Mechanism of Selective Desulphurization in Iron Ore Sintering Process by Adding Urea
-
Hongming Long
 ,
Jiaxin Li
,
Jiaxin Li

Abstract
Iron ore sintering is an important part during the ironmaking process, and a large amount of SO2 is also generated. Our previous research shows that it is an effective way to reduce SO2 content of flue gas by adding urea to a special sintering material zone position. In this paper, the mechanism of selective desulphurization by adding urea during the iron ore sintering was carried out. The results show that 88.14 % desulphurization rate was obtained with the addition of 0.05 % urea particles at 100 mm height from the feed bottom. During the sintering process, when drying zone reached the added position of urea, large amounts of NH3 were generated by urea decomposition, and then reacted with SO2 to produce (NH4)2SO4 in the wetting zone. With the accumulated desulphurization reactions during the sintering, the low SO2 emission in the flue gas was achieved. Moreover, the addition of urea in the bottom zone avoided the ammonia present in the sintering ore and promoted the urea utilization efficiency.
Introduction
Iron ore sintering process is an important sector for iron production as well as major pollution emission source of SO2 NOx and Polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs). The emission of SO2 produced by iron and steel production is about 10 % with respect to the national total emission, and sintering process accounts for more than 60 % of steel industry [1, 2]. Sintering flue gas owes the following characteristics, including large amount, remarkable flow fluctuations and lower pollutant concentration, which seriously restrict the effective removal of pollutants. As environmental issues become increasingly acute and strict environmental policies are being conducted, the desulphurization, denitrification and dioxin removal from sintering flue gas with high efficiency become key technological innovations. The emission standard of sintering flue gas pollutant has been clearly pointed out in the “2011–2020 Chinese Steel Industry Science and Technology Development Guide”.
The end treatment of SO2 in sintering flue gas is the principal method for reducing SO2 emission in steel industry. According to the process characteristics, flue gas desulphurization (FGD) process can be classified into three categories, namely wet process, semi-dry process and dry process [3–5]. Currently, these processes have been widely applied but huge cost and serious financial burden should be paid. Under this background, it is critical and urgent to develop innovative online (source treatment) desulphurization method with higher efficiency and lower cost.
The former research reported that the concentration of SO2 was decreased sharply when flue gas of boiler was injected through urea solution [6, 7]. Our previous research found that urea mixing in sintering raw materials presented good desulphurization effects through the sintering pot tests [8]. However, the mechanism of desulphurization in sintering process by adding urea has been fully unclear. In this paper, the mechanism of urea desulphurization in the sintering process was investigated, which provides theoretical support for improvement and application of the innovative FGD process.
Experiment
Materials
Raw materials for sintering tests were provided by a commercially running sintering plant. The commercially available urea contained 46.7 % nitrogen that was used as desulphurizer, and its average particle granularity was 1–2 mm. The chemical compositions of raw materials were listed in Table 1. The mass ratio of raw materials in the sintering process was listed in Table 2.
Chemical compositions of the raw materials in percentage.
| Raw material | TFe | FeO | SiO2 | CaO | Al2O3 | MgO | P | S | C |
|---|---|---|---|---|---|---|---|---|---|
| Pilbara blend ore | 60.96 | 0.25 | 4.06 | 0.06 | 2.15 | 0.04 | 0.087 | 0.018 | – |
| Newman ore | 59.15 | 0.25 | 4.89 | 0.10 | 1.87 | 0.05 | 0.063 | 0.01 | – |
| Kara ore | 65.19 | 0.18 | 1.93 | 0.05 | 1.26 | 0.01 | 0.037 | 0.007 | – |
| Yandi ore | 58.56 | 0.31 | 4.11 | 0.14 | 1.74 | 0.03 | 0.051 | 0.007 | – |
| SFIT ore | 64.30 | 0.74 | 4.55 | 0.03 | 0.85 | 0.01 | 0.064 | 0.007 | – |
| FMG ore | 57.32 | 0.62 | 6.28 | 0.13 | 1.51 | 0.04 | 0.065 | 0.022 | – |
| Blast furnace dust | 38.45 | – | 5.57 | 6.02 | 2.28 | 1.11 | – | 0.44 | 19.22 |
| Iron oxide scale | 69.02 | 1.91 | 2.73 | 1.31 | 0.68 | 0.23 | – | 0.25 | |
| Lime | – | – | 1.08 | 81.95 | 0.4 | 0.47 | – | 0.003 | – |
| Limestone | – | – | 2.88 | 51.20 | 0.64 | 0.95 | – | 0.116 | – |
| Dolomite | – | – | 1.15 | 30.68 | 0.34 | 20.31 | – | 0.024 | – |
| Coal fines | 1.14 | – | 3.03 | 0.60 | 3.48 | – | – | 0.38 | 80.82 |
| Coke fines | 1.63 | – | 6.20 | 0.60 | 4.32 | – | – | 0.64 | 82.44 |
Mass ratio of raw materials in percentage.
