Determination of membrane protein orientation upon liposomal reconstitution down to the single vesicle level
-
Sarina Veit
Sarina Veit is working on a PhD thesis at the Ruhr University Bochum, Germany which is funded by the
Studienstiftung des deutschen Volkes . The topic of her PhD thesis is focused on the exploration of P4-ATPasesin vivo as well as purified and reconstituted into model membrane systems. Her special interest lies in the transfer from bulk to single-vesicle experiments based on TIRF microscopy. During her PhD, she spent a research stay at the research group of Anant Menon at the Weill Cornell University in New York, USA and the group of Nikos Hatzakis at the Copenhagen University in Copenhagen, Denmark. ,
Laura Charlotte Paweletz
,
Laura Charlotte Paweletz
Laura Charlotte Paweletz studied biochemistry at the Ruhr University Bochum, Germany and spend an Erasmus term at Stockholm University, Stockholm, Sweden. She received her PhD in 2022 on the interplay of lipids and membrane transporters and was funded by the
Studienstiftung des deutschen Volkes . During this time, she gained experiences in single vesicle microscopy visiting in the labs of Anant Menon at the Weill Cornell University in New York, USA and Nikos Hatzakis at the Copenhagen University in Copenhagen, Denmark. Currently, she holds a researcher and teaching position at the Ruhr University Bochum.
und
Thomas Günther Pomorski
Thomas Günther Pomorski received his Ph.D. in Biophysics in 1996 from the Humboldt University of Berlin, Germany. After postdoctoral studies at the Max-Planck-Institute of Infectious Diseases in Berlin and the Academic Medical Center Amsterdam in the Netherlands, he became a Junior Professor for Cell Biophysics at the Humboldt University of Berlin. In 2009 he was appointed as Associate Professor and in 2016 as Professor at University of Copenhagen, Denmark. Since 2016 he has been Chair Professor in Molecular Biochemistry at the Ruhr University Bochum, Germany. His lab develops and applies molecular methods to study organization, function and regulation of membrane transporters.


Abstract
Reconstitution of membrane proteins into liposomal membranes represents a key technique in enabling functional analysis under well-defined conditions. In this review, we provide a brief introduction to selected methods that have been developed to determine membrane protein orientation after reconstitution in liposomes, including approaches based on proteolytic digestion with proteases, site-specific labeling, fluorescence quenching and activity assays. In addition, we briefly highlight new strategies based on single vesicle analysis to address the problem of sample heterogeneity.
Introduction
Membrane proteins play an essential role in controlling bioenergetics, the movement of ions, lipids, and metabolites across cellular membranes, and initializing signaling pathways. Additionally, membrane trafficking is based on the concerted action of multiple membrane proteins, wherefore malfunction of these transport and signaling systems leads to various diseases (Marinko et al. 2019). Given the pivotal role of membrane proteins in cell homeostasis, a thorough understanding of their functioning and dynamics is highly desirable.
The study of membrane proteins at the molecular level using in vivo experimental setups is hampered by the complexity of the cell. A single membrane is composed of hundreds of proteins and thousands of lipids continuously interacting in time and space, complicating the dissection of the molecular function and contributions of each component. To reduce this high complexity, a wide variety of membrane platforms has been developed for the investigation of membrane protein function under well-defined conditions outside the complex cellular environment. The boom of structural biology boosting the number of resolved structures of transmembrane proteins and the development of computational biology, linking the snapshot of proteins in different states via molecular dynamic simulation, gives rise to new hypotheses on detailed molecular mechanisms of transporters or receptors. To prove and evolve those computational presumptions, experiments on the molecular level are required. The most common approach for membrane-mimicking systems is based on the reconstitution of membrane proteins into lipid bilayers to create proteoliposomes (Amati et al. 2020; Murray et al. 2014; Rigaud et al. 1995; Rigaud and Lévy 2003; Skrzypek et al. 2018). The approach offers a well-defined experimental system where the protein of interest can be investigated individually. Moreover, proteoliposomes can be customized in a variety of ways by finetuning the lipid composition. On the one hand, lipids can be utilized that promote protein stability and/or control protein function (Corradi et al. 2019; Lasitza-Male et al. 2020; Natarajan et al. 2009) and on the other hand, modified lipids such as pH-sensors, fluorophores, or immobilization anchors to tailor the vesicle for a variety of applications can be included (Kemmer et al. 2015; Schwamborn et al. 2017).
Different methods have evolved for the rapid and efficient reconstitution of membrane proteins into lipid membranes, whereas the most common method for proteoliposome preparation is based on detergent-mediated reconstitution (Rigaud et al. 1995; Rigaud and Lévy 2003). After solubilization and purification, the membrane protein is supplemented with an excess of lipids and detergent molecules, leading to a mixture of lipid-protein-detergent and lipid-detergent micelles. Subsequently, the detergent is removed resulting in the progressive vesicle bilayer formation and thereby embedding the protein. While the membrane protein’s topology in vivo is determined and ensured by the cellular machinery, membrane proteins reconstituted in liposomal model membrane systems have either the cytosolic (in-side-out orientation) or the extracellular site (out-side-out orientation) exposed. However, the orientation of the reconstituted membrane protein appears to be specific to each protein and dependent on the utilized reconstitution conditions (Rigaud et al. 1995). In the case of mitochondrial respiratory complex I, for example, the type of detergent and the method of its removal affect the unidirectional incorporation (Biner et al. 2020). Furthermore, the protein itself might prefer a specific orientation in the vesicle, due to bulky domains and/or the curvature of the vesicle. For instance, the F0F1 ATPase tends to be inserted into vesicles with its large cytosolic domain outside, unlike the bo3 oxidase which shows a more random insertion (Biner et al. 2020; Deutschmann et al. 2022). Thus, the protein orientation is hardly predictable, which limits the quantitative and functional analysis of vesicle preparations. Typically, only enzymes exposing their substrate binding site to the outside have access to substrates in the buffer and therefore contribute to the sample’s activity. This resulting discrepancy between the measured total protein amount and the actual protein amount contributing to the recorded activity of a proteoliposome sample emphasizes the importance of protein orientation determination.
Several methods have been developed to analyze the distribution of orientation for the reconstituted protein. In this review, we will introduce selected methods to determine membrane protein orientation after reconstitution into liposomes. The main approaches are all based on the differences in accessibility of inside-out and outside-out oriented protein together with impermeable and permeable modifying reagents (Figure 1). This review focuses on approaches based on proteolytic digestion, site-specific labeling, fluorescence quenching, and activity-based assays. In addition, we will highlight new strategies to transfer the common approaches from bulk to single vesicle analysis, to address the problem of sample heterogeneity.
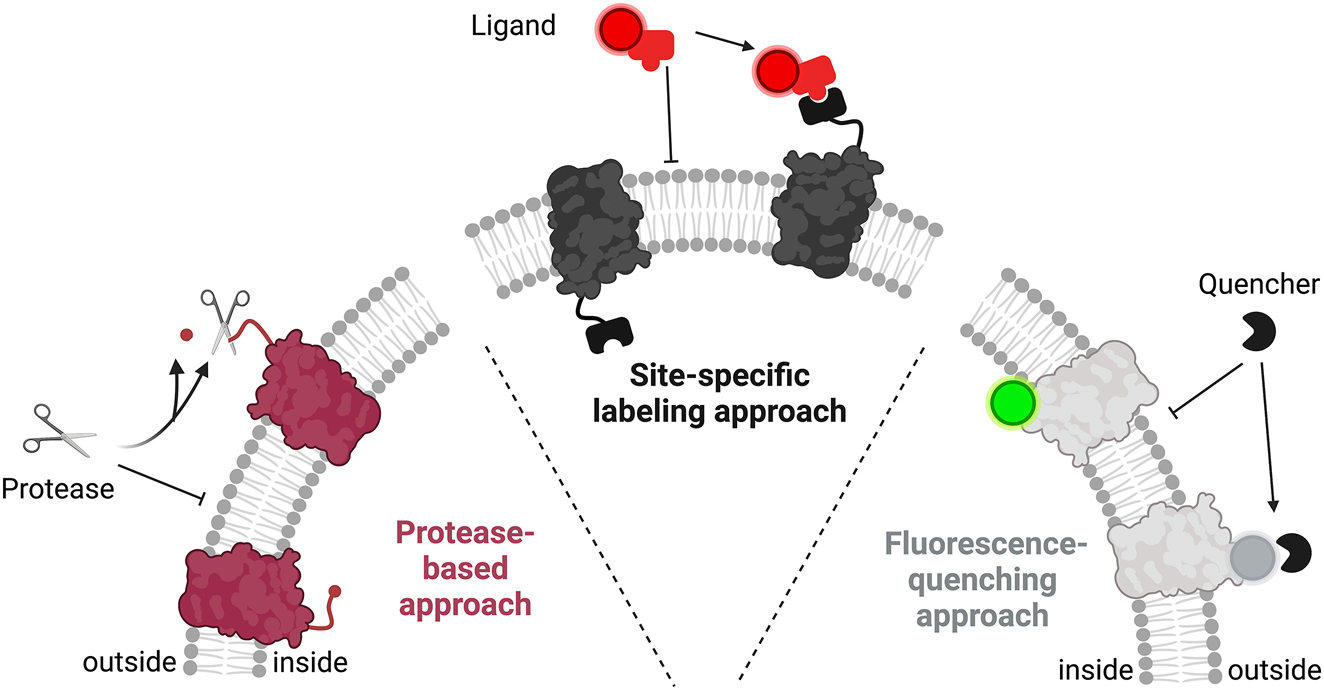
Schematic overview of the three most common approaches for protein orientation determination in liposomes. All three approaches are based on different accessibilities of outwards and lumen-facing protein portions. The protease-based approach relies on digestion of the outwards-facing portion/tag by an impermeable protease. The site-specific labeling approach exploits differences in the permeability of protein-binding ligands. Fluorescence quenching-based approaches take advantage of a fluorescent tag that is attached to the protein and whose fluorescence is diminished by an impermeable quenching reagent.
Protease-based approaches
A common approach to determine the orientation of transmembrane proteins in liposomes is based on proteolytic digestion of the accessible outward-facing portion. For each protein in proteoliposomes, either the cytosolic or extracellular site is exposed to the extraluminal solution and thus accessible for cleavage, resulting in a unique average ratio of inside to outside oriented proteins for each sample preparation. Consequently, protease-induced cleavage of proteoliposome-embedded proteins produces protein fragments depending on the protein orientation. Hereby, the specificity of the used protease gives rise to different, distinct sets of protein fragments, which are usually separated by SDS-PAGE for subsequent analysis of the digestion products (Figure 2). The comparison of the protease-treated sample with an untreated sample allows to determine the orientation of the reconstituted protein. Additionally, a control for the complete digest is needed using a sample treated in the presence of detergent to show that the digestion conditions (time, protease concentration, temperature) are sufficient for the digestion of the entire protein population in the sample.
