
Research progress in quinazoline derivatives as multi-target tyrosine kinase inhibitors
-
Hao Jin


Abstract
Receptor tyrosine kinases (RTKs), such as epidermal growth factor receptor (EGFR), are involved in multiple human tumors. Therefore, RTKs are attractive targets for various antitumor strategies. Two classes of tyrosine kinase antagonists were applied in the clinic for monoclonal antibodies and small-molecule tyrosine kinase inhibitors. A well-studied class of small-molecule inhibitors is represented by 4-anilinoquinazolines, exemplified by gefitinib and erlotinib as mono-targeted EGFR inhibitors, which were approved for the treatment of non-small-cell lung cancer. Mono-target drugs may result in drug resistance and the innovation of multi-target drugs has grown up to be an active field. Recent advances in research on antitumor bioactivity of 4-anilino(or phenoxy)quinazoline derivatives with multiple targets are reviewed in this paper. At the same time, synthetic methods of quinazolines were introduced from the point of building the ring skeleton and based on the types of reaction.
Introduction
Quinazolines (Figure 1) show extensive biological activities [1], [2], [3], [4], [5], [6]. Their synthetic methods are important topics for pharmaceutical research. The 4(3H)-quinazolinone core plays a significant role in the most commonly applied synthetic methods. Microwave-assisted synthetic methods, acid-catalyzed synthetic methods and metal-catalyzed synthetic methods are also applied in their synthesis. Quinazoline derivatives are important antitumor drugs as small-molecule inhibitors especially in the area of receptor tyrosine kinase (RTK) inhibitors. The downstream signaling pathways of RTKs are closely related to the occurrence of the tumor, which can induce several oncogenic effects including enhancing cell proliferation, aberrant differentiation, malignant transformation and migration in solid tumor cells. Inhibiting the activity of the receptors could block the downstream signaling pathways of growth factors and effectively inhibit the growth of tumors [7]. Cancer is a disease usually accompanied by a disorder of multiple signaling pathways. Mono-target drugs may result in drug resistance [8], and the innovation of multi-target drugs has grown up to be an important field. Developing multi-target RTK inhibitors acting on multiple signal pathways could regulate multiple signaling pathways at different levels resulting in better antineoplastic activity.
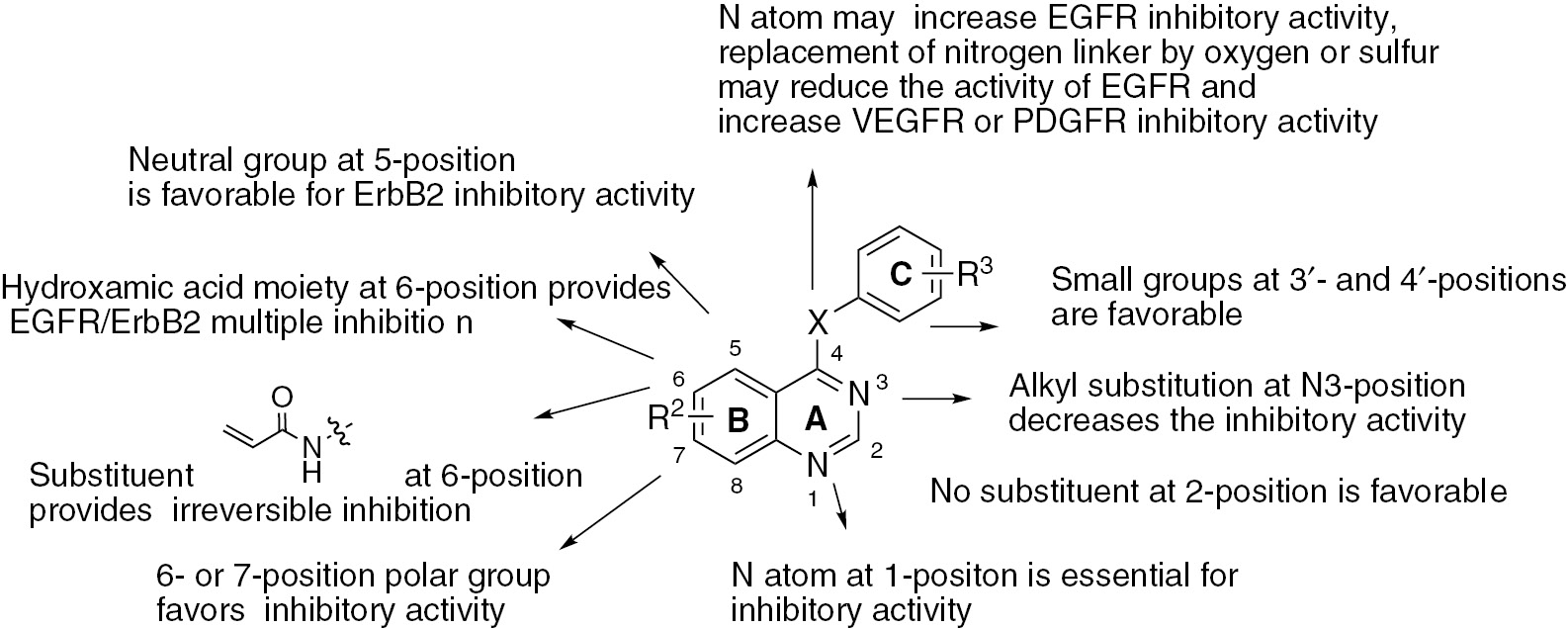
Structure-activity relationship of quinazolines.
