, BH, Wein, MN, Mohammed, W, Alkharafi, L. A homozygous SP7/OSX mutation causes osteogenesis and dentinogenesis imperfecta with craniofacial anomalies. JBMR Plus 2024;8. https://doi.org/10.1093/JBMRPL/ZIAE026.Search in Google Scholar PubMed PubMed Central
55. Land, C, Rauch, F, Travers, R, Glorieux, FH. Osteogenesis imperfecta type VI in childhood and adolescence: effects of cyclical intravenous pamidronate treatment. Bone 2007;40:638–44. https://doi.org/10.1016/j.bone.2006.10.010.Search in Google Scholar PubMed
56. Ward, L, Bardai, G, Moffatt, P, Al-Jallad, H, Trejo, P, Glorieux, FH, et al.. Osteogenesis imperfecta type VI in individuals from northern Canada. Calcif Tissue Int 2016;98:566–72. https://doi.org/10.1007/s00223-016-0110-1.Search in Google Scholar PubMed
57. Glorieux, FH, Rauch, F, Plotkin, H, Ward, L, Travers, R, Roughley, P, et al.. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res 2000;15:1650–8. https://doi.org/10.1359/JBMR.2000.15.9.1650.Search in Google Scholar PubMed
58. Dubail, J, Brunelle, P, Baujat, G, Huber, C, Doyard, M, Michot, C, et al.. Homozygous loss‐of‐function mutations in CCDC134 are responsible for a severe form of osteogenesis imperfecta. J Bone Miner Res 2020;35:1470–80. https://doi.org/10.1002/JBMR.4011.Search in Google Scholar
59. Ali, TM, Linnenkamp, BDW, Yamamoto, GL, Honjo, RS, Cabral de Menezes Filho, H, Kim, CA, et al.. The recurrent homozygous translation start site variant in CCDC134 in an individual with severe osteogenesis imperfecta of non‐Morrocan ancestry. Am J Med Genet A 2022;188:1545–9. https://doi.org/10.1002/AJMG.A.62651.Search in Google Scholar
60. Holick, MF, Shirvani, A, Charoenngam, N. Fetal fractures in an infant with maternal ehlers-danlos syndrome, CCDC134 pathogenic mutation and a negative genetic test for osteogenesis imperfecta. Children 2021;8. https://doi.org/10.3390/CHILDREN8060512.Search in Google Scholar
61. Tuysuz, B, Uludag Alkaya, D, Geyik, F, Alaylloǧlu, M, Kasap, B, Kurugoǧlu, S, et al.. Biallelic frameshift variants in PHLDB1 cause mild-type osteogenesis imperfecta with regressive spondylometaphyseal changes. J Med Genet 2023;60:819–26. https://doi.org/10.1136/JMG-2022-108763.Search in Google Scholar
62. Ma, DL, Hu, J, Fang, K. Olmsted syndrome. J Clin Dermatol 2012;35:637–9. https://doi.org/10.1155/2012/927305.Search in Google Scholar PubMed PubMed Central
63. Elise Tonoli, R, De Villa, D, Hübner Frainer, R, Pizzarro Meneghello, L, Ricachnevsky, N, de Quadros, M. Olmsted syndrome. Case Rep Dermatol Med 2012;2012:927305. https://doi.org/10.1155/2012/927305. 23320205.Search in Google Scholar PubMed PubMed Central
64. Zhou, QL, Jiang, ZY, Mabardy, AS, Del Campo, CM, Lambright, DG, Holik, J, et al.. A novel Pleckstrin homology domain-containing protein enhances insulin-stimulated Akt phosphorylation and GLUT4 translocation in adipocytes. JBC 2010;285:27581–9. https://doi.org/10.1074/jbc.M110.146886.Search in Google Scholar PubMed PubMed Central
65. Nabavizadeh, N, Bressin, A, Shboul, M, Moreno Traspas, R, Chia, PH, Bonnard, C, et al.. A progeroid syndrome caused by a deep intronic variant in TAPT1 is revealed by RNA/SI-NET sequencing. EMBO Mol Med 2023;15. https://doi.org/10.15252/EMMM.202216478.Search in Google Scholar PubMed PubMed Central
66. Etich, J, Semler, O, Stevenson, NL, Stephan, A, Besio, R, Garibaldi, N, et al.. TAPT1 – at the crossroads of extracellular matrix and signaling in Osteogenesis imperfecta. EMBO Mol Med 2023;15. https://doi.org/10.15252/emmm.202317528.Search in Google Scholar PubMed PubMed Central