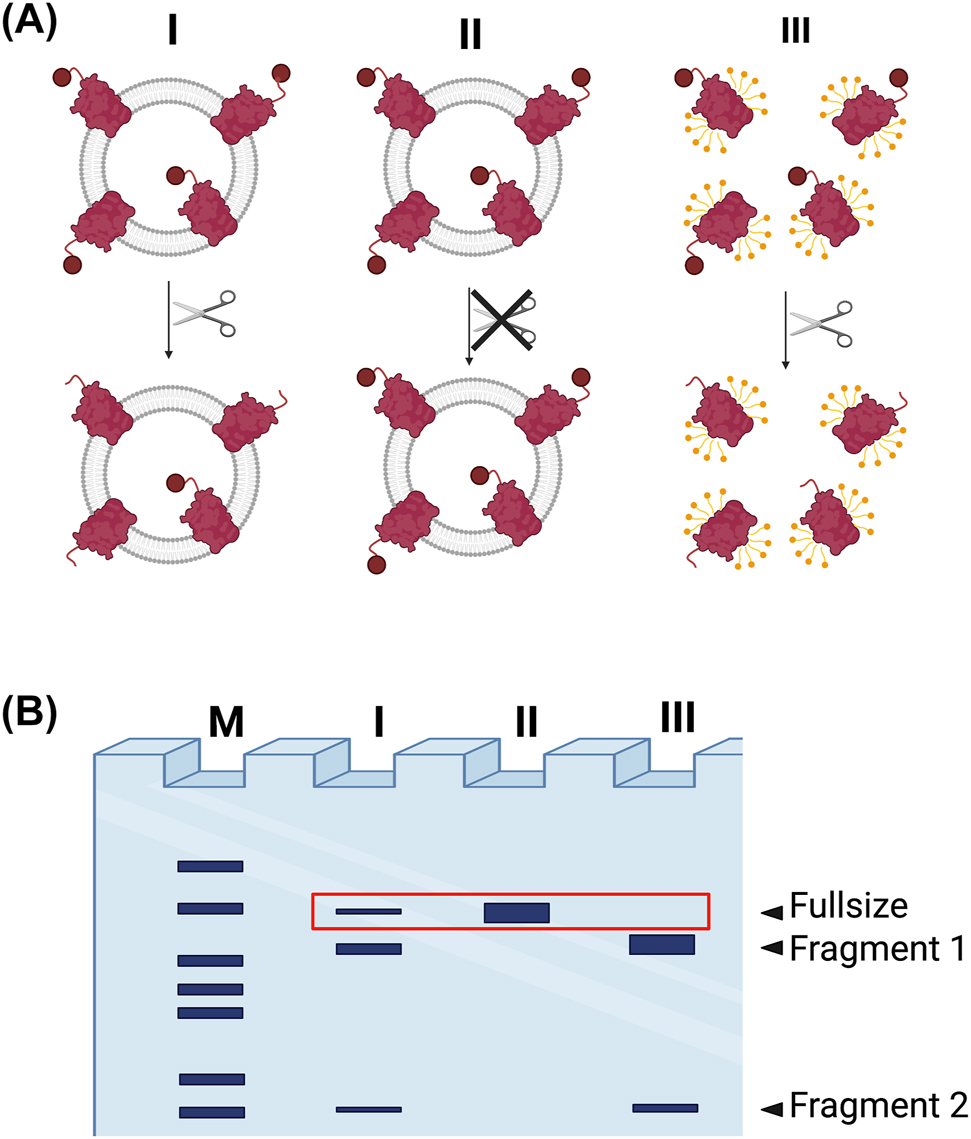
Protease-based assay for determining membrane protein orientation using a site-specific protease (e.g., TEV protease). (A) Proteoliposomes are incubated with a protease, resulting in the generation of proteolytic fragments depending on which side of the protein is exposed (Sample I). Controls include experiments performed in the absence of protease, maintaining the undigested protein (Sample II) and cleavage in presence of detergent, disrupting the vesicles and allowing access for the protease to all protein (Sample III). (B) SDS-PAGE gel analysis of the digestion products. Lane I: digested protein in intact proteoliposomes; Lane II: undigested protein in proteoliposomes; Lane III: completely digested protein in solubilized proteoliposomes; M, marker lane. Band of the non-cleaved form (red box) can be used for quantification. Note, this is a simplified gel result for site-specific cleavage. The use of non-specific proteases will result in a more complex digestion pattern. Secondly, the solubilized sample could show incomplete digestion due to aggregation.
The first decision when setting up a protease-based orientation assay is the choice of protease. An overview of frequently used proteases and their cleavage sites is provided in Table 1. Depending on the published work, digest conditions vary substantially regarding incubation temperature (4–37 °C) and incubation time (minutes to overnight). Proteolytic digestion with trypsin, a non-specific serine protease (Polgár 2005) that cleaves at the carboxylic side of lysine and arginine residues (Olsen et al. 2004; Rodriguez et al. 2008), has been successfully used to characterize many reconstituted proteins. The more aggressive Proteinase K, a serine protease like trypsin, cuts preferentially after hydrophobic amino acids (Kraus et al. 1976) and is often used if trypsin digestion is not efficient enough (Lorenz et al. 2006; Rigaud and Lévy 2003). The high abundance of their cleavage sites leads to the digestion of almost all outwards-facing protein portions, leaving the membrane integral and inwards-facing portions intact. After digestion, the sample is loaded on an SDS-PAGE with subsequent Coomassie staining to analyze digestion fragment ratios, which allows a conclusion about protein orientation. Here, ratios need to be compared quantitatively to translate the pattern in a ratio of inside-out to outside-out orientation. However, if outwards facing loops are digested, the leftover portions could lack connection and the digestion pattern can be complex and thereby not straightforward to analyze. In rare cases, destabilization of the vesicles upon protease treatment has been reported (Hu et al. 1986) that might compromise the assay results.
Common proteases and their cleavage sites.
| Protease | Cleavage sitea | References | |
|---|---|---|---|
| Serine proteases | Trypsin | Basic AA (Lys, Arg) | Bayburt et al. (1998), Cuello et al. (1998), Gennarini et al. (1984), Ishijima et al. (2012), Iwahashi and Nakamura (1989), Lévy et al. (1992), Schuette et al. (2004), Serek et al. (2004), Surrey and Jahnig (1992) |
| Chymotrypsin | Aromatic AA and Leu | Ishijima et al. (2012), Schwarz and Steinem (2022), Ueki and Iwamoto (2021) | |
| Proteinase K | Hydrophobic AA, (Pro) | Fimmel et al. (1989), Gerber et al. (1977), Kalmbach et al. (2007), Ohnishi and Kamiya (2021), Ritzmann et al. (2017), Tunuguntla et al. (2013) | |
| Thrombin | LVPR\GS | Richards et al. (2016) | |
| Human rhinovirus (HRV) 3C Protease | LEVLFQ\GP | Taylor et al. (2017) | |
| Cysteine proteases | Tobacco etch virus (TEV) protease | ENLYFQ\G, ENLYFQ\S | Eisinger et al. (2018), Islam et al. (2013), Martín et al. (2009), Periasamy et al. (2013), Stefan et al. (2020) |
| Papain | Hydrophobic AA followed by Arg and Lysb | Engelhard et al. (1978) | |
-
aAA – amino acid. bWith complex environmental requirements.
The introduction of a specific cleavage site in the protein of interest allows to use the highly sequence-specific cysteine protease from Tobacco Etch Virus (TEV) (Kapust et al. 2001; Kostallas et al. 2011; Parks et al. 1994), resulting in two fragments only, thereby facilitating the analysis of the fragments, as shown in Figure 2. This approach is often combined with a fluorescent or epitope tag, which further facilitates a comparison of cleaved and intact proteins, referring to inside-out and outside-out oriented proteins, respectively.
Although the protease-based approach is often used for protein orientation determination after reconstitution, this method has several potential pitfalls. Starting with the digestion step, a prerequisite of the assay is that the protein is susceptible to a selected protease in general and especially under the used in vitro conditions; otherwise, no conclusions regarding its orientation can be drawn from these experiments. However, the actual accessibility of the exposed cleavage site can be hampered sterically by the surrounding lipid bilayer, large protein domains, high protein density, or even oligomer formation, which could lead to an underestimation of outwards oriented protein.
The second prerequisite is a control to show that in principle the digestion conditions were suitable for complete digestion, which is usually addressed with a detergent-solubilized sample. On the one hand, the solubilized state differs significantly from the reconstituted system, regarding protein density and thereby protease accessibility as mentioned before. In fact, the comparison with the detergent-solubilized sample could be misleading, since steric hindrance might be released in presence of detergent. On the other hand, detergents might alter the fluorescent characteristics of fluorescent tags which are sensitive to their environment (Galbán et al. 2009), influencing the subsequent intensity analysis of the fragments.
The third prerequisite of the assay concerns the successful termination of the cleavage at the end of incubation since the digest must be stopped before analysis via SDS-PAGE. Otherwise, luminal protein gets accessible after the destruction of the liposomes by SDS which might lead to ongoing cleavage of the previously protected pool. In some protocols, the addition of SDS itself is assumed to be enough to inactivate the protease and prohibit further cleavage (Ishijima et al. 2012; Schuette et al. 2004). In contrast to soluble protein samples, heat inactivation of the protease is normally not considered since membrane proteins tend to aggregate upon heating. Two strategies are used instead – often in combination – to inactivate the protease: (i) treatment with an irreversible protease inhibitor (Cuello et al. 1998; Jeter and Escalante-Semerena 2021; Kalmbach et al. 2007; Tunuguntla et al. 2013) and/or (ii) a fast denaturing protein precipitation step using trichloroacetic acid or acetone (Ohnishi and Kamiya 2021; Serek et al. 2004). However, the termination efficiency must be verified for a reliable read-out e.g., with a control sample in which the protease is added at the same time as the stopping reagent which should result in no cleavage at all. Alternatively, and especially for large transmembrane proteins, samples can be undertaken a centrifugation step to pellet the liposomes containing undigested membrane protein and to prevent further digestion. The supernatant containing the cleaved, soluble fragments can be loaded on an SDS-PAGE together with a sample digested under solubilization conditions and analyzed by Coomassie staining. In combination with protease cleavable fluorescent tags, the released fluorophores such as GFP or YFP (Islam et al. 2013; Taylor et al. 2017) can simplify the detection by in-gel fluorescence and might allow lower detection limits.
Moreover, protease-based approaches can be combined with blotting and epitope detection. If an antibody is available specifically for one site of the protein, the destruction of the epitope can be achieved with unspecific protease treatment and the intensity of the band after western blotting can be compared to a non-digested sample and/or a completely digested sample (Cvjetkovic et al. 2016). On the other hand, if site-specific proteases are used to cleave an epitope tag, the ratio of full-size protein and free tag can be compared in the western blot.
Overall, the common advantage of the protease-based approaches is their simplicity, requiring just a few specific reagents (i.e., a protease with a suitable inhibitor) and common protein analysis equipment (e.g., SDS-PAGE and Western-blotting apparatus). On the contrary, optimization of the cleavage conditions can be time-consuming since the concentration of the protease, the incubation time and temperature, as well as the termination of the digestion must be optimized for every sample. More precisely, potential steric hindrance and incomplete cleavage due to the membrane environment must be considered. Furthermore, special care needs to be taken, since many fluorophores are highly sensitive to their environment and not only the detergent-solubilized protein, but the cleaved, soluble tag might have altered fluorescence properties compared to the protein-linked tag (Galbán et al. 2009; Veshaguri et al. 2016).
Site-specific labeling approaches
For site-specific labeling assays, many labeling methods have been developed and utilized, all based on the binding of a non-permeable factor to the exposed side of the membrane protein only. The main advantage of these assays is their high sensitivity and thus, only a small amount of sample is required. Herein we highlight three common approaches for site-specific labeling assays based on: labeling with antibodies, cysteine labeling using thiol-reactive dyes, and self-labeling enzyme tags.
Site-specific labeling with antibodies
A frequently used approach is based on affinity labeling, using antibodies raised against a specific epitope located at the termini or loops of the protein (Knol et al. 1998), or antibodies specific for a tag introduced into the membrane protein of interest such as poly-histidine or herpes simplex virus tags (Tunuguntla et al. 2013). The protein copies with exposed epitopes are labeled by incubation with an antibody solution, allowing subsequent immunodetection of labeled proteoliposomes. The labeling of the intact proteoliposomes will depend on the orientation of proteins within the bilayer, as binding sites facing the internal compartment will be inaccessible to the non-permeable antibodies. Upon removal of the unbound antibody fraction by centrifugation or size exclusion chromatography (Knol et al. 1998; Tunuguntla et al. 2013), the ratio of tag fluorescence to protein absorption is measured, or the proteoliposomes are subjected to immunoblotting techniques. Limitations of the approach include restricted antigen accessibility to the antibody and the potential impact of the tag on protein structure and/or function as well as orientation.
Site-specific labeling via cysteines
Another common approach is based on site-specific labeling via cysteines using thiol-reactive reagents, as reviewed by Bogdanov et al. (2005). Notably, labeling with reagents of different permeability allows analysis under nondestructive conditions for the vesicle and the protein. For this, the vesicles are first incubated in the presence of a membrane-impermeable thiol-reactive reagent (e.g., 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid) to block outwards oriented cysteines followed by the addition of a membrane-permeable thiol-reactive reagent to label inwards facing cysteines (Fang et al. 1999; Knol et al. 1996), respectively. Here, labeling of protein portions orientated to the inside of the vesicle is achieved using labels, which are membrane permeable in general (benzophe-none-4-maleimide), in a time and concentration-dependent manner (3-(N-maleimido-propionyl) biocytin) or when combined with membrane lysis or permeabilization. Importantly, the reaction can be stopped easily by the addition of reducing agents such as dithiothreitol (DTT) prior to SDS-PAGE analysis and thus vesicle destruction (Loo and Clarke 1995; Poelarends and Konings 2002). The subsequent analysis is achieved by antibody detection (Harris et al. 2017), biotin/avidin interaction for biotinylated substrates (Bayer et al. 1987; Knol et al. 1996), or by in-gel fluorescence when using cysteine-reactive fluorophores such as 5-Iodoacetamidofluorescein during the labeling (Vecino et al. 2010; Wang et al. 2013). However, the membrane permeability or impermeability of the probes needs to be validated. For instance, thiol responder, such as thionitrobenzoate, can be encapsulated in the proteoliposomes to test the permeability of the cysteine labeling agents (Holmgren et al. 1996).