The structure-activity relationship of quinazolines (Figure 1) can be summarized as follows:
Modifications in the A and C rings affect the ability of the compounds to inhibit epidermal growth factor receptor (EGFR)-tyrosine kinases. Both of the nitrogen atoms of quinazoline are essential for inhibitory activity. The phenyl ring with a small lipophilic substituent such as a chloro, bromo or trifluoromethyl group is important as it occupies the lipophilic pocket. The character of the linking group between the quinazoline ring and phenyl side chain makes a big difference in the inhibitory activity. Replacing the nitrogen linker by an oxygen or sulfur atom may result in the reduction of EGFR inhibitory activity. On the other hand, it may increase vascular endothelial growth factor receptor (VEGFR) inhibitory activity. Derivatives of the urea or thiourea group linked to the phenyl side chain are more potent.
Modifications in the B ring affect the inhibitory activity. The presence of a polar group at the 6- or 7-position of the quinazoline ring is beneficial. The absence of a substituent at the 2- or 3-position of quinazoline favors the inhibitory activity. Otherwise, the presence of a propylene amide or acetylene group at the 6-position of quinazoline can result in EGFR/ErbB2 irreversible inhibition. Introducing the hydroxamic acid moiety at the 6-position of quinazoline can result in EGFR/ErbB2 multiple kinase inhibition. The presence of a large neutral substituent at the 5-position can enhance ErbB2 inhibitory activity.
Quinazoline derivatives as multi-target EGFR family kinase inhibitors
EGFR is overexpressed in the majority of non-small-cell lung cancers and its expression is inversely related to survival outcome. EGFR and ErbB2 are two important validated target molecules in cancer treatment. Several single-target EGFR 4-aminoquinazoline compounds are applied in the clinic. Drug resistance is inevitable among the first generation [9]. Research on 4-anilinoquinazoline mono-target drugs shows that introducing a bulky lipophilic group at the 4-position of quinazoline can improve multiple inhibitory activities of the EGFR family, hence the occurrence of drug resistance can be reduced. Based on this finding, lapatinib (1a) in Table 1 was developed as a new kind of selective EGFR/ErbB2 dual inhibitor with an inhibitory concentration (IC50) value of 10.2 nm and 9.8 nm, respectively. The downstream cell proliferation signal is blocked by the reversible combination of EGFR/ErbB2 and adenosine triphosphate (ATP) active sites [10]. Our research group [11] also designed and synthesized several highly effective compounds, some of which are active in the micromolar range of IC50 values against MCF-7 and A-549 cells. Propylene amide or acetylene groups were introduced at the 6-position of quinazoline to afford a series of EGFR/ErbB2 irreversible inhibitors 1b–e with the IC50 at the nanomolar level for EGFR kinase and HER2 kinase. Structure-activity and molecular modeling studies suggested that positioning of the acrylamide moiety at the 6-position can effectively reduce the resistance to the first generation of EGFR inhibitors [12], [13], [14], [15]. A neutral hydroxyacylamino moiety was introduced into the anilinoquinazoline to furnish a RTK inhibitor 1f. Its IC50 values for the EGFR and ErbB2 kinases are around 2 nm [16]. Compound AZD8931 (1g) is a reversible inhibitor of phosphorylation of EGFR, HER2 and HER3 and demonstrates potent in vitro inhibition against isolated HER2 and EGFR tyrosine kinases with the respective IC50 values of 14 nm and 12 nm against HER2 and EGFR [17]. Compound 1h with an oxazine ring shows significant cytotoxic activities with the IC50 values of 3 nm and 250 nm against HER2 and EGFR, respectively. It shows better inhibitory activities against several cell lines compared to gefitinib and erlotinib, with extensive antiproliferative activities [18].