67. Khan, SI, Yonko, EA, Carter, EM, Dyer, D, Sandhaus, RA, Raggio, CL. Cardiopulmonary status in adults with osteogenesis imperfecta: intrinsic lung disease may contribute more than scoliosis. Clin Orthop Relat Res 2020;478:2833–43. https://doi.org/10.1097/CORR.0000000000001400.Search in Google Scholar PubMed PubMed Central
68. Wallace, MJ, Kruse, RW, Shah, SA. The spine in patients with osteogenesis imperfecta. J Am Acad Orthop Surg 2017;25:100–9. https://doi.org/10.5435/JAAOS-D-15-00169.Search in Google Scholar PubMed
69. Sienko, S, Tucker, C, Welborn, MC. Surgical outcomes for spinal deformity in osteogenesis imperfecta. Spine Deform 2023;11:391–8. https://doi.org/10.1007/S43390-022-00600-X.Search in Google Scholar
70. Frank, E, Berger, T, Tew, JM. Basilar impression and platybasia in osteogenesis imperfecta tarda. Surg Neurol 1982;17:116–9. https://doi.org/10.1016/S0090-3019(82)80033-5.Search in Google Scholar
71. Scheiber, AL, Wilkinson, KJ, Suzuki, A, Enomoto-Iwamoto, M, Kaito, T, Cheah, KSE, et al.. 4PBA reduces growth deficiency in osteogenesis imperfecta by enhancing transition of hypertrophic chondrocytes to osteoblasts. JCI Insight 2022;7. https://doi.org/10.1172/jci.insight.149636.Search in Google Scholar PubMed PubMed Central
72. Rodriguez Celin, M, Kruger, KM, Caudill, A, Murali, CN, Nagamani, SCS, Smith, PA, et al.. A multicenter study to evaluate pain characteristics in osteogenesis imperfecta. Am J Med Genet A 2023;191:160–72. https://doi.org/10.1002/ajmg.a.63009.Search in Google Scholar PubMed PubMed Central
73. Zack, P, Franck, L, Devile, C, Clark, C. Fracture and non‐fracture pain in children with osteogenesis imperfecta. Acta Paediatr 2005;94:1238–42. https://doi.org/10.1111/j.1651-2227.2005.tb02082.x.Search in Google Scholar PubMed
74. Westerheim, I, Hart, T, van Welzenis, T, Wekre, LL, Semler, O, Raggio, C, et al.. The IMPACT survey: a mixed methods study to understand the experience of children, adolescents and adults with osteogenesis imperfecta and their caregivers. Orphanet J Rare Dis 2024;19:128. https://doi.org/10.1186/s13023-024-03126-9.Search in Google Scholar PubMed PubMed Central
75. Treurniet, S, Burger, P, Ghyczy, EAE, Verbraak, FD, Curro‐ Tafili, KR, Micha, D, et al.. Ocular characteristics and complications in patients with osteogenesis imperfecta: a systematic review. Acta Ophthalmol 2022;100. https://doi.org/10.1111/aos.14882.Search in Google Scholar PubMed PubMed Central
76. Forlino, A, Marini, JC. Osteogenesis imperfecta. Lancet 2016;387:1657–71. https://doi.org/10.1016/S0140-6736(15)00728-X.Search in Google Scholar PubMed PubMed Central
77. Kuptanon, C, Srichomthong, C, Sangsin, A, Kovitvanitcha, D, Suphapeetiporn, K, Shotelersuk, V. The most 5′ truncating homozygous mutation of WNT1 in siblings with osteogenesis imperfecta with a variable degree of brain anomalies: a case report. BMC Med Genet 2018;19. https://doi.org/10.1186/S12881-018-0639-0.Search in Google Scholar PubMed PubMed Central
78. Ugarteburu, M, Cardoso, L, Richter, C-P, Carriero, A. Treatments for hearing loss in osteogenesis imperfecta: a systematic review and meta-analysis on their efficacy. Sci Rep 2022;12:17125. https://doi.org/10.1038/s41598-022-20169-9.Search in Google Scholar PubMed PubMed Central
79. Folkestad, L, Hald, JD, Gram, J, Langdahl, BL, Hermann, AP, Diederichsen, AC, et al.. Cardiovascular disease in patients with osteogenesis imperfecta — a nationwide, register-based cohort study. Int J Cardiol 2016;225:250–7. https://doi.org/10.1016/j.ijcard.2016.09.107.Search in Google Scholar PubMed