Cysteines are well suited for site-specific conjugation because they are relatively rare throughout the proteome and can be easily inserted at a specific position in the amino acid chain of the protein. However, introducing a reactive cysteine residue into proteins with multiple disulfide bonds is often a challenging task as it may interfere with the structural and functional properties of the protein. Additional bottlenecks in this approach include the different accessibility and modification rates of cysteines depending on the microenvironment of the membrane, the maintenance of reactive thiol groups without oxidation before the reaction, and the effective removal of unreacted labeling molecules prior to analysis. To overcome some of these limitations, non-natural, non-canonical amino acids can be incorporated into membrane proteins to introduce reactive chemical groups for direct protein labeling with small organic fluorophores (Lee et al. 2019). However, the success rate and efficiency of non-natural, non-canonical amino acid mutagenesis vary considerably with the type of membrane protein being investigated, the expression system, the choice of amino acid, and the target site in the protein (Leisle et al. 2015).
Site-specific labeling using self-labeling enzyme tags
Labeling methods based on enzyme reactions take advantage of the specific interactions between enzymes and their substrates. In this approach, self-labeling enzyme tags are genetically attached to the target proteins, and the fluorophores are then incorporated into the tag by enzyme reactions with the substrates carrying those labels, such as SNAP, HALO, ACP, or CLIP tag (Andra et al. 2018; Cole 2013; England et al. 2015; Gautier et al. 2008; Keppler et al. 2003; Tirat et al. 2006). Recently, this approach has been used in combination with membrane permeable and impermeable dyes to determine membrane protein topology upon reconstitution (Figure 3; Paweletz et al. 2022; Veit et al. 2022). In contrast to cysteine labeling, side specific, stoichiometric 1:1 labeling without change of the amino acid sequence is ensured. Stepwise incubation with non-permeable and permeable dyes or blocking reagents is used for the protein orientation determination, which can be quantified by comparison of the in-gel fluorescence with control samples in which no blocking/outside only label was applied. In contrast to the protease-based approach, all controls can be performed on the intact liposome sample and allow comparability since detergent solubilization is not needed when using labels with different permeability. Yet, membrane permeability or impermeability of the probes might depend on the liposomal lipid composition and need to be validated and labeling conditions have to be optimized for each sample.
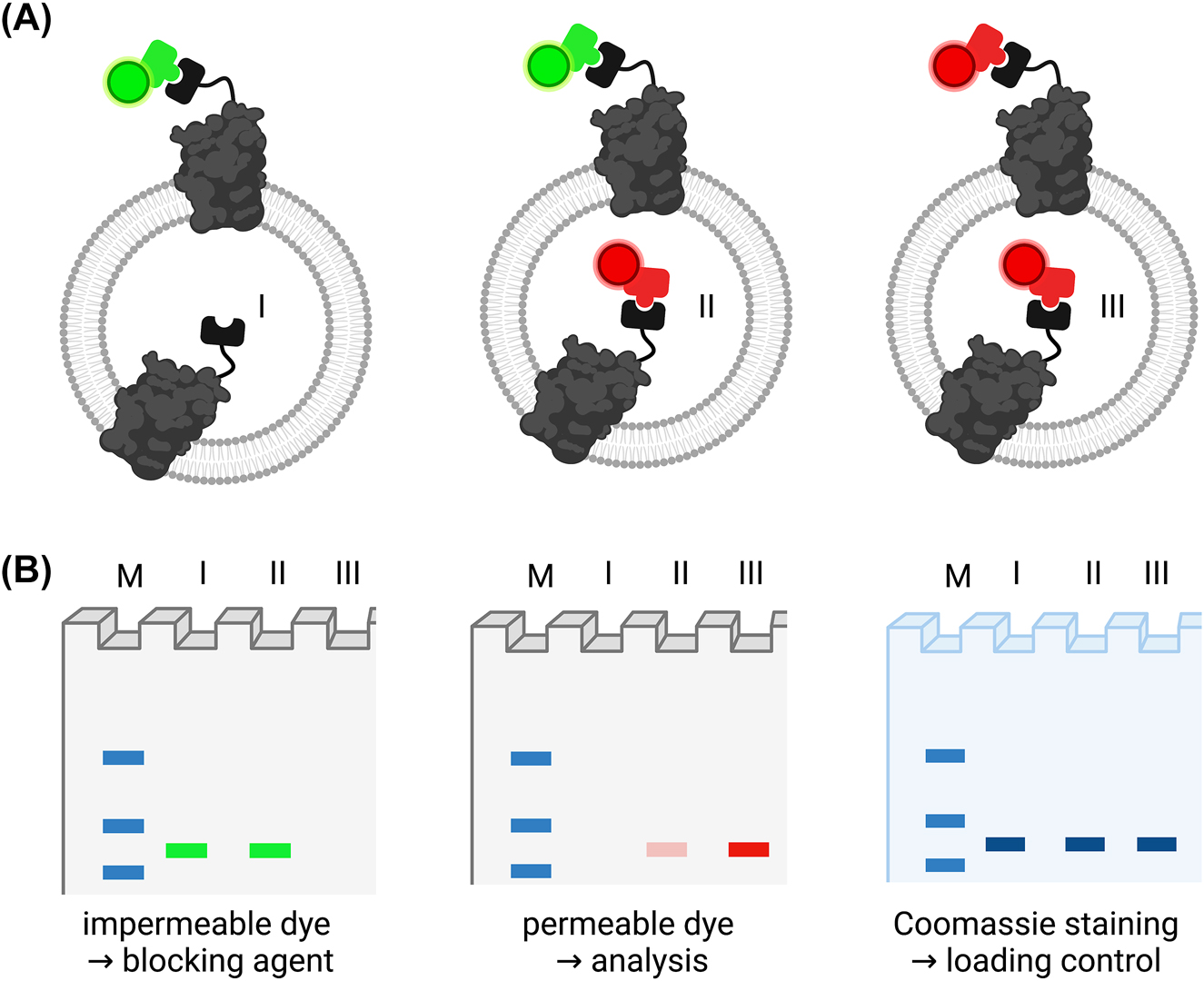
Site-specific labeling approach. (A) Proteoliposomes are labeled with an impermeable dye or blocking agent (green) only (I), a permeable dye (red) only (III) or sequentially with the both (II) and analyzed via SDS-PAGE followed by in-gel fluorescence imaging and subsequent Coomassie staining. (B) Exemplary read-out from SDS-PAGE analysis. All samples are normalized to their Coomassie signal as control for equal protein loading. A sample only incubated with blocking agent/impermeable dye (I) is used for bleed-through correction in case of partially overlapping emission spectra of the two dyes. The signal from inside labeled protein (II) is quantified using complete labeling (III) as 100% value. The actual orientation can be calculated as (F ii − F i)/F iii, where F i, F ii, and F iii are the fluorescence intensities of sample I, II, and III, respectively.
Fluorescence quenching-based assays
Fluorescence quenching-based assays employ a membrane-impermeable agent that efficiently suppresses the fluorescence of a fluorophore, which is attached to a specific site of the protein of interest in a fixed stoichiometry. The focus of this section lies on fluorescent quenching, however, the assay can be adapted to other spectroscopic methods such as UV–vis absorption for tryptophan quenching analysis and electron paramagnetic resonance spectroscopy for spin-labeled protein (Pan et al. 2021). Upon reconstitution of the fluorescent-tagged membrane protein, the fluorophore will either be protected in the vesicle lumen or exposed to the outer solution containing the quencher. By comparing the extent of fluorescence reduction on intact and permeabilized vesicles, the orientation of the fluorescent-tagged membrane protein can be estimated (Figure 4). These assays allow for live monitoring by fluorescence spectrometry because they do not require a separation step like the previously presented approaches. As efficient quenchers for GFP-tagged proteins, copper (Isarankura-Na-Ayudhya et al. 2010) and trypan blue (Jarvela et al. 2021; Karpowicz et al. 2017) have been reported; for RFP- and Cy5-tagged proteins, copper and tris(2-carboxyethyl)phosphine) have been used, respectively (Deutschmann et al. 2022; Marek et al. 2011). Furthermore, potassium quenching has been applied on membrane proteins labeled with the Atto-520 dye to monitor protein orientation (Kuhn et al. 2017). Since several lipid transport assays use nitrobenzoxadiazole (NBD)-labeled lipids in combination with reductants and quenchers (Chmyrov et al. 2010; McIntyre and Sleight 1991), this well-established technique has been adapted to protein orientation assays. Labeling of small peptides with NBD and analysis of orientation via dithionite reduction has been shown upon self-insertion of the tested peptides (Drin et al. 2003; Reshetnyak et al. 2006; Thévenin et al. 2009). Furthermore, dithionite reduction might be applied to dyes other than NBD, as recently demonstrated for Atto-488 and Alexa647 labeled membrane proteins (Andra et al. 2018, Veit et al. 2022).
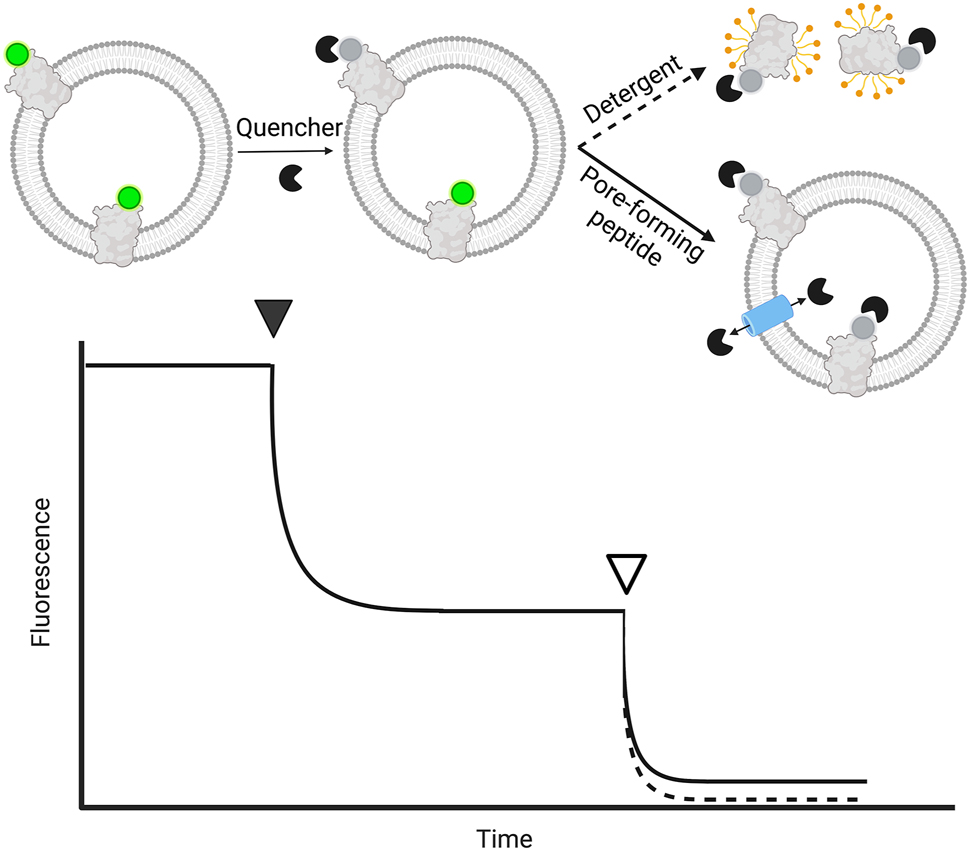
Quencher-based assay for determining membrane protein orientation. The fluorescence of tagged membrane protein reconstituted into liposomes is monitored. Outside-oriented fluorophores are quenched with a membrane-impermeable quencher (solid arrowhead), before permeabilization of liposomes by a pore-forming peptide (solid) or detergent (dashed), indicated by the non-filled arrowhead, leading to maximal quenching. As liposomes contribute to the signal read-out via unspecific scattering, the end plateau depends on the permeabilization strategy as the vesicles stay intact (pore-forming peptide, small loss in scattering), or are solubilized (use of detergent, loss of scattering). Therefore, the contribution of scattering to the signal can be verified by non-labeled liposomes. Membrane protein orientation corresponds to the percentage of quenching after the addition of an agent normalized to the initial signal.