Quinazoline derivatives as multi-target tyrosine kinase inhibitors.
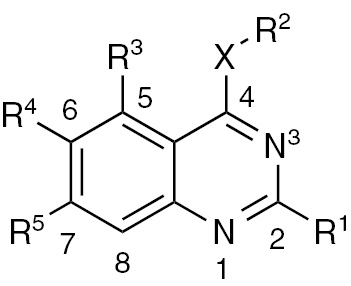
| Compd. | Target | X | R1 | R2 | R3 | R4 | R5 |
|---|---|---|---|---|---|---|---|
| 1a | EGFR/ErbB2 | NH | H | 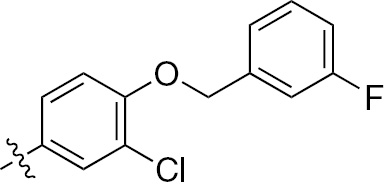 | – | 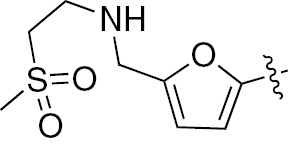 | – |
| 1b | EGFR/ErbB2 | NH | H | 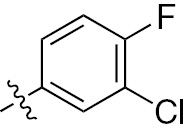 | – | 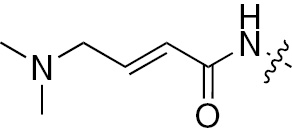 | 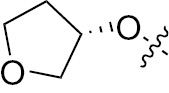 |
| 1c | EGFR/ErbB2 | NH | H | 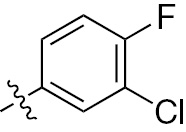 | – | 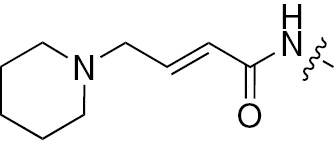 | 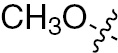 |
| 1d | EGFR/ErbB2 | NH | H | 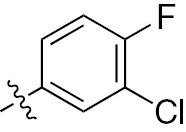 | – | 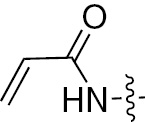 | 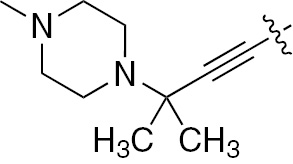 |
| 1e | EGFR/ErbB2 | NH | H | 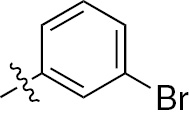 | – | 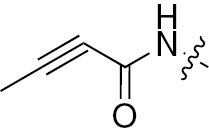 | – |
| 1f | EGFR/ErbB2 | NH | H | 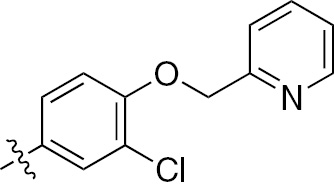 | 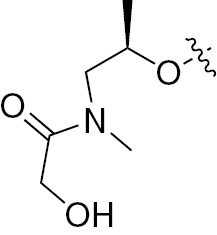 | – | – |
| 1g | EGFR/ErbB2/ErbB3 | NH | H | 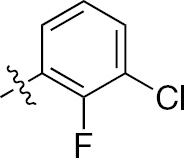 | – | 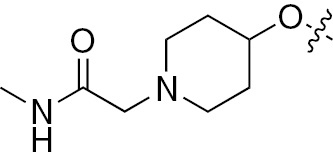 | 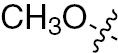 |
| 1h | EGFR/ErbB2 | NH | H | 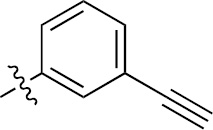 | – | 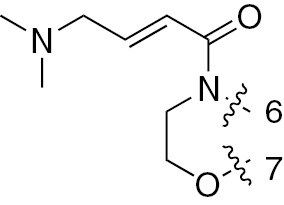 | |
| 1i | EGFR/VEGFR | NH | H | 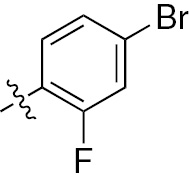 | – | 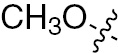 | 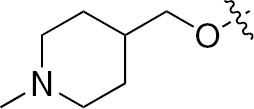 |