80. Gatto, G, et al.. Osteogenesis imperfecta and related diseases: collagen I alterations in skeletal and extraskeletal tissues. In: Rossi, A, Zaucke, F, editors. The extracellular matrix in genetic skeletal disorders. Cham: Springer; 2025. In press.10.1007/978-3-031-70835-0_2Search in Google Scholar
81. Hoyer-Kuhn, H, Bartz-Seel, J, Blickheuser, R, Deimling, U, Stücker, R, Wirth, T, et al.. Diagnostik und Therapie der Osteogenesis imperfecta. Monatsschr Kinderh 2017;165:333–46. https://doi.org/10.1007/s00112-016-0189-5.Search in Google Scholar
82. Renaud, A, Aucourt, J, Weill, J, Bigot, J, Dieux, A, Devisme, L, et al.. Radiographic features of osteogenesis imperfecta. Insights Imag 2013;4:417–29. https://doi.org/10.1007/s13244-013-0258-4.Search in Google Scholar PubMed PubMed Central
83. Obafemi, AA, Bulas, DI, Troendle, J, Marini, JC. Popcorn calcification in osteogenesis imperfecta: incidence, progression, and molecular correlation. Am J Med Genet A 2008;146A:2725. https://doi.org/10.1002/AJMG.A.32508.Search in Google Scholar
84. Morlino, S, Micale, L, Ritelli, M, Rohrbach, M, Zoppi, N, Vandersteen, A, et al.. COL1‐related overlap disorder: a novel connective tissue disorder incorporating the osteogenesis imperfecta/Ehlers‐Danlos syndrome overlap. Clin Genet 2020;97:396–406. https://doi.org/10.1111/cge.13683.Search in Google Scholar PubMed
85. Semler, O, Hoyer-Kuhn, H, Netzer, C. Osteogenesis imperfecta. Med Genet 2012;24:297–311. https://doi.org/10.1007/s11825-012-0358-4.Search in Google Scholar
86. Tsimicalis, A, Denis-Larocque, G, Michalovic, A, Lepage, C, Williams, K, Yao, T-R, et al.. The psychosocial experience of individuals living with osteogenesis imperfecta: a mixed-methods systematic review. Qual Life Res 2016;25:1877–96. https://doi.org/10.1007/s11136-016-1247-0.Search in Google Scholar PubMed
87. Mueller, B, Engelbert, R, Baratta-Ziska, F, Bartels, B, Blanc, N, Brizola, E, et al.. Consensus statement on physical rehabilitation in children and adolescents with osteogenesis imperfecta. Orphanet J Rare Dis 2018;13:1–14. https://doi.org/10.1186/S13023-018-0905-4/FIGURES/3.Search in Google Scholar
88. Antičević, D, Jeleč, Ž, Zagreb, H. Osteogenesis imperfecta: surgical treatment options with emphasis on today’s orthopedic approach. Paediatr Croat 2017;61:129–62. https://doi.org/10.13112/PC.2017.18.Search in Google Scholar
89. Kasahun, AE, Sendekie, AK, Abebe, RB. Assessment of pain management adequacy among hospitalized pediatric patients: institutional-based cross-sectional study. Front Pediatr 2023;11:1195416. https://doi.org/10.3389/FPED.2023.1195416.Search in Google Scholar
90. Hoyer-Kuhn, H, Semler, O, Stark, C, Struebing, N, Goebel, O, Schoenau, E. A specialized rehabilitation approach improves mobility in children with osteogenesis imperfecta. J Musculoskelet Neuronal Interact 2014;14:445–53.Search in Google Scholar
91. Semler, O, Fricke, O, Vezyroglou, K, Stark, C, Stabrey, A, Schoenau, E. Results of a prospective pilot trial on mobility after whole body vibration in children and adolescents with osteogenesis imperfecta. Clin Rehabil 2008;22:387–94. https://doi.org/10.1177/0269215507080763.Search in Google Scholar PubMed
92. Glorieux, FH, Bishop, NJ, Plotkin, H, Chabot, G, Lanoue, G, Travers, R. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med 1998;339:947–52. https://doi.org/10.1056/NEJM199810013391402.Search in Google Scholar PubMed
93. Land, C, Rauch, F, Munns, CF, Sahebjam, S, Glorieux, FH. Vertebral morphometry in children and adolescents with osteogenesis imperfecta: effect of intravenous pamidronate treatment. Bone 2006;39:901–6. https://doi.org/10.1016/j.bone.2006.04.004.Search in Google Scholar PubMed