Despite its simplicity, several aspects need to be considered when performing a fluorescence quenching-based assay for the determination of protein orientation. First, the labeling of the protein needs to be homogenous to allow a straightforward analysis. Furthermore, the impermeability of the quenching reagent is crucial: If the membrane is permeable for the quenching reagent, the fluorophore fraction facing the inside of the vesicles will also be quenched and the number of outwards facing fluorophores will be overestimated. Moreover, the heterogeneity of the proteoliposome sample implicates another level of complexity, since different subpopulations can vary in their leakiness. Furthermore, membrane permeability of a quencher reagent might differ for liposomes of different lipid compositions and should therefore be tested for every new sample, e.g., by asymmetric labeled proteoliposomes or encapsulation of soluble fluorophores into the vesicle lumen (Langner and Hui 1993; Moreno et al. 2006).
Another important factor regards the complete quenching control to show that in theory, the quencher concentration and reaction time would have been sufficient to quench all fluorophores. Therefore, the liposomes are permeabilized by the addition of either a pore-forming peptide or detergent to allow complete access of the quencher to all fluorophores, resulting in a baseline. As visualized in Figure 4, pore-forming peptides have the advantage that the liposomes stay intact and contribute consistently with their scattering to the measured signal, while detergent solubilization removes the contribution of the vesicle’s scattering from the measured signal, leading to a lower baseline. Pore-forming peptides or ionophores can be broadly used to change the permeability of reagents and can be applied to several assay set-ups allowing for the quenching of inward-facing dyes (Biner et al. 2020). In contrast to fluorescence-based lipid transport assays, the signal-to-noise ratio is lower for orientation studies as the number of protein copies, and thus fluorophores, is lower compared to the used number of labeled lipid probes. Therefore, additional controls with unlabeled proteoliposomes are needed as scattering control.
In general, quenching-based approaches are fast and easy to perform, when the reconstituted membrane protein contains a fluorescent tag anyways, commonly used for localization studies and further applications. However, controls for scattering and complete quenching have to be designed carefully. Notably, tagging the protein of interest with an additional fluorophore can change the membrane proteins’ orientation from the beginning due to changes in e.g., bulkiness or charge, so it should be taken into consideration when designing the construct (Ritzmann et al. 2017). However, the effects of labeling are not yet sufficiently known and cannot be reliably predicted.
Activity assays with inhibitors
In contrast to the approaches presented previously, where the assays were solely designed for orientation determination to support the interpretation of activity data, the assays presented in this section are combinational approaches. For membrane proteins with enzymatic activities, the activity measurements can be combined in some cases with the determination of the protein orientation. These approaches are customized for each protein and have integrated controls by membrane-impermeable substrates or inhibitors. Often these activities are energy-coupled (ATP or NADH consumption, light-induced activity). Thereby, the orientation can be determined based on the limited access of the membrane-impermeable substrate, such as ATP or NADH, to the proteins with the substrate binding site oriented to the outside (Dröse et al. 2005). Those experiments are often accompanied by an detergent-solubilized control to determine the activity of all enzymes independent from their orientation (Wiedenmann et al. 2008) or the use of pore-forming peptides such as alamethicin making substrates available on both sides of the membrane (Biner et al. 2020). Additionally, the set-up is expanded by the usage of site-specific inhibitors to allow for conclusions on the orientation (Santos et al. 2005; Wiedenmann et al. 2008; Young et al. 1997). Commonly used inhibitors are vanadate (Santos et al. 2005) and fluorescein 5-isothiocyanate (Young et al. 1997) for P-type ATPases or ouabain more specifically for the Na+/K+-pump (Bramkamp et al. 2004). Alternatively, Iwahashi and Nakamura (1989) titrated different amounts of antibodies against the enzyme’s substrate-binding site in reconstituted and solubilized state to inhibit the outwards-oriented enzymes. In these assays, one has to consider, that the use of detergent for permeabilization can alter protein activity, complicating the comparison of the activity before and after solubilization. Likewise, inhibitors are only affecting the protein activity to a specific degree, requiring titrations of detergent and inhibitor amounts (Santos et al. 2005).
In contrast, enzymes whose activity is not limited by substrate permeability such as the light-driven bacteriorhodopsin (Rigaud et al. 1988; Seigneuret and Rigaud 1985) or ion channels where ion permeability in both directions can be measured (Wang and Sigworth 2009) require adaptation of the analysis. Here, the protein orientation distributions can be derived from the addition of the inhibitor itself, comparing the residual activity to the non-treated sample, since no detergent treatment is needed. However, special care is required in case of bidirectional transport, since in and outward transport cannot be differentiated (Li et al. 2015). Furthermore, activity-based measurements can be applied and customized for most enzyme types, e.g., redox enzymes such as cytochrome C oxidase and site-specific binding of cytochrome (Öjemyr et al. 2012), potassium channels with site-specific pore blocking agents (Wang and Sigworth 2009) or removing of attached subunits by washing with low ionic strength buffer (Wiedenmann et al. 2008).
From bulk to single vesicle analysis
Classical analysis of reconstituted membrane protein orientation is based on ensemble-averaged biophysical or biochemical studies as described previously. A drawback of ensemble measurements arises from the compositional heterogeneity of proteoliposome reconstitutions which hampers quantitative analysis of vesicle properties and their correlation with the protein activity (Larsen et al. 2011; Veshaguri et al. 2016). Further technical issues with commonly used ensemble measurements include potential, heterogenous leakiness of proteoliposomes, and aggregation. Such heterogeneities can skew the results and lead to misinterpretation, especially if only a distinct subpopulation shows e.g., leakiness. The solution to this problem lies in single vesicle experiments where the lipid and protein content of individual vesicles within an ensemble can be precisely determined.
Electron microscopy has been used to visualize single lipid vesicles, as it allows imaging with nanometer resolution (Figure 5A). In combination with immuno-gold staining it is possible to detect membrane proteins in the vesicle’s membrane surface (Pal et al. 1987; Trépout et al. 2007). In particular, cryogenic electron microscopy (Cryo-EM) allows for the analysis of liposomes in their most native state. This technique can provide detailed information on vesicle morphology, but its ability to accurately determine vesicle sizes of all subpopulation fractions is limited by low throughput and artifacts that may occur during the preparation (Wang and Sigworth 2009; Yao et al. 2020).
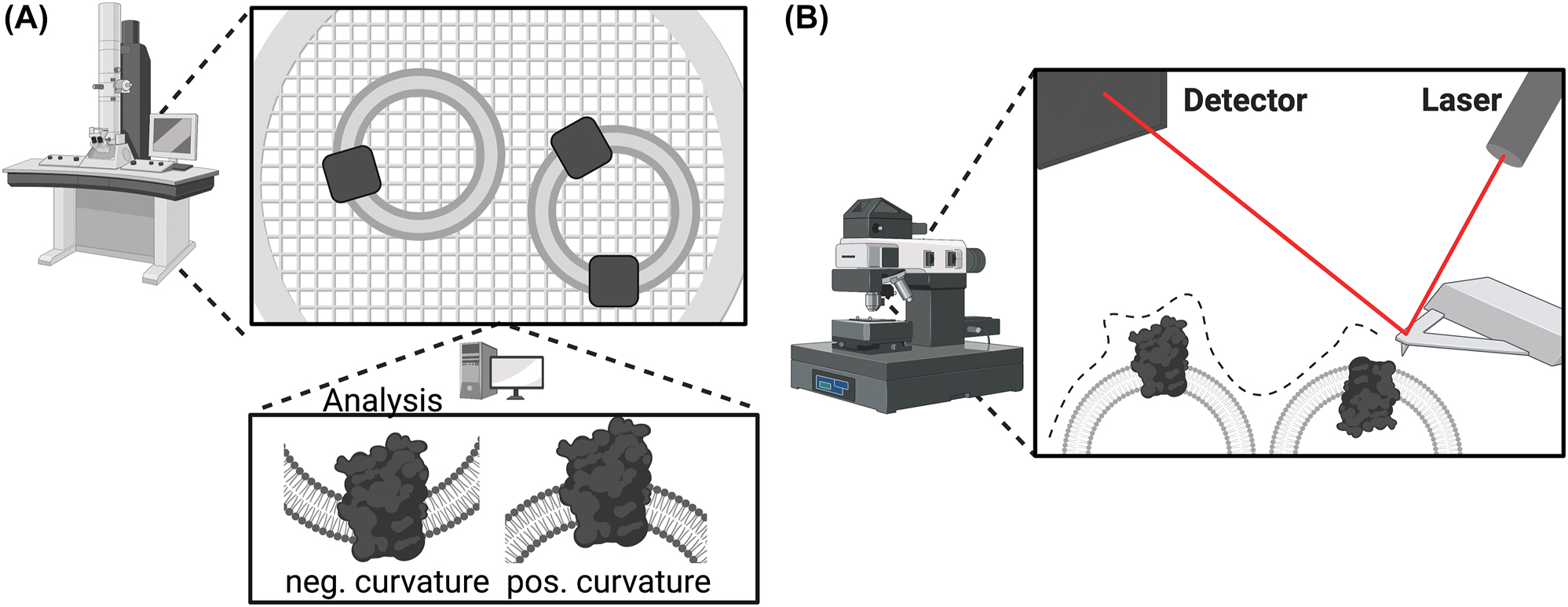
Electron and atomic force microscopy-based techniques. (A) Electron microscopy. Fixed samples of the vesicle preparation are imaged by electron microscopy, and computational analysis of the densities allows not only to resolve structural details but also analysis of the protein orientation, depending on the curvature of the membrane. (B) Atomic force microscopy. Vesicles are scanned with the tip of the atomic force microscope, allowing determination of the surface morphology. Protrusions of the membrane can be assigned to bulky protein domains oriented toward the outside of the vesicle. To allow accurate determination of the orientation, the protein of interest must contain extracellular and intracellular domains of different sizes.
Furthermore, atomic force microscopy (AFM) of giant vesicles with a size of around 10 µm was used to measure the protrusion of the enzyme from the membrane (Figure 5B). Due to a difference in enzyme height reaching out of the membrane in the outside-out compared to inside-out orientation, a conclusion about protein orientation is possible (Bhatia et al. 2016), revealing a suitable approach for AFM experts.
More recently, the presented bulk assays were transferred to the single-vesicle level utilizing fluorescence microscopy. This technique allows for monitoring individual vesicles as diffraction limited spots with diameters ranging from 50 µm down to 50 nm using fluorescent lipid probes and fluorescently labeled membrane proteins within. The first example is a protease-based assay that was established based on the imaging of single fluorescent proteins in vesicles prepared by blebbing plasma membranes (Richards et al. 2016). In this approach, the protein fluorescence intensity of each immobilized bleb or bleb bilayer is measured followed by site-specific cleavage between the protein and the fluorescent tag. Subsequently, the free tag is washed away, and the same bleb or bleb bilayer is imaged again. The comparison of fluorescence intensities before and after cleavage reveals a loss of fluorescence if the protein had an outside-oriented fluorescent tag. This approach might be applied to immobilized liposomes.
Furthermore, the fluorescence-bleaching approach was transferred to the single vesicle level, allowing a fluorescence intensity comparison of hundreds of individual vesicles before and after quencher addition (Veit et al. 2022). Here, a simultaneously quenchable lipid marker can be used as leakiness control (Figure 6), allowing a more reliable interpretation of the results compared to the bulk assay. Furthermore, with a combination of fluorescently tagged membrane proteins, lipids, and luminal markers, this approach permits parallel analysis of multiple parameters besides the protein orientation (vesicle size, tightness, unilamellarity, membrane protein numbers, activity) of individual proteoliposomes. Applying single vesicle experiments to proteins facilitating transport enables deducing the orientation from the derived activity trace itself without the use of inhibitors (Li et al. 2015), since the activity of a single protein is recorded directly. In general, single vesicle experiments allow exploiting the heterogeneity between individual vesicles to monitor thousands of individual vesicles with single membrane proteins simultaneously. Thereby, the main drawback of bulk analysis can be overcome by the transfer of protein orientation assays to the single vesicle level. However, bulk experiments like dynamic light scattering for vesicle size distribution or quantitative Coomassie staining for the determination of total protein amount can be used to complement single vesicle data.