| Raw materials | Pilbara blend ore | Newman ore | Kara ore | Yandi ore | SFIT ore | FMG ore | Blast furnace dust |
|---|---|---|---|---|---|---|---|
| Mass ratio | 12.75 | 15.93 | 15.93 | 11.93 | 11.93 | 7.95 | 1.60 |
| Raw materials | Iron oxide scale | Lime | Limestone | Dolomite | Coal fines | Coke fines | |
| Mass ratio | 1.60 | 3.00 | 4.83 | 7.85 | 2.35 | 2.35 |
Experimental methods
The laboratory sintering pot with 200 mm in diameter and 700 mm in height is shown in Figure 1. During each experiment, first, 2 kg hearth layer was added into the sintering pot, and then the sintering mixture was put into the sintering pot via the distributing device. The weight of the sintering mixture was about 44 kg at 6 % moisture. After mixture materials were charged into sintering pot, the igniter began to heat up with 6 kPa negative pressure at 1,050 °C for 90 s. Simultaneously, testing devices were conducted. After the ignition process, negative pressure was adjusted to 15 kPa and the sintering process was started. When the temperature of sintering flue gas reaches the peak value, testing devices were closed and the sintering test was ended.

The apparatus for sinter pot test.
Our previous research presented that the optimum addition ratio of urea was 0.05 % [8]. So, the ratio of urea is 0.05 % in this study. The studies of SO2 emission rules of single iron ore and raw materials show that significant temperature of SO2 emission is in the range of 500–1,200 °C [9, 10]. According to the characteristics of sintering process, SO2 is mainly generated in the sintering zone and drying zone [11]. Thus, the experimental scheme was designed to investigate the effect of urea adding methods and adding positions on desulphurization efficiency, as listed in Table 3. The addition of the special position is shown in Figure 2. During the desulphurization test, the SO2 concentration was detected by Testo350 flue gas analyser.
In addition, the dust of flue gas was filtrated by a filter bottle (200 ml H2O) connected to a pump. At the beginning of sintering, the vacuum pump was kept open to maintain exhaust pipe pressure similar to the main gas flue, and the flow was adjusted to be 0.03 m3/min. When sintering was completed, the vacuum pump was closed. The SO42– and NH4+ ionic concentrations of filter liquor were detected using inductively coupled plasma mass spectrometry (ICP-MS).

The position of adding urea.
The desulphurization scheme for the sintering pot test by adding urea.
| No. | Adding methods of urea | Mass of urea/% |
|---|---|---|
| Test 1 | Without adding urea | – |
| Test 2 | Adding urea particle to 100 mm height from the feed zone bottom | 0.05 |
| Test 3 | Mixed urea solution with mixture material | 0.05 |
The mass of SO2 (
where
The desulphurization rate (δ) using urea was calculated according to the following equation:
where M is the SO2 mass in the absence of urea (mg) and m is the SO2 mass in the presence of urea (mg).
Results and discussion
SO2 emission of flue gas
The emission concentration of SO2 and the mass of SO2 in the flue gas are shown in Figures 3 and 4, respectively.