| 1j | EGFR/VEGFR | NH | H | 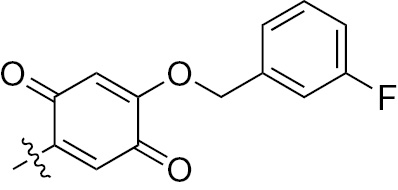 | – | 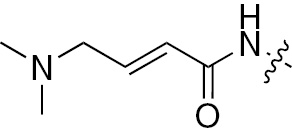 | 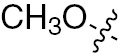 |
| 1k | EGFR/VEGFR | NH | Cl | 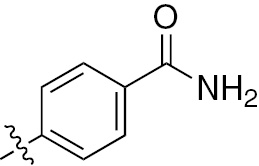 | – | 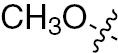 | 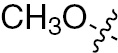 |
| 1l | EGFR/VEGFR | NH | H | 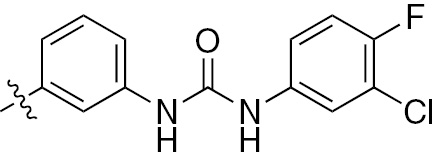 | – | – | 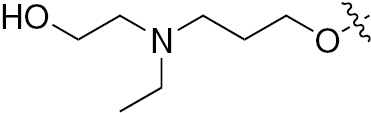 |
| 1m | EGFR/VEGFR | NH | H | 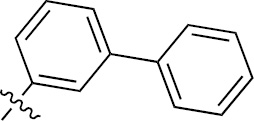 | – | 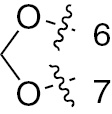 | |
| 1n | EGFR/VEGFR | NH | H | 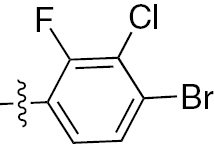 | – | 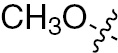 | 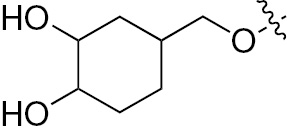 |
| 1o | VEGFR/PDGFR | O | H | 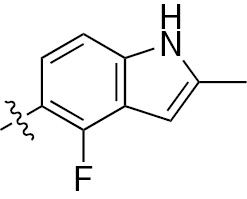 | – | 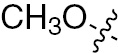 | 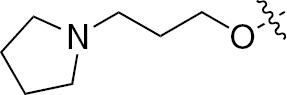 |
| 1p | VEGFR/PDGFR | O | H | 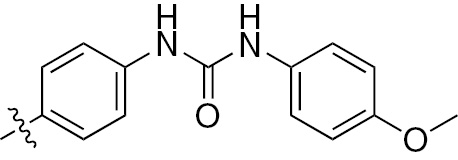 | – | 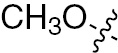 | 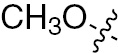 |
| 1q | VEGFR/PDGFR | O | H | 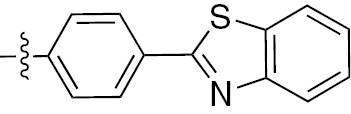 | – | 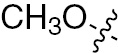 | 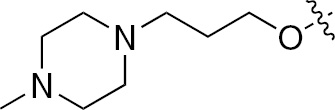 |
| 1r | VEGFR/PDGFR | O | H | 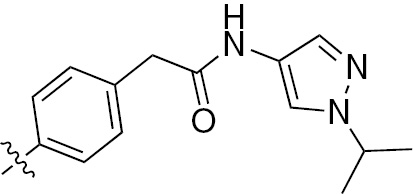 | – | 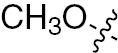 | 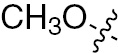 |