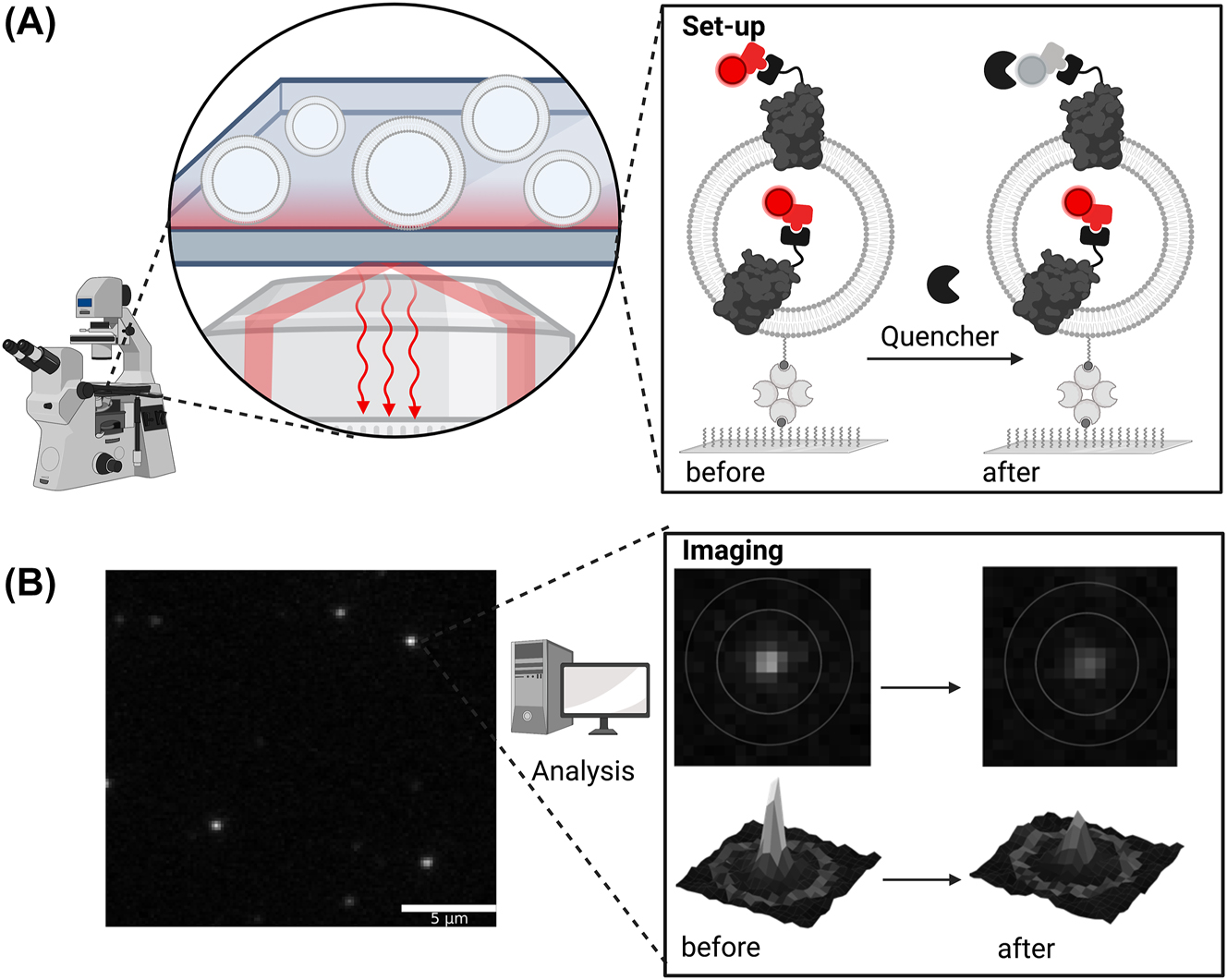
Fluorescence-bleaching approach on the single vesicle level. (A) Proteoliposomes are immobilized on a surface-inactivated dish via biotin-avidin interaction. Liposomes can be detected via lipid marker (not shown) and proteins via protein marker (red), respectively. The addition of dithionite reduces accessible fluorophores of the protein marker, which allows determination of orientation; (B) exemplary microscopy images of the protein marker as an overview (left) and zoom-in (before and after dithionite addition, right). Depending on the number of proteins with an attached fluorophore, the intensities of the single liposomes spots vary. Upon addition of dithionite, outward-facing dyes are quenched resulting in the loss of overall fluorescence when all protein copies are orientated outwards, partial loss if orientation is mixed, or no loss when the tagged side is facing the vesicle lumen.
While surface-based detection allows for sensitive analysis of vesicle populations, surface-immobilization has the drawback of often requiring optimized surface chemistries and several sample preparation steps, which is time-consuming and could damage the sample. Furthermore, single molecule microscopy is a technique that requires a suitable setup for high signal-to-noise ratios. Due to the low measured signal intensities of single molecules, data analysis requires customized, often home-written programs for signal extraction and advanced data analysis skills (Mathiasen et al. 2014; Thomsen et al. 2019; Veit et al. 2022).
Outlook
There is a wide range of techniques available for determining membrane protein orientation upon reconstitution into liposomes, limited by the proteins’ modifications (Table 2). Each analytical method presented here has particular advantages and technical limitations, and optimization is required for each system. In particular, the heterogeneity of proteoliposome preparations is a well-known phenomenon, which limits quantitative analysis by ensemble average measurements and prevents their correlation with protein activity (Larsen et al. 2011; Lohse et al. 2008; Mathiasen et al. 2014). Consequently, efforts have been taken to obtain more homogeneous vesicle preparations by tuning the reconstitution conditions and performing liposome flotation assays. Such alternative designs of workflows have not been considered in this review, such as removing non-incorporated protein (Althoff et al. 2012; Harris et al. 2017), enriching proteoliposomes of specific orientation (Madden and Cullis 1985) or aiming for oriented reconstitution (Dezi et al. 2013; Schadauer et al. 2015; Zheng et al. 2015). As mentioned briefly, a promising approach is to characterize intrasample compositional variations at the level of single vesicles (Christensen et al. 2012; Guha et al. 2021; Kuyper et al. 2006; Veit et al. 2022; Veshaguri et al. 2016). Combined with new technical advances that allow to generate liposome models with defined lipid composition of the inner and outer leaflets (Markones et al. 2020) and to analyze single vesicles without immobilization (Górecki et al. 2022; Mutch et al. 2011), this will open enormous possibilities for bioanalysis and biotechnological applications involving unprecedented miniaturization at the nanometer and attoliter range. These studies will boost our understanding of essential biological processes such as membrane transport, vesiculation and fusion, signaling, and cell-cell recognition. Future challenges remain concerning the development of new, commercially available fluorescent dyes and membrane probes.
Summary of important approaches for the determination of protein orientation upon liposomal reconstitution discussed in this review based on the protein of interest.
| Membrane protein of interest | Approaches | |||||||
|---|---|---|---|---|---|---|---|---|
| Protease-based | Site-specific labeling | Quenching | Activity-based | |||||
| Unspecific | Site-specific | Cysteine based | Antibody based | Self labelling | ||||
| Unmodified | X | Xf | ||||||
| Genetically modified | Specific cleavage site | X | ||||||
| Fluorescent tag | Xc | Xc,e | Xg | Xa,d | ||||
| Self-labeling tag | Xc | Xc,e | Xg | X | Xa,d | |||
| Site specific cysteine | X | Xa,d | ||||||
| Epitope tagb | Xc | Xc,e | X | |||||
| Enzyme activity | Non-permeable substrates/inhibitors | X | ||||||
-
aFor fluorescent proteins suitable for quenching, see text. bOften used for purification, such as FLAG- or Poly-His-tag. cApproach is commonly used for proteins, having additional fluorescent tags or antibody epitopes, thereby simplifying fragment analysis. dThe chromophore can be introduced in different ways, e.g., via fusion to fluorescent proteins, via fluorescent tagging of cysteines or self-labeling tags. eRequires proteases cleavage site between protein and fluorescent tag. fRequires antibody against protein of interest. gFor many fluorophores, antibodies are available commercially.
Funding source: Deutsche Forschungsgemeinschaft
Award Identifier / Grant number: GU 1133/11-1
Award Identifier / Grant number: INST 213/886-1 FUGG
Award Identifier / Grant number: INST 213/985-1 FUGG
About the authors

Sarina Veit is working on a PhD thesis at the Ruhr University Bochum, Germany which is funded by the Studienstiftung des deutschen Volkes. The topic of her PhD thesis is focused on the exploration of P4-ATPases in vivo as well as purified and reconstituted into model membrane systems. Her special interest lies in the transfer from bulk to single-vesicle experiments based on TIRF microscopy. During her PhD, she spent a research stay at the research group of Anant Menon at the Weill Cornell University in New York, USA and the group of Nikos Hatzakis at the Copenhagen University in Copenhagen, Denmark.

Laura Charlotte Paweletz studied biochemistry at the Ruhr University Bochum, Germany and spend an Erasmus term at Stockholm University, Stockholm, Sweden. She received her PhD in 2022 on the interplay of lipids and membrane transporters and was funded by the Studienstiftung des deutschen Volkes. During this time, she gained experiences in single vesicle microscopy visiting in the labs of Anant Menon at the Weill Cornell University in New York, USA and Nikos Hatzakis at the Copenhagen University in Copenhagen, Denmark. Currently, she holds a researcher and teaching position at the Ruhr University Bochum.

Thomas Günther Pomorski received his Ph.D. in Biophysics in 1996 from the Humboldt University of Berlin, Germany. After postdoctoral studies at the Max-Planck-Institute of Infectious Diseases in Berlin and the Academic Medical Center Amsterdam in the Netherlands, he became a Junior Professor for Cell Biophysics at the Humboldt University of Berlin. In 2009 he was appointed as Associate Professor and in 2016 as Professor at University of Copenhagen, Denmark. Since 2016 he has been Chair Professor in Molecular Biochemistry at the Ruhr University Bochum, Germany. His lab develops and applies molecular methods to study organization, function and regulation of membrane transporters.
Acknowledgments
We thank Richard Callaghan (University of Leeds, U.K) for valuable comments on the manuscript. L.P. and S.V. appreciate funding from Studienstiftung des Deutschen Volkes. T.G.P. acknowledges support from the DAAD (57386621) and the German Research Foundation (INST 213/886-1 FUGG, INST 213/985-1, FUGG; GU 1133/11-1). Figures were created in BioRender.com
-
Author contributions: S.V., L.C.P. and T.G.P. wrote the article. All authors read and approved the final manuscript.
-
Research funding: This work was financially supported by the Deutsche Forschungsgemeinschaft (GU 1133/11-1, INST 213/886-1 FUGG, INST 213/985-1 FUGG).