Effect of urea adding methods on SO2 concentration of flue gas.
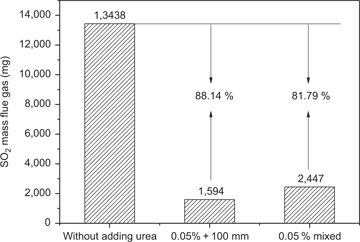
Effect of urea adding methods on flue gas desulphurization.
As shown in Figures 3 and 4, adding urea is an effective technology to reduce SO2 emission during the iron ore sintering. Compared with the result without adding urea, the desulphurization rate of adding urea by “0.05 % + 100 mm” and “0.05 % mixed” are 88.14 % and 81.79 %, respectively. Table 4 presents the SO42– and NH4+ concentration of filter liquor. During the test without adding urea, SO42– and NH4+ concentration are 106 and 32 mg/L, respectively. The SO42– and NH4+ concentrations increase significantly when urea was added, which indicates the formation of (NH4)2SO4 during the sintering.
SO42– and NH4+ concentration of filter liquor.
| Item | SO42– (mg/L) | NH4+ (mg/L) |
|---|---|---|
| Without adding urea | 106 | 32 |
| 0.05 % + 100 mm | 2,468 | 706 |
| 0.05 % mixed | 2,283 | 647 |
Desulphurization mechanism discussion
Generation and migration characteristics of SO2
Air draft sintering is carried out from the surface of sintering raw materials zone and gradually conducted down. Along with the height direction, materials zone is divided into sintered zone, sintering zone, drying zone and wetting zone, respectively, from the surface to the bottom of zone according to the different temperatures and reactions, as shown in Figure 5.

Material bed and temperature distribution of the sintering process.
Sintering zone is the place of the highest temperature (800–1,350 °C) in sintering process, in which sulphate is decomposed and sulphide is oxidized. The temperature of drying zone is between 60 and 800 °C. SO2 adsorbed in wetting zone is released again because of water evaporation when drying zone is coming. About 90 % sulphur of sintering process is transformed into SO2 in the drying zone or sintering zone [10], which is taken to the wetting zone by negative pressure air draft and easily adsorbed by free water since wetting zone has great resistance with low temperature (less than 100 °C). Simultaneously, there are some alkaline flux (CaO) and weak acid (CaCO3, MgCO3, etc.) in the wetting zone [9, 10], which can interact with SO2 in wet condition. With free water, alkaline flux and weak acid existing, wetting zone owes great adsorption. Therefore, SO2 concentration in the sintering flue gas is low from the initial stage to the middle stage of sintering process. Figure 6 [8] shows the relationship between SO2 emission concentration as well as wind box temperature and different sintering wind boxes in a sinter plant. It is clear from Figure 6 that wind box temperature is lower in front half part of sintering machine as well as SO2 concentration, which proves the reasonability of above inference.

SO2 emission concentration curve and temperature curve on different sintering wind boxes.
As shown in Figure 6, the temperature of flue gas is a parabolic curve and reached the highest value in the 20# wind box, and then declines. The SO2 concentration of flue gas reached the peak value from 12# box to 16# box. The main reason is that the absorption and containing capability of wetting zone was weakened on account of water evaporation with the above drying zone coming down, resulting to SO2 adsorbed in wetting zone releasing again. When sintering zone came down to this zone, sintering raw materials containing sulphur in this zone were decomposed to generate SO2. SO2 deriving from above two parts was taken to the below wetting zone by air draft and adsorbed by it. The above process of SO2 absorbing and releasing is recycled until sintering ending. In addition, SO2 was enriched in wetting zone at the bottom of materials zone in sintering middle or final stage. When the wetting zone disappeared, SO2 was released concentratedly along with the bottom materials containing sulphur oxidizing and decomposing, resulting in the peak value of SO2 emission concentration. SO2 concentration attained the maximum value in the place from 12# box to 16# box in Figure 6, which stated that drying zone was arrived at the bottom of bed and that SO2 in the wetting zone was released in accumulated.