94. Ott, SM. Long-term safety of bisphosphonates. J Clin Endocrinol Metab 2005;90:1897–9. https://doi.org/10.1210/jc.2005-0057.Search in Google Scholar PubMed
95. Dwan, K, Phillipi, CA, Steiner, RD, Basel, D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev 2016;2016. https://doi.org/10.1002/14651858.CD005088.pub4.Search in Google Scholar PubMed PubMed Central
96. Maines, E, Tadiotto, E, Morandi, G, Fedrizzi, M, Gaudino, R, Cavarzere, P, et al.. Hypocalcemia following neridronate administration in pediatric patients with osteogenesis imperfecta: a prospective observational study. J Pediatr Genet 2020;09:093–100. https://doi.org/10.1055/s-0039-1700972.Search in Google Scholar PubMed PubMed Central
97. Hennedige, AA, Jayasinghe, J, Khajeh, J, Macfarlane, TV. Systematic review on the incidence of bisphosphonate related osteonecrosis of the jaw in children diagnosed with osteogenesis imperfecta. J Oral Maxillofac Res 2013;4. https://doi.org/10.5037/jomr.2013.4401.Search in Google Scholar PubMed PubMed Central
98. Duarte, NT, Rech, Bde O, Martins, IG, Franco, JB, Ortega, KL. Can children be affected by bisphosphonate-related osteonecrosis of the jaw? A systematic review. Int J Oral Maxillofac Surg 2020;49:183–91. https://doi.org/10.1016/j.ijom.2019.08.004.Search in Google Scholar PubMed
99. Sun, L, Hu, J, Liu, J, Zhang, Q, Wang, O, Jiang, Y, et al.. Relationship of pathogenic mutations and responses to zoledronic acid in a cohort of osteogenesis imperfecta children. J Clin Endocrinol Metab 2022;107:2571–9. https://doi.org/10.1210/clinem/dgac366.Search in Google Scholar PubMed
100. Lacey, DL, Tan, HL, Lu, J, Kaufman, S, Van, G, Qiu, W, et al.. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am J Pathol 2000;157:435–48. https://doi.org/10.1016/S0002-9440(10)64556-7.Search in Google Scholar PubMed PubMed Central
101. Feng, X. RANKing intracellular signaling in osteoclasts. IUBMB Life 2005;57:389–95. https://doi.org/10.1080/15216540500137669.Search in Google Scholar PubMed
102. Burgess, TL, Qian, Y, Kaufman, S, Ring, BD, Van, G, Capparelli, C, et al.. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J Cell Biol 1999;145:527–38. https://doi.org/10.1083/jcb.145.3.527.Search in Google Scholar PubMed PubMed Central
103. Jimi, E, Akiyama, S, Tsurukai, T, Okahashi, N, Kobayashi, K, Udagawa, N, et al.. Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J Immunol 1999;163:434–42. https://doi.org/10.4049/jimmunol.163.1.434.Search in Google Scholar
104. Papapoulos, SE, Cremers, SCLM. Prolonged bisphosphonate release after treatment in children. N Engl J Med 2007;356:1075–6. https://doi.org/10.1056/NEJMc062792.Search in Google Scholar PubMed
105. Chen, Q, Hu, C, Liu, Y, Song, R, Zhu, W, Zhao, H, et al.. Pharmacokinetics, pharmacodynamics, safety, and tolerability of single-dose denosumab in healthy Chinese volunteers: a randomized, single-blind, placebo-controlled study. PLoS One 2018;13:e0197984. https://doi.org/10.1371/journal.pone.0197984.Search in Google Scholar PubMed PubMed Central
106. Cummings, SR, Martin, JS, McClung, MR, Siris, ES, Eastell, R, Reid, IR, et al.. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009;361:756–65. https://doi.org/10.1056/NEJMOA0809493.Search in Google Scholar PubMed
107. Gnant, M, Pfeiler, G, Dubsky, PC, Hubalek, M, Greil, R, Jakesz, R, et al.. Adjuvant denosumab in breast cancer (ABCSG-18): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015;386:433–43. https://doi.org/10.1016/S0140-6736(15)60995-3.Search in Google Scholar PubMed