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
Althoff, T., Davies, K.M., Schulze, S., Joos, F., and Kühlbrandt, W. (2012). GRecon: a method for the lipid reconstitution of membrane proteins. Angew. Chem. Int. Ed. 51: 8343–8347, https://doi.org/10.1002/anie.201202094.Suche in Google Scholar PubMed PubMed Central
Amati, A.M., Graf, S., Deutschmann, S., Dolder, N., and von Ballmoos, C. (2020). Current problems and future avenues in proteoliposome research. Biochem. Soc. Trans. 48: 1473–1492, https://doi.org/10.1042/bst20190966.Suche in Google Scholar PubMed
Andra, K.K., Dorsey, S., Royer, C.A., and Menon, A.K. (2018). Structural mapping of fluorescently-tagged, functional nhTMEM16 scramblase in a lipid bilayer. J. Biol. Chem. 293: 12248–12258, https://doi.org/10.1074/jbc.ra118.003648.Suche in Google Scholar
Bayburt, T.H., Carlson, J.W., and Sligar, S.G. (1998). Reconstitution and imaging of a membrane protein in a nanometer-size phospholipid bilayer. J. Struct. Biol. 123: 37–44, https://doi.org/10.1006/jsbi.1998.4007.Suche in Google Scholar PubMed
Bayer, E.A., Safars, M., and Wilchek, M. (1987). Selective labeling of sulfhydryls and disulfides on blot transfers using avidin-biotin technology: studies on purified proteins and erythrocyte membranes. Anal. Biochem. 161: 262–271, https://doi.org/10.1016/0003-2697(87)90450-7.Suche in Google Scholar PubMed
Bhatia, T., Cornelius, F., Brewer, J., Bagatolli, L.A., Simonsen, A.C., Ipsen, J.H., and Mouritsen, O.G. (2016). Spatial distribution and activity of Na+/K+ -ATPase in lipid bilayer membranes with phase boundaries. Biochim. Biophys. Acta Biomembr. 1858: 1390–1399, https://doi.org/10.1016/j.bbamem.2016.03.015.Suche in Google Scholar PubMed
Biner, O., Fedor, J.G., Yin, Z., and Hirst, J. (2020). Bottom-up construction of a minimal system for cellular respiration and energy regeneration. ACS Synth. Biol. 9: 1450–1459, https://doi.org/10.1021/acssynbio.0c00110.Suche in Google Scholar PubMed PubMed Central
Bogdanov, M., Zhang, W., Xie, J., and Dowhan, W. (2005). Transmembrane protein topology mapping by the substituted cysteine accessibility method (SCAMTM): application to lipid-specific membrane protein topogenesis. Methods 36: 148–171, https://doi.org/10.1016/j.ymeth.2004.11.002.Suche in Google Scholar PubMed PubMed Central
Bramkamp, M., Gassel, M., and Altendorf, K. (2004). FITC binding site and p-nitrophenyl phosphatase activity of the kdp-ATPase of Escherichia coli. Biochemistry 43: 4559–4567, https://doi.org/10.1021/bi030198a.Suche in Google Scholar PubMed
Chmyrov, A., Sandén, T., and Widengren, J. (2010). Iodide as a fluorescence quencher and promoter—mechanisms and possible implications. J. Phys. Chem. B 114: 11282–11291, https://doi.org/10.1021/jp103837f.Suche in Google Scholar PubMed
Christensen, S.M., Bolinger, P.-Y., Hatzakis, N.S., Mortensen, M.W., and Stamou, D. (2012). Mixing subattolitre volumes in a quantitative and highly parallel manner with soft matter nanofluidics. Nat. Nanotechnol. 7: 51–55, https://doi.org/10.1038/nnano.2011.185.Suche in Google Scholar PubMed
Cole, N.B. (2013). Site-specific protein labeling with SNAP-tags. Curr. Protoc. Protein Sci. 73: 30.1.1–30.1.16, https://doi.org/10.1002/0471140864.ps3001s73.Suche in Google Scholar PubMed PubMed Central
Corradi, V., Sejdiu, B.I., Mesa-Galloso, H., Abdizadeh, H., Noskov, S.Y., Marrink, S.J., and Tieleman, D.P. (2019). Emerging diversity in lipid–protein interactions. Chem. Rev. 119: 5775–5848, https://doi.org/10.1021/acs.chemrev.8b00451.Suche in Google Scholar PubMed PubMed Central
Cuello, L.G., Romero, J.G., Cortes, D.M., and Perozo, E. (1998). pH-dependent gating in the Streptomyces lividans K+ channel. Biochemistry 37: 3229–3236, https://doi.org/10.1021/bi972997x.Suche in Google Scholar PubMed
Cvjetkovic, A., Jang, S.C., Konečná, B., Höög, J.L., Sihlbom, C., Lässer, C., and Lötvall, J. (2016). Detailed analysis of protein topology of extracellular vesicles–evidence of unconventional membrane protein orientation. Sci. Rep. 6: 36338, https://doi.org/10.1038/srep36338.Suche in Google Scholar PubMed PubMed Central
Deutschmann, S., Rimle, L., and von Ballmoos, C. (2022). Rapid estimation of membrane protein orientation in liposomes. Chembiochem 23: e202100543, https://doi.org/10.1002/cbic.202100543.Suche in Google Scholar PubMed PubMed Central
Dezi, M., Di Cicco, A., Bassereau, P., and Lévy, D. (2013). Detergent-mediated incorporation of transmembrane proteins in giant unilamellar vesicles with controlled physiological contents. Proc. Natl. Acad. Sci. U. S. A. 110: 7276–7281, https://doi.org/10.1073/pnas.1303857110.Suche in Google Scholar PubMed PubMed Central
Drin, G., Cottin, S., Blanc, E., Rees, A.R., and Temsamani, J. (2003). Studies on the internalization mechanism of cationic cell-penetrating peptides. J. Biol. Chem. 278: 31192–31201, https://doi.org/10.1074/jbc.m303938200.Suche in Google Scholar PubMed
Dröse, S., Galkin, A., and Brandt, U. (2005). Proton pumping by complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica reconstituted into proteoliposomes. Biochim. Biophys. Acta, Bioenerg. 1710: 87–95, https://doi.org/10.1016/j.bbabio.2005.10.001.Suche in Google Scholar PubMed
Eisinger, M.L., Nie, L., Dörrbaum, A.R., Langer, J.D., and Michel, H. (2018). The xenobiotic extrusion mechanism of the MATE transporter NorM_PS from Pseudomonas stutzeri. J. Mol. Biol. 430: 1311–1323, https://doi.org/10.1016/j.jmb.2018.03.012.Suche in Google Scholar PubMed
Engelhard, V.H., Guild, B.C., Helenius, A., Terhorst, C., and Strominger, J.L. (1978). Reconstitution of purified detergent-soluble HLA-A and HLA-B antigens into phospholipid vesicles. Proc. Natl. Acad. Sci. U. S. A. 75: 3230–3234, https://doi.org/10.1073/pnas.75.7.3230.Suche in Google Scholar PubMed PubMed Central
England, C.G., Luo, H., and Cai, W. (2015). HaloTag technology: a versatile platform for biomedical applications. Bioconjugate Chem. 26: 975–986, https://doi.org/10.1021/acs.bioconjchem.5b00191.Suche in Google Scholar PubMed PubMed Central
Fang, G., Friesen, R., Lanfermeijer, F., Hagting, A., Poolman, B., and Konings, W.N. (1999). Manipulation of activity and orientation of membrane-reconstituted di-tripeptide transport protein DtpT of Lactococcus lactis. Mol. Membr. Biol. 16: 297–304, https://doi.org/10.1080/096876899294517.Suche in Google Scholar PubMed
Fimmel, S., Choli, T., Dencher, N.A., Büldt, G., and Wittmann-Liebold, B. (1989). Topography of surface-exposed amino acids in the membrane protein bacteriorhodopsin determined by proteolysis and micro-sequencing. Biochim. Biophys. Acta Biomembr. 978: 231–240, https://doi.org/10.1016/0005-2736(89)90120-x.Suche in Google Scholar PubMed
Galbán, J., Mateos, E., Cebolla, V., Domínguez, A., Delgado-Camón, A., de Marcos, S., Sanz-Vicente, I., and Sanz, V. (2009). The environmental effect on the fluorescence intensity in solution. An analytical model. Analyst 134: 2286–2292, https://doi.org/10.1039/b912063g.Suche in Google Scholar PubMed
Gautier, A., Juillerat, A., Heinis, C., Corrêa, I.R., Kindermann, M., Beaufils, F., and Johnsson, K. (2008). An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 15: 128–136, https://doi.org/10.1016/j.chembiol.2008.01.007.Suche in Google Scholar PubMed
Gennarini, G., Hirn, M., Deagostini-Bazin, H., and Goridis, C. (1984). Studies on the transmembrane disposition of the neural cell adhesion molecule N-CAM. The use of liposome-inserted radioiodinated N-CAM to study its transbilayer orientation. Eur. J. Biochem. 142: 65–73, https://doi.org/10.1111/j.1432-1033.1984.tb08251.x.Suche in Google Scholar PubMed
Gerber, G.E., Gray, C.P., Wildenauer, D., and Khorana, H.G. (1977). Orientation of bacteriorhodopsin in Halobacterium halobium as studied by selective proteolysis. Proc. Natl. Acad. Sci. U. S. A. 74: 5426–5430, https://doi.org/10.1073/pnas.74.12.5426.Suche in Google Scholar PubMed PubMed Central
Górecki, K., Hansen, J.S., Li, P., Nayeri, N., Lindkvist-Petersson, K., and Gourdon, P. (2022). Microfluidic-derived detection of protein-facilitated copper flux across lipid membranes. Anal. Chem. 94: 11831–11837, https://doi.org/10.1021/acs.analchem.2c02081.Suche in Google Scholar PubMed PubMed Central
Guha, A., McGuire, M.L., Leriche, G., Yang, J., and Mayer, M. (2021). A single-liposome assay that enables temperature-dependent measurement of proton permeability of extremophile-inspired lipid membranes. Biochim. Biophys. Acta Biomembr. 1863: 183567, https://doi.org/10.1016/j.bbamem.2021.183567.Suche in Google Scholar PubMed
Harris, N.J., Reading, E., Ataka, K., Grzegorzewski, L., Charalambous, K., Liu, X., Schlesinger, R., Heberle, J., and Booth, P.J. (2017). Structure formation during translocon-unassisted co-translational membrane protein folding. Sci. Rep. 7: 8021, https://doi.org/10.1038/s41598-017-08522-9.Suche in Google Scholar PubMed PubMed Central
Holmgren, M., Liu, Y., Xu, Y., and Yellen, G. (1996). On the use of thiol-modifying agents to determine channel topology. Neuropharmacology 35: 797–804, https://doi.org/10.1016/0028-3908(96)00129-3.Suche in Google Scholar PubMed
Hu, L.R., Ho, R.J., and Huang, L. (1986). Trypsin induced destabilization of liposomes composed of dioleoylphosphatidylethanolamine and glycophorin. Biochem. Biophys. Res. Commun. 141: 973–978, https://doi.org/10.1016/s0006-291x(86)80139-5.Suche in Google Scholar PubMed
Isarankura-Na-Ayudhya, C., Tantimongcolwat, T., Galla, H.-J., and Prachayasittikul, V. (2010). Fluorescent protein-based optical biosensor for copper ion quantitation. Biol. Trace Elem. Res. 134: 352–363, https://doi.org/10.1007/s12011-009-8476-9.Suche in Google Scholar PubMed
Ishijima, S., Shigemi, Z., Adachi, H., Makinouchi, N., and Sagami, I. (2012). Functional reconstitution and characterization of the Arabidopsis Mg2+ transporter AtMRS2-10 in proteoliposomes. Biochim. Biophys. Acta Biomembr. 1818: 2202–2208, https://doi.org/10.1016/j.bbamem.2012.04.015.Suche in Google Scholar PubMed
Islam, S.T., Eckford, P.D.W., Jones, M.L., Nugent, T., Bear, C.E., Vogel, C., and Lam, J.S. (2013). Proton-dependent gating and proton uptake by Wzx support O-antigen-subunit antiport across the bacterial inner membrane. mBio 4: e00678-13, https://doi.org/10.1128/mbio.00678-13.Suche in Google Scholar PubMed PubMed Central
Iwahashi, Y. and Nakamura, T. (1989). Orientation and reactivity of NADH kinase in proteoliposomes. J. Biochem. 105: 922–926, https://doi.org/10.1093/oxfordjournals.jbchem.a122780.Suche in Google Scholar PubMed
Jarvela, T.S., Chaplot, K., and Lindberg, I. (2021). A protease protection assay for the detection of internalized alpha-synuclein pre-formed fibrils. PLoS One 16: e0241161, https://doi.org/10.1371/journal.pone.0241161.Suche in Google Scholar PubMed PubMed Central
Jeter, V.L. and Escalante-Semerena, J.C. (2021). Insights into the relationship between cobamide synthase and the cell membrane. mBio 12: e00215–e00221, https://doi.org/10.1128/mbio.00215-21.Suche in Google Scholar