Reaction of urea in the sintering zone
The distance from the up plane of wetting zone (less than 100 °C) to the bottom of sintering zone (more than 1,000 °C), namely drying zone, is only dozens of millimeters. Drying zone lying between the sintering zone and the wetting zone is heated rapidly from the top to the bottom by high-temperature exhaust gas, namely from wetting zone disappearing to 700 °C, the time of which is very short and the staying time of drying zone is about 1 min [12].
The hydrolysis rate of urea at room temperature is low. Chen et al. [13] found that urea started to decompose largely at melting temperature (132 °C) by using thermal gravity analysis–mass spectrometry technology to research hydrolysis process of urea. Koebel and Strutz [14] and Koebel et al. [15] pointed out that urea began to decompose at 80 °C and significant decomposition phenomenon of urea was attained at melting temperature. It was found [16] that urea was heated slowly to generate biuret (C2H5N3O2) and ammonia gas (NH3), which interacted with each other to produce cyanic acid (HOCN) and ammonia gas (NH3) when reaction temperature was more than 193 °C. However, with the condition of rapid heating, urea was decomposed to create cyanuric acid (C3N3(OH)3) and ammonia gas (NH3) first and then to produce cyanic acid (HOCN) and ammonia gas (NH3). Figure 7 shows the schematic diagram of urea decomposing with heating. Research [16, 17] was studied to state that the effective decomposing rate of urea was 100 % at 600 °C. Therefore, urea was rapidly decomposed to HOCN and NH3 with drying zone coming down to the place where urea was added. In addition, HOCN was disintegrated further to NH3, which was taken to the below wetting zone by negative pressure air draft.
![Figure 7: Urea decomposing reactions with different heating conditions [16].](/document/doi/10.1515/htmp-2015-0172/asset/graphic/htmp-2015-0172_figure7.gif)
Urea decomposing reactions with different heating conditions [16].
Mechanism of SO2 interaction with NH3
NH3 and SO2 barely interacted with each other in the drying zone with only dozens of millimeters height, which were mainly happened in the wetting zone. The reasons were that reaction between NH3 and SO2 was carried out slowly with reaction temperature more than 60 °C [18, 19]. Simultaneously, NH3 and SO2 produced in the drying zone were rapidly taken to the below wetting zone by air draft, resulting in little time of gas-phase reaction between NH3 and SO2. NH3 was adsorbed by water or reacted with SO2 in the wetting zone. With abundant free water and some O2 existing in wetting zone [10], some reactions happened as follows:
Oxidizing reaction taking place with SO2 dissolved in water:
NH3 decomposed by urea interacting with NH3 possibly in the wetting zone:
Figure 8 shows the Gibbs free energy of desulphurization reactions in the wetting zone. It is known thermodynamically from Figure 8 that Gibbs free energy of above reactions are all of negative value, at 273–393 K, which declare that reactions (1)–(6) can take place in the wetting zone. In tests 2 and 3, sintering time was about 34 and 13–18 min of which appeared plenty of “rime fog” in experimental filter bottle. From Table 4, it is shown that the SO42– and NH4+ ionic concentrations of adding urea increase subsequently compared to 106 and 32 mg/L free of urea, which declared the “rime fog” above was ammonium sulphate and reactions (1)–(6) happened as well. And the SO42– and NH4+ ionic concentrations of “0.05 % + 100 mm” adding method were 2,468 and 706 mg/L, while the SO42– and NH4+ ionic concentrations of “0.05 % mixed” adding method were 2,283 and 647 mg/L, respectively.
It was reported that ammonium sulphate powder starts to decompose at 213 °C and decomposes completely at 419 °C [20]. Therefore, ammonium sulphate powder, taken away by strong negative pressure air draft before decomposing in sintering process, could not exist in sintering products and increase the sulphur content of sinter.

Gibbs free energy of desulphurization reactions in the wetting zone.