108. Liu, J, Lin, X, Sun, L, Zhang, Q, Jiang, Y, Wang, O, et al.. Safety and efficacy of denosumab in children with osteogenesis imperfecta—the first prospective comparative study. J Clin Endocrinol Metab 2024;109:1827–36. https://doi.org/10.1210/CLINEM/DGAD732.Search in Google Scholar
109. Horiuchi, K, Kobayashi, E, Mizuno, T, Susa, M, Chiba, K. Hypercalcemia following discontinuation of denosumab therapy: a systematic review. Bone Rep 2021;15:101148. https://doi.org/10.1016/j.bonr.2021.101148.Search in Google Scholar PubMed PubMed Central
110. Anastasilakis, AD, Evangelatos, G, Makras, P, Iliopoulos, A. Rebound-associated vertebral fractures may occur in sequential time points following denosumab discontinuation: need for prompt treatment re-initiation. Bone Rep 2020;12:100267. https://doi.org/10.1016/j.bonr.2020.100267.Search in Google Scholar PubMed PubMed Central
111. Cummings, SR, Ferrari, S, Eastell, R, Gilchrist, N, Jensen, JEB, McClung, M, et al.. Vertebral fractures after discontinuation of denosumab: a post hoc analysis of the randomized placebo-controlled freedom trial and its extension. J Bone Miner Res 2018;33:190–8. https://doi.org/10.1002/JBMR.3337.Search in Google Scholar PubMed
112. Raux, S, Bouhamama, A, Gaspar, N, Brugières, L, Entz-Werlé, N, Mallet, C, et al.. Denosumab for treating aneurysmal bone cysts in children. Orthop Traumatol Surg Res 2019;105:1181–5. https://doi.org/10.1016/J.OTSR.2019.04.028.Search in Google Scholar PubMed
113. Grasemann, C, Schündeln, MM, Hövel, M, Schweiger, B, Bergmann, C, Herrmann, R, et al.. Effects of RANK-ligand antibody (denosumab) treatment on bone turnover markers in a girl with juvenile paget’s disease. J Clin Endocrinol Metab 2013;98:3121–6. https://doi.org/10.1210/JC.2013-1143.Search in Google Scholar
114. Multicenter, single-arm study to evaluate efficacy, safety, & pharmacokinetics of denosumab in children w/OI. n.d. https://clinicaltrials.gov/study/NCT02352753 [Accessed 6 Oct 2024].Search in Google Scholar
115. Grafe, I, Yang, T, Alexander, S, Homan, EP, Lietman, C, Jiang, MM, et al.. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat Med 2014;20:670–5. https://doi.org/10.1038/nm.3544.Search in Google Scholar PubMed PubMed Central
116. Infante, A, Cabodevilla, L, Gener, B, Rodríguez, CI. Circulating TGF-β pathway in osteogenesis imperfecta pediatric patients subjected to MSCs-based cell therapy. Front Cell Dev Biol 2022;10. https://doi.org/10.3389/FCELL.2022.830928.Search in Google Scholar PubMed PubMed Central
117. Song, I-W, Nagamani, SCS, Nguyen, D, Grafe, I, Sutton, VR, Gannon, FH, et al.. Targeting TGF-β for treatment of osteogenesis imperfecta. J Clin Invest 2022;132. https://doi.org/10.1172/JCI152571.Search in Google Scholar PubMed PubMed Central
118. Single ascending dose study of SAR439459 in adults with osteogenesis imperfecta (OI). n.d. https://clinicaltrials.gov/study/NCT02352753 [Accessed 6 Oct 2024].Search in Google Scholar
119. Rubin, MR, Cosman, F, Cosman, F, Lindsay, R, Lindsay, R, Bilezikian, JP, et al.. The anabolic effects of parathyroid hormone. Clin Geriatr Med 2002;13:267–77. https://doi.org/10.1007/s001980200026.Search in Google Scholar PubMed
120. Orwoll, ES, Shapiro, J, Veith, S, Wang, Y, Lapidus, J, Vanek, C, et al.. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest 2014;124:491–8. https://doi.org/10.1172/JCI71101.Search in Google Scholar PubMed PubMed Central
121. Leali, PT, Balsano, M, Maestretti, G, Brusoni, M, Amorese, V, Ciurlia, E, et al.. Efficacy of teriparatide vs neridronate in adults with osteogenesis imperfecta type I: a prospective randomized international clinical study. Clin Cases Miner Bone Metab 2017;14:153–6. https://doi.org/10.11138/CCMBM/2017.14.1.153.Search in Google Scholar