Kalmbach, R., Chizhov, I., Schumacher, M.C., Friedrich, T., Bamberg, E., and Engelhard, M. (2007). Functional cell-free synthesis of a seven helix membrane protein: in situ insertion of bacteriorhodopsin into liposomes. J. Mol. Biol. 371: 639–648, https://doi.org/10.1016/j.jmb.2007.05.087.Suche in Google Scholar PubMed
Kapust, R.B., Tözsér, J., Fox, J.D., Anderson, D.E., Cherry, S., Copeland, T.D., and Waugh, D.S. (2001). Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. Des. Sel. 14: 993–1000, https://doi.org/10.1093/protein/14.12.993.Suche in Google Scholar PubMed
Karpowicz, R.J., Haney, C.M., Mihaila, T.S., Sandler, R.M., Petersson, E.J., and Lee, V.M.-Y. (2017). Selective imaging of internalized proteopathic α-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J. Biol. Chem. 292: 13482–13497, https://doi.org/10.1074/jbc.m117.780296.Suche in Google Scholar
Kemmer, G.C., Bogh, S.A., Urban, M., Palmgren, M.G., Vosch, T., Schiller, J., and Günther Pomorski, T. (2015). Lipid-conjugated fluorescent pH sensors for monitoring pH changes in reconstituted membrane systems. Analyst 140: 6313–6320, https://doi.org/10.1039/c5an01180a.Suche in Google Scholar PubMed
Keppler, A., Gendreizig, S., Gronemeyer, T., Pick, H., Vogel, H., and Johnsson, K. (2003). A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21: 86–89, https://doi.org/10.1038/nbt765.Suche in Google Scholar PubMed
Knol, J., Sjollema, K., and Poolman, B. (1998). Detergent-mediated reconstitution of membrane proteins. Biochemistry 37: 16410–16415, https://doi.org/10.1021/bi981596u.Suche in Google Scholar PubMed
Knol, J., Veenhoff, L., Liang, W.-J., Henderson, P.J.F., Leblanc, G., and Poolman, B. (1996). Unidirectional reconstitution into detergent-destabilized liposomes of the purified lactose transport system of Streptococcus thermophilus. J. Biol. Chem. 271: 15358–15366, https://doi.org/10.1074/jbc.271.26.15358.Suche in Google Scholar PubMed
Kostallas, G., Löfdahl, P.-Å., and Samuelson, P. (2011). Substrate profiling of tobacco etch virus protease using a novel fluorescence-assisted whole-cell assay. PLoS One 6: e16136, https://doi.org/10.1371/journal.pone.0016136.Suche in Google Scholar PubMed PubMed Central
Kraus, E., Kiltz, H.H., and Femfert, U.F. (1976). The specificity of proteinase K against oxidized insulin B chain. Hoppe-Seyler’s Z. für Physiol. Chem. 357: 233–237.10.1515/bchm2.1976.357.2.937Suche in Google Scholar PubMed
Kuhn, A., Haase, M., and Leptihn, S. (2017). Assisted and unassisted protein insertion into liposomes. Biophys. J. 113: 1187–1193, https://doi.org/10.1016/j.bpj.2017.03.027.Suche in Google Scholar PubMed PubMed Central
Kuyper, C.L., Kuo, J.S., Mutch, S.A., and Chiu, D.T. (2006). Proton permeation into single vesicles occurs via a sequential two-step mechanism and is heterogeneous. J. Am. Chem. Soc. 128: 3233–3240, https://doi.org/10.1021/ja057349c.Suche in Google Scholar PubMed
Langner, M. and Hui, S.W. (1993). Dithionite penetration through phospholipid bilayers as a measure of defects in lipid molecular packing. Chem. Phys. Lipids 65: 23–30, https://doi.org/10.1016/0009-3084(93)90078-h.Suche in Google Scholar PubMed
Larsen, J., Hatzakis, N.S., and Stamou, D. (2011). Observation of inhomogeneity in the lipid composition of individual nanoscale liposomes. J. Am. Chem. Soc. 133: 10685–10687, https://doi.org/10.1021/ja203984j.Suche in Google Scholar PubMed
Lasitza-Male, T., Bartels, K., Jungwirth, J., Wiggers, F., Rosenblum, G., Hofmann, H., and Löw, C. (2020). Membrane chemistry tunes the structure of a peptide transporter. Angew. Chem. Int. Ed. 59: 19121–19128, https://doi.org/10.1002/anie.202008226.Suche in Google Scholar PubMed PubMed Central
Lee, K.J., Kang, D., and Park, H.-S. (2019). Site-specific labeling of proteins using unnatural amino acids. Mol Cells 42: 386–396, https://doi.org/10.14348/molcells.2019.0078.Suche in Google Scholar PubMed PubMed Central
Leisle, L., Valiyaveetil, F., Mehl, R.A., and Ahern, C.A. (2015). Incorporation of non-canonical amino acids. Adv. Exp. Med. Biol. 869: 119–151, https://doi.org/10.1007/978-1-4939-2845-3_7.Suche in Google Scholar PubMed PubMed Central
Lévy, D., Gulik, A., Bluzat, A., and Rigaud, J.-L. (1992). Reconstitution of the sarcoplasmic reticulum Ca2+-ATPase: mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. Biochim. Biophys. Acta Biomembr. 1107: 283–298, https://doi.org/10.1016/0005-2736(92)90415-i.Suche in Google Scholar PubMed
Li, M., Jørgensen, S.K., McMillan, D.G.G., Krzemiński, Ł., Daskalakis, N.N., Partanen, R.H., Tutkus, M., Tuma, R., Stamou, D., Hatzakis, N.S., et al.. (2015). Single enzyme experiments reveal a long-lifetime proton leak state in a heme-copper oxidase. J. Am. Chem. Soc. 137: 16055–16063, https://doi.org/10.1021/jacs.5b08798.Suche in Google Scholar PubMed PubMed Central
Lohse, B., Bolinger, P.-Y., and Stamou, D. (2008). Encapsulation efficiency measured on single small unilamellar vesicles. J. Am. Chem. Soc. 130: 14372–14373, https://doi.org/10.1021/ja805030w.Suche in Google Scholar PubMed
Loo, T.W. and Clarke, D.M. (1995). Membrane topology of a cysteine-less mutant of human P-glycoprotein. J. Biol. Chem. 270: 843–848, https://doi.org/10.1074/jbc.270.2.843.Suche in Google Scholar PubMed
Lorenz, H., Hailey, D.W., Wunder, C., and Lippincott-Schwartz, J. (2006). The fluorescence protease protection (FPP) assay to determine protein localization and membrane topology. Nat. Protoc. 1: 276–279, https://doi.org/10.1038/nprot.2006.42.Suche in Google Scholar PubMed
Madden, T.D. and Cullis, P.R. (1985). Preparation of reconstituted cytochrome oxidase vesicles with defined trans-membrane protein orientations employing a cytochrome c affinity column. Biochim. Biophys. Acta, Bioenerg. 808: 219–224, https://doi.org/10.1016/0005-2728(85)90002-7.Suche in Google Scholar PubMed
Marek, M., Milles, S., Schreiber, G., Daleke, D.L., Dittmar, G., Herrmann, A., Müller, P., and Pomorski, T.G. (2011). The yeast plasma membrane ATP binding cassette (ABC) transporter Aus1: purification, characterization, and the effect of lipids on its activity. J. Biol. Chem. 286: 21835–21843, https://doi.org/10.1074/jbc.m111.244525.Suche in Google Scholar
Marinko, J.T., Huang, H., Penn, W.D., Capra, J.A., Schlebach, J.P., and Sanders, C.R. (2019). Folding and misfolding of human membrane proteins in health and disease: from single molecules to cellular proteostasis. Chem. Rev. 119: 5537–5606, https://doi.org/10.1021/acs.chemrev.8b00532.Suche in Google Scholar PubMed PubMed Central
Markones, M., Fippel, A., Kaiser, M., Drechsler, C., Hunte, C., and Heerklotz, H. (2020). Stairway to asymmetry: five steps to lipid-asymmetric proteoliposomes. Biophys. J. 118: 294–302, https://doi.org/10.1016/j.bpj.2019.10.043.Suche in Google Scholar PubMed PubMed Central
Martín, M., Albanesi, D., Alzari, P.M., and de Mendoza, D. (2009). Functional in vitro assembly of the integral membrane bacterial thermosensor DesK. Protein Expr. Purif. 66: 39–45, https://doi.org/10.1016/j.pep.2009.02.006.Suche in Google Scholar PubMed
Mathiasen, S., Christensen, S.M., Fung, J.J., Rasmussen, S.G.F., Fay, J.F., Jorgensen, S.K., Veshaguri, S., Farrens, D.L., Kiskowski, M., Kobilka, B., et al.. (2014). Nanoscale high-content analysis using compositional heterogeneities of single proteoliposomes. Nat. Methods 11: 931–934, https://doi.org/10.1038/nmeth.3062.Suche in Google Scholar PubMed PubMed Central
McIntyre, J.C. and Sleight, R.G. (1991). Fluorescence assay for phospholipid membrane asymmetry. Biochemistry 30: 11819–11827, https://doi.org/10.1021/bi00115a012.Suche in Google Scholar PubMed
Moreno, M.J., Estronca, L.M.B.B., and Vaz, W.L.C. (2006). Translocation of phospholipids and dithionite permeability in liquid-ordered and liquid-disordered membranes. Biophys. J. 91: 873–881, https://doi.org/10.1529/biophysj.106.082115.Suche in Google Scholar PubMed PubMed Central
Murray, D.T., Griffin, J., and Cross, T.A. (2014). Detergent optimized membrane protein reconstitution in liposomes for solid state NMR. Biochemistry 53: 2454–2463, https://doi.org/10.1021/bi500144h.Suche in Google Scholar PubMed PubMed Central
Mutch, S.A., Kensel-Hammes, P., Gadd, J.C., Fujimoto, B.S., Allen, R.W., Schiro, P.G., Lorenz, R.M., Kuyper, C.L., Kuo, J.S., Bajjalieh, S.M., et al.. (2011). Protein quantification at the single vesicle level reveals that a subset of synaptic vesicle proteins are trafficked with high precision. J. Neurosci. 31: 1461–1470, https://doi.org/10.1523/jneurosci.3805-10.2011.Suche in Google Scholar
Natarajan, P., Liu, K., Patil, D.V., Sciorra, V.A., Jackson, C.L., and Graham, T.R. (2009). Regulation of a Golgi flippase by phosphoinositides and an ArfGEF. Nat. Cell Biol. 11: 1421–1426, https://doi.org/10.1038/ncb1989.Suche in Google Scholar PubMed PubMed Central
Ohnishi, S. and Kamiya, K. (2021). Formation of giant lipid vesicle containing dual functions facilitates outer membrane phospholipase. ACS Synth. Biol. 10: 1837–1846, https://doi.org/10.1021/acssynbio.0c00468.Suche in Google Scholar PubMed
Öjemyr, L.N., von Ballmoos, C., Faxén, K., Svahn, E., and Brzezinski, P. (2012). The membrane modulates internal proton transfer in cytochrome c oxidase. Biochemistry 51: 1092–1100, https://doi.org/10.1021/bi201795c.Suche in Google Scholar PubMed
Olsen, J.V., Ong, S.-E., and Mann, M. (2004). Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3: 608–614, https://doi.org/10.1074/mcp.t400003-mcp200.Suche in Google Scholar
Pal, R., Barenholz, Y., and Wagner, R.R. (1987). Vesicular stomatitis virus membrane proteins and their interactions with lipid hilayers. Biochim. Biophys. Acta Biomembr. 19: 175–193, https://doi.org/10.1016/0304-4157(87)90011-6.Suche in Google Scholar PubMed
Pan, Y., Li, H., Li, Q., Lenertz, M., Zhu, X., Chen, B., and Yang, Z. (2021). Site-directed spin labeling-electron paramagnetic resonance spectroscopy in biocatalysis: enzyme orientation and dynamics in nanoscale confinement. Chem Catalysis 1: 207–231, https://doi.org/10.1016/j.checat.2021.03.005.Suche in Google Scholar
Parks, T.D., Leuther, K.K., Howard, E.D., Johnston, S.A., and Dougherty, W.G. (1994). Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 216: 413–417, https://doi.org/10.1006/abio.1994.1060.Suche in Google Scholar PubMed
Paweletz, L.C., Veit, S., and Pomorski, T.G. (2022). A fluorescence-based approach utilizing self-labeling enzyme tags to determine protein orientation in large unilamellar vesicles. Bio-protocol 12: e4542, https://doi.org/10.21769/bioprotoc.4542.Suche in Google Scholar
Periasamy, A., Shadiac, N., Amalraj, A., Garajová, S., Nagarajan, Y., Waters, S., Mertens, H.D.T., and Hrmova, M. (2013). Cell-free protein synthesis of membrane (1,3)-β-d-glucan (curdlan) synthase: co-translational insertion in liposomes and reconstitution in nanodiscs. Biochim. Biophys. Acta Biomembr. 1828: 743–757, https://doi.org/10.1016/j.bbamem.2012.10.003.Suche in Google Scholar PubMed
Poelarends, G.J. and Konings, W.N. (2002). The transmembrane domains of the ABC multidrug transporter LmrA form a cytoplasmic exposed, aqueous chamber within the membrane. J. Biol. Chem. 277: 42891–42898, https://doi.org/10.1074/jbc.m206508200.Suche in Google Scholar PubMed