The influence of different methods of adding urea to the sintering raw materials on FGD was investigated in this paper. The desulphurization rate of “0.05 %+ 100 mm” adding method is 88.14 %, while the “0.05 % mixed” adding method is only 81.79 %. The main reasons were that urea added location (100 mm height from feed zone bottom) was the drying zone when SO2 was just released concentratedly. In other words, when SO2 adsorbed in the wetting zone was released concentratedly, drying zone just came down to the addition position of urea. At the same time, urea was decomposed accumulatedly to NH3, which was taken to wetting zone by negative pressure air draft and interacted with SO2 in the wetting zone. Hence, desulphurization reactions were conducted concentratedly. Simultaneously, addition of urea in the bottom zone avoided ammonia existing in sintering ore, which increased the effective availability of urea desulphurization and made it reasonable to investigate urea-selective desulphurization in sintering process.
Conclusions
When 0.05 % urea was added, the emission concentration of SO2 in flue gas reduces sharply. The desulphurization rate of adding 0.05 % urea particles to 100 mm height from the feed zone bottom can reach 88.14 %, which is better than that with urea mixed into sintering raw materials in which the desulphurization rate is 81.79 %.
The results of mechanism analysis shows that, when drying zone moves down to the urea added location in the sintering zone, the urea starts decomposing and generates large amounts of NH3, and most of the NH3 is taken to the wetting zone by negative pressure air draft. As the concentration of SO2 is relatively high in the wetting zone, those NH3 will react with SO2, which makes the desulphurization reactions to be conducted concentratedly. In addition, as the urea is added at the bottom of sintering zone, the ammonia generated by the decomposition of urea can be avoided existing in sintering ore, which improves the desulphurization efficiency and emissions reduction effect.
Funding statement: Funding: The authors are thankful to the Union Project of National Natural Science Foundation and Baosteel (U1260101) and Project of Science and Technology Development Anhui Province (1501041126) for sponsoring the research work.
References
[1] C. Zhao, T. Wu, X. Bo and Y. Su, Environ. Eng., 10 (2014) 76–78.Search in Google Scholar
[2] T. Chun and D. Zhu, Metall. Mater. Trans. B, 46 (2015) 1–4.10.1007/s11663-014-0243-4Search in Google Scholar
[3] J. Pan, Ph.D. Thesis Cent. South. Univ. Changsha (2007).Search in Google Scholar
[4] J. Chen, World Iron Steel, 3 (2010) 1–6.10.1016/S1006-706X(10)60104-5Search in Google Scholar
[5] Y. Qu, Y. Mao and D. Zhang, Energy Metall. Ind., 29 (2010) 51–56.Search in Google Scholar
[6] C. Cen and D. Zhang, J. Environ. Eng., 3 (2005) 25–28.10.1002/ceat.200407050Search in Google Scholar
[7] S. Wang, X. Bi and D. Weng, Adv. Mater. Res., 726 (2013) 2284–2290.10.4028/www.scientific.net/AMR.726-731.2284Search in Google Scholar
[8] J. Xiao, M.S. Thesis. An. Hui. Univ. Tech. Ma’anshan (2014).Search in Google Scholar
[9] K. Chen, Sinter. Pellet., 32 (2007) 13–17.10.1590/S0104-71832007000200002Search in Google Scholar
[10] Y. Zhang, S. Hao, W. Jiang and Y. Long, Adv. Mater. Res., 284 (2011) 1279–1283.10.4028/www.scientific.net/AMR.284-286.1279Search in Google Scholar
[11] H. Long, J. Li and P. Wang, J. Cent. South. Univ. T., 19 (2012) 1359–1363.10.1007/s11771-012-1150-ySearch in Google Scholar
[12] H. Long, J. Li and P. Wang, Ironmak. Steelmak., 38 (2011) 258–262.10.1179/1743281210Y.0000000008Search in Google Scholar