122. Vahle, JL, Sato, M, Long, GG, Young, JK, Francis, PC, Engelhardt, JA, et al.. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1-34) for 2 Years and relevance to human safety. Toxicol Pathol 2002;30:312–21. https://doi.org/10.1080/01926230252929882.Search in Google Scholar PubMed
123. Hald, JD, Keerie, C, Weir, CJ, Javaid, MK, Lam, W, Osborne, P, et al.. Protocol: protocol of a randomised trial of teriparatide followed by zoledronic acid to reduce fracture risk in adults with osteogenesis imperfecta. BMJ Open 2023;13:78164. https://doi.org/10.1136/BMJOPEN-2023-078164.Search in Google Scholar PubMed PubMed Central
124. Poole, KES, Van Bezooijen, RL, Loveridge, N, Hamersma, H, Papapoulos, SE, Löwik, CW, et al.. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 2005;19:1842–4. https://doi.org/10.1096/fj.05-4221fje.Search in Google Scholar PubMed
125. Lewiecki, EM, Shah, A, Shoback, D. Sclerostin inhibition: a novel therapeutic approach in the treatment of osteoporosis. Int J Womens Health 2015;565. https://doi.org/10.2147/IJWH.S73244.Search in Google Scholar PubMed PubMed Central
126. Cardinal, M, Dessain, A, Roels, T, Lafont, S, Ominsky, MS, Devogelaer, J-P, et al.. Sclerostin-antibody treatment decreases fracture rates in axial skeleton and improves the skeletal phenotype in growing oim/oim mice. Calcif Tissue Int 2020;106:494–508. https://doi.org/10.1007/s00223-019-00655-5.Search in Google Scholar PubMed
127. Ominsky, MS, Niu, Q-T, Li, C, Li, X, Ke, HZ. Tissue-level mechanisms responsible for the increase in bone formation and bone volume by sclerostin antibody. J Bone Miner Res 2014;29:1424–30. https://doi.org/10.1002/jbmr.2152.Search in Google Scholar PubMed
128. Glorieux, FH, Langdahl, B, Chapurlat, R, De Beur, SJ, Sutton, VR, Poole, KES, et al.. Setrusumab for the treatment of osteogenesis imperfecta: 12-month results from the phase 2b asteroid study. J Bone Miner Res 2024;39:1215–28. https://doi.org/10.1093/jbmr/zjae112.Search in Google Scholar PubMed PubMed Central
129. Study to evaluate efficacy and safety of Romosozumab compared with bisphosphonates in children and adolescents with osteogenesis imperfecta. n.d. https://clinicaltrials.gov/study/NCT05972551 [Accessed 7 Oct 2024).Search in Google Scholar
130. Setrusumab vs bisphosphonates in pediatric subjects with osteogenesis imperfecta. n.d. https://clinicaltrials.gov/study/NCT05768854 [Accessed 7 Oct 2024].Search in Google Scholar
131. Jones, GN, Moschidou, D, Abdulrazzak, H, Kalirai, BS, Vanleene, M, Osatis, S, et al.. Potential of human fetal chorionic stem cells for the treatment of osteogenesis imperfecta. Stem Cells Dev 2014;23:262. https://doi.org/10.1089/SCD.2013.0132.Search in Google Scholar PubMed PubMed Central
132. Horwitz, EM, Gordon, PL, Koo, WKK, Marx, JC, Neel, MD, McNall, RY, et al.. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci U S A 2002;99:8932–7. https://doi.org/10.1073/pnas.132252399.Search in Google Scholar PubMed PubMed Central
133. Götherström, C, Westgren, M, Shaw, SWS, Åström, E, Biswas, A, Byers, PH, et al.. Pre- and postnatal transplantation of fetal mesenchymal stem cells in osteogenesis imperfecta: a two-center experience. Stem Cells Transl Med 2014;3:255. https://doi.org/10.5966/SCTM.2013-0090.Search in Google Scholar PubMed PubMed Central