Polgár, L. (2005). The catalytic triad of serine peptidases. Cell. Mol. Life Sci. 62: 2161–2172, https://doi.org/10.1007/s00018-005-5160-x.Suche in Google Scholar PubMed
Reshetnyak, Y.K., Andreev, O.A., Lehnert, U., and Engelman, D.M. (2006). Translocation of molecules into cells by pH-dependent insertion of a transmembrane helix. Proc. Natl. Acad. Sci. U. S. A. 103: 6460–6465, https://doi.org/10.1073/pnas.0601463103.Suche in Google Scholar PubMed PubMed Central
Richards, M.J., Hsia, C.-Y., Singh, R.R., Haider, H., Kumpf, J., Kawate, T., and Daniel, S. (2016). Membrane protein mobility and orientation preserved in supported bilayers created directly from cell plasma membrane blebs. Langmuir 32: 2963–2974, https://doi.org/10.1021/acs.langmuir.5b03415.Suche in Google Scholar PubMed
Rigaud, J.-L. and Lévy, D. (2003). Reconstitution of membrane proteins into liposomes. Methods Enzymol. 372: 65–86, https://doi.org/10.1016/S0076-6879(03)72004-7.Suche in Google Scholar PubMed
Rigaud, J.L., Paternostre, M.T., and Bluzat, A. (1988). Mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. 2. Incorporation of the light-driven proton pump bacteriorhodopsin. Biochemistry 27: 2677–2688, https://doi.org/10.1021/bi00408a007.Suche in Google Scholar PubMed
Rigaud, J.-L., Pitard, B., and Levy, D. (1995). Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim. Biophys. Acta, Bioenerg. 1231: 223–246, https://doi.org/10.1016/0005-2728(95)00091-v.Suche in Google Scholar PubMed
Ritzmann, N., Thoma, J., Hirschi, S., Kalbermatter, D., Fotiadis, D., and Müller, D.J. (2017). Fusion domains guide the oriented insertion of light-driven proton pumps into liposomes. Biophys. J. 113: 1181–1186, https://doi.org/10.1016/j.bpj.2017.06.022.Suche in Google Scholar PubMed PubMed Central
Rodriguez, J., Gupta, N., Smith, R.D., and Pevzner, P.A. (2008). Does trypsin cut before proline? J. Proteome Res. 7: 300–305, https://doi.org/10.1021/pr0705035.Suche in Google Scholar PubMed
Santos, H.de L., Lopes, M.L., Maggio, B., and Ciancaglini, P. (2005). Na,K-ATPase reconstituted in liposomes: effects of lipid composition on hydrolytic activity and enzyme orientation. Colloids Surf. B Biointerfaces 41: 239–248, https://doi.org/10.1016/j.colsurfb.2004.12.013.Suche in Google Scholar PubMed
Schadauer, F., Geiss, A.F., Srajer, J., Siebenhofer, B., Frank, P., Reiner-Rozman, C., Ludwig, B., Richter, O.-M.H., Nowak, C., and Naumann, R.L.C. (2015). Silica nanoparticles for the oriented encapsulation of membrane proteins into artificial bilayer lipid membranes. Langmuir 31: 2511–2516, https://doi.org/10.1021/la504417j.Suche in Google Scholar PubMed
Schuette, C.G., Hatsuzawa, K., Margittai, M., Stein, A., Riedel, D., Kuster, P., Konig, M., Seidel, C., and Jahn, R. (2004). Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc. Natl. Acad. Sci. U. S. A. 101: 2858–2863, https://doi.org/10.1073/pnas.0400044101.Suche in Google Scholar PubMed PubMed Central
Schwamborn, M., Schumacher, J., Sibold, J., Teiwes, N.K., and Steinem, C. (2017). Monitoring ATPase induced pH changes in single proteoliposomes with the lipid-coupled fluorophore Oregon Green 488. Analyst 142: 2670–2677, https://doi.org/10.1039/c7an00215g.Suche in Google Scholar PubMed
Schwarz, P. and Steinem, C. (2022). The role of the transmembrane domain of silicanin-1: reconstitution of the full-length protein in artificial membranes. Biochim. Biophys. Acta Biomembr. 1864: 183921, https://doi.org/10.1016/j.bbamem.2022.183921.Suche in Google Scholar PubMed
Seigneuret, M. and Rigaud, J.-L. (1985). Use of the fluorescent pH probe pyranine to detect heterogeneous directions of proton movement in bacteriorhodopsin reconstituted large liposomes. FEBS Lett. 188: 101–106, https://doi.org/10.1016/0014-5793(85)80883-8.Suche in Google Scholar
Serek, J., Bauer-Manz, G., Struhalla, G., van den Berg, L., Kiefer, D., Dalbey, R., and Kuhn, A. (2004). Escherichia coli YidC is a membrane insertase for Sec-independent proteins. EMBO J. 23: 294–301, https://doi.org/10.1038/sj.emboj.7600063.Suche in Google Scholar PubMed PubMed Central
Skrzypek, R., Iqbal, S., and Callaghan, R. (2018). Methods of reconstitution to investigate membrane protein function. Methods 147: 126–141, https://doi.org/10.1016/j.ymeth.2018.02.012.Suche in Google Scholar PubMed
Stefan, E., Hofmann, S., and Tampé, R. (2020). A single power stroke by ATP binding drives substrate translocation in a heterodimeric ABC transporter. Elife 9: e55943, https://doi.org/10.7554/elife.55943.Suche in Google Scholar
Surrey, T. and Jahnig, F. (1992). Refolding and oriented insertion of a membrane protein into a lipid bilayer. Proc. Natl. Acad. Sci. U. S. A. 89: 7457–7461, https://doi.org/10.1073/pnas.89.16.7457.Suche in Google Scholar PubMed PubMed Central
Taylor, N.M.I., Manolaridis, I., Jackson, S.M., Kowal, J., Stahlberg, H., and Locher, K.P. (2017). Structure of the human multidrug transporter ABCG2. Nature 546: 504–509, https://doi.org/10.1038/nature22345.Suche in Google Scholar PubMed
Thévenin, D., An, M., and Engelman, D.M. (2009). pHLIP-mediated translocation of membrane-impermeable molecules into cells. Chem. Biol. 16: 754–762, https://doi.org/10.1016/j.chembiol.2009.06.006.Suche in Google Scholar PubMed PubMed Central
Thomsen, R.P., Malle, M.G., Okholm, A.H., Krishnan, S., Bohr, S.S.-R., Sørensen, R.S., Ries, O., Vogel, S., Simmel, F.C., Hatzakis, N.S., et al.. (2019). A large size-selective DNA nanopore with sensing applications. Nat. Commun. 10: 5655, https://doi.org/10.1038/s41467-019-13284-1.Suche in Google Scholar PubMed PubMed Central
Tirat, A., Freuler, F., Stettler, T., Mayr, L.M., and Leder, L. (2006). Evaluation of two novel tag-based labelling technologies for site-specific modification of proteins. Int. J. Biol. Macromol. 39: 66–76, https://doi.org/10.1016/j.ijbiomac.2006.01.012.Suche in Google Scholar PubMed
Trépout, S., Mornet, S., Benabdelhak, H., Ducruix, A., Brisson, A.R., and Lambert, O. (2007). Membrane protein selectively oriented on solid support and reconstituted into a lipid membrane. Langmuir 23: 2647–2654, https://doi.org/10.1021/la062227z.Suche in Google Scholar PubMed
Tunuguntla, R., Bangar, M., Kim, K., Stroeve, P., Ajo-Franklin, C.M., and Noy, A. (2013). Lipid bilayer composition can influence the orientation of proteorhodopsin in artificial membranes. Biophys. J. 105: 1388–1396, https://doi.org/10.1016/j.bpj.2013.07.043.Suche in Google Scholar PubMed PubMed Central
Ueki, M. and Iwamoto, M. (2021). Fluorescent labeling in size‐controlled liposomes reveals membrane curvature‐induced structural changes in the KcsA potassium channel. FEBS Lett. 595: 1914–1919, https://doi.org/10.1002/1873-3468.14141.Suche in Google Scholar PubMed
Vecino, A.J., Segura, R.L., Ugarte-Uribe, B., Águila, S., Hormaeche, I., de la Cruz, F., Goñi, F.M., and Alkorta, I. (2010). Reconstitution in liposome bilayers enhances nucleotide binding affinity and ATP-specificity of TrwB conjugative coupling protein. Biochim. Biophys. Acta Biomembr. 1798: 2160–2169, https://doi.org/10.1016/j.bbamem.2010.07.005.Suche in Google Scholar PubMed
Veit, S., Paweletz, L.C., Bohr, S.S.-R., Menon, A.K., Hatzakis, N.S., and Pomorski, T.G. (2022). Single vesicle fluorescence-bleaching assay for multi-parameter analysis of proteoliposomes by total internal reflection fluorescence microscopy. ACS Appl. Mater. Interfaces 14: 29659–29667, https://doi.org/10.1021/acsami.2c07454.Suche in Google Scholar PubMed
Veshaguri, S., Christensen, S.M., Kemmer, G.C., Ghale, G., Møller, M.P., Lohr, C., Christensen, A.L., Justesen, B.H., Jørgensen, I.L., Schiller, J., et al.. (2016). Direct observation of proton pumping by a eukaryotic P-type ATPase. Science 351: 1469–1473, https://doi.org/10.1126/science.aad6429.Suche in Google Scholar PubMed PubMed Central
Wang, L., Quan, C., Liu, B., Wang, J., Xiong, W., Zhao, P., and Fan, S. (2013). Functional reconstitution of Staphylococcus aureus truncated AgrC histidine kinase in a model membrane system. PLoS One 8: e80400, https://doi.org/10.1371/journal.pone.0080400.Suche in Google Scholar PubMed PubMed Central
Wang, L. and Sigworth, F.J. (2009). Structure of the BK potassium channel in a lipid membrane from electron cryomicroscopy. Nature 461: 292–295, https://doi.org/10.1038/nature08291.Suche in Google Scholar PubMed PubMed Central
Wiedenmann, A., Dimroth, P., and von Ballmoos, C. (2008). Δψ and ΔpH are equivalent driving forces for proton transport through isolated F0 complexes of ATP synthases. Biochim. Biophys. Acta, Bioenerg. 1777: 1301–1310, https://doi.org/10.1016/j.bbabio.2008.06.008.Suche in Google Scholar PubMed
Yao, X., Fan, X., and Yan, N. (2020). Cryo-EM analysis of a membrane protein embedded in the liposome. Proc. Natl. Acad. Sci. U. S. A. 117: 18497–18503, https://doi.org/10.1073/pnas.2009385117.Suche in Google Scholar PubMed PubMed Central
Young, H.S., Rigaud, J.L., Lacapère, J.J., Reddy, L.G., and Stokes, D.L. (1997). How to make tubular crystals by reconstitution of detergent-solubilized Ca2+-ATPase. Biophys. J. 72: 2545–2558, https://doi.org/10.1016/s0006-3495(97)78898-2.Suche in Google Scholar
Zheng, H., Lee, S., Llaguno, M.C., and Jiang, Q.-X. (2015). bSUM: a bead-supported unilamellar membrane system facilitating unidirectional insertion of membrane proteins into giant vesicles. J. Gen. Physiol. 147: 77–93, https://doi.org/10.1085/jgp.201511448.Suche in Google Scholar PubMed PubMed Central
© 2023 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- Highlight: Membrane Proteins from Structure to Function
- The Rauischholzhausen Transport Colloquium: membrane proteins from structure to function
- Determination of membrane protein orientation upon liposomal reconstitution down to the single vesicle level
- Interaction of RTX toxins with the host cell plasma membrane
- Interactions of Na+/taurocholate cotransporting polypeptide with host cellular proteins upon hepatitis B and D virus infection: novel potential targets for antiviral therapy
- Mycobacterial type VII secretion systems
- Lipid exchange among electroneutral Sulfo-DIBMA nanodiscs is independent of ion concentration
- Membrane-anchored substrate binding proteins are deployed in secondary TAXI transporters
- ATP binding and ATP hydrolysis in full-length MsbA monitored via time-resolved Fourier transform infrared spectroscopy
Artikel in diesem Heft
- Frontmatter
- Highlight: Membrane Proteins from Structure to Function
- The Rauischholzhausen Transport Colloquium: membrane proteins from structure to function
- Determination of membrane protein orientation upon liposomal reconstitution down to the single vesicle level
- Interaction of RTX toxins with the host cell plasma membrane
- Interactions of Na+/taurocholate cotransporting polypeptide with host cellular proteins upon hepatitis B and D virus infection: novel potential targets for antiviral therapy
- Mycobacterial type VII secretion systems
- Lipid exchange among electroneutral Sulfo-DIBMA nanodiscs is independent of ion concentration
- Membrane-anchored substrate binding proteins are deployed in secondary TAXI transporters
- ATP binding and ATP hydrolysis in full-length MsbA monitored via time-resolved Fourier transform infrared spectroscopy