[13] J. Chen and K. Isa, J. Mass. Spectrom. Soc. Jpn., 46 (1998) 299–303.10.5702/massspec.46.299Search in Google Scholar
[14] M. Koebel and E. Strutz, Ind. Eng. Chem. Res., 42 (2003) 2093–2100.10.1021/ie020950oSearch in Google Scholar
[15] M. Koebel, M. Elsener and M. Kleemann, Catal. Today, 59 (2000) 335–345.10.1016/S0920-5861(00)00299-6Search in Google Scholar
[16] K. Eiki, K. Shunsuke, H. Goto and T. Murakami, ISIJ Int., 48 (2008) 1305–1310.10.2355/isijinternational.48.1305Search in Google Scholar
[17] H. Lv, W. Yang, J. Zhou, J. Liu, M. Zhang and F. Li, Proc. CSEE, 30 (2010) 35–40.Search in Google Scholar
[18] B. He, X. Zheng, Y. Wen, H. Tong, M. Chen and C. Chen, Energ. Convers. Manage., 44 (2003) 2175–2188.10.1016/S0196-8904(02)00230-3Search in Google Scholar
[19] H. Bai, P. Biswas and T. Keener, Ind. Eng. Chem. Res., 33 (1994) 1231–1236.10.1021/ie00029a019Search in Google Scholar
[20] Y. Fan and F. Cao, J. Chem. Eng. Chin. Univ., 25 (2011) 341–346.Search in Google Scholar
©2017 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Research Articles
- Preparation of Co3O4 Nanostructures via a Hydrothermal- Assisted Thermal Treatment Method by Using of New Precursors
- Transformation Temperatures, Shape Memory and Magnetic Properties of Hafnium Modified Ti-Ta Based High Temperature Shape Memory Alloys
- Modified Sol-Gel Processing of NiCr2O4 Nanoparticles; Structural Analysis and Optical Band Gap
- Effect of Proeutectoid Ferrite Morphology on the Microstructure and Mechanical Properties of Hot Rolled 60Si2MnA Spring Steel
- Uniaxial Properties versus Temperature, Creep and Impact Energy of an Austenitic Steel
- Effect of Rare Earth Cerium Addition on Microstructures and Mechanical Properties of Low Carbon High Manganese Steels
- Geometry and Material Constraint Effects on Creep Crack Growth Behavior in Welded Joints
- Preparation and Properties of ZrO2/Mo Alloys
- The Mechanical Properties of the Mo-0.5Ti and Mo-0.1Zr Alloys at Room Temperature and High Temperature Annealing
- Diffusion Kinetics of Chromium in a Novel Super304H Stainless Steel
- Mechanism of Selective Desulphurization in Iron Ore Sintering Process by Adding Urea
- The Characteristics and Generating Mechanism of Large Precipitates in Ti-Containing H13 Tool Steel
Articles in the same Issue
- Frontmatter
- Research Articles
- Preparation of Co3O4 Nanostructures via a Hydrothermal- Assisted Thermal Treatment Method by Using of New Precursors
- Transformation Temperatures, Shape Memory and Magnetic Properties of Hafnium Modified Ti-Ta Based High Temperature Shape Memory Alloys
- Modified Sol-Gel Processing of NiCr2O4 Nanoparticles; Structural Analysis and Optical Band Gap
- Effect of Proeutectoid Ferrite Morphology on the Microstructure and Mechanical Properties of Hot Rolled 60Si2MnA Spring Steel
- Uniaxial Properties versus Temperature, Creep and Impact Energy of an Austenitic Steel
- Effect of Rare Earth Cerium Addition on Microstructures and Mechanical Properties of Low Carbon High Manganese Steels
- Geometry and Material Constraint Effects on Creep Crack Growth Behavior in Welded Joints
- Preparation and Properties of ZrO2/Mo Alloys
- The Mechanical Properties of the Mo-0.5Ti and Mo-0.1Zr Alloys at Room Temperature and High Temperature Annealing
- Diffusion Kinetics of Chromium in a Novel Super304H Stainless Steel
- Mechanism of Selective Desulphurization in Iron Ore Sintering Process by Adding Urea
- The Characteristics and Generating Mechanism of Large Precipitates in Ti-Containing H13 Tool Steel