134. Sagar, RL, Åström, E, Chitty, LS, Crowe, B, David, AL, Devile, C, et al.. An exploratory open-label multicentre phase I/II trial evaluating the safety and efficacy of postnatal or prenatal and postnatal administration of allogeneic expanded fetal mesenchymal stem cells for the treatment of severe osteogenesis imperfecta in infants and fetuses: the BOOSTB4 trial protocol. BMJ Open 2024;14. https://doi.org/10.1136/BMJOPEN-2023-079767.Search in Google Scholar
135. Schindeler, A, Lee, LR, O’Donohue, AK, Ginn, SL, Munns, CF. Curative cell and gene therapy for osteogenesis imperfecta. J Bone Miner Res 2020;37:826–36. https://doi.org/10.1002/jbmr.4549.Search in Google Scholar PubMed PubMed Central
© 2024 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Review
- Osteogenesis imperfecta: shifting paradigms in pathophysiology and care in children
- Opinion Paper
- CRH receptor antagonist crinecerfont – a promising new treatment option for patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
- Original Articles
- Age and sex mark clinical differences in the presentation of pediatric type 1 diabetes mellitus
- Geographic information system mapping and predictors of glycemic control in children and youth with type 1 diabetes: a study from Western India
- Body composition assessment measured via bioelectrical impedance analysis in euthyroid children with newly diagnosed Hashimoto’s thyroiditis
- Outcomes of newborns screened for congenital hypothyroidism in Turkey – a single center experience
- High yield of congenital hypothyroidism among infants attending Children Hospital, Nairobi, Kenya. Facility based study in the absence of newborn screening
- Immune checkpoint inhibitors and endocrinopathies in pediatric brain tumor patients
- Assessment of quality of life in families affected by maple syrup urine disease: a cross sectional study
- Case Reports
- Reninoma: an unusual cause of growth failure
- Persistent hypoglycemia in congenital syphilis: hyperinsulinemic hypoglycemia with a focal pancreatic lesion
- Diagnostic challenges in pediatric Cushing’s disease associated with chronic renal failure: a report of three patients
- A novel de novo missense OTC mutation in an Iranian girl: a case report
Articles in the same Issue
- Frontmatter
- Review
- Osteogenesis imperfecta: shifting paradigms in pathophysiology and care in children
- Opinion Paper
- CRH receptor antagonist crinecerfont – a promising new treatment option for patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
- Original Articles
- Age and sex mark clinical differences in the presentation of pediatric type 1 diabetes mellitus
- Geographic information system mapping and predictors of glycemic control in children and youth with type 1 diabetes: a study from Western India
- Body composition assessment measured via bioelectrical impedance analysis in euthyroid children with newly diagnosed Hashimoto’s thyroiditis
- Outcomes of newborns screened for congenital hypothyroidism in Turkey – a single center experience
- High yield of congenital hypothyroidism among infants attending Children Hospital, Nairobi, Kenya. Facility based study in the absence of newborn screening
- Immune checkpoint inhibitors and endocrinopathies in pediatric brain tumor patients
- Assessment of quality of life in families affected by maple syrup urine disease: a cross sectional study
- Case Reports
- Reninoma: an unusual cause of growth failure
- Persistent hypoglycemia in congenital syphilis: hyperinsulinemic hypoglycemia with a focal pancreatic lesion
- Diagnostic challenges in pediatric Cushing’s disease associated with chronic renal failure: a report of three patients
- A novel de novo missense OTC mutation in an Iranian girl: a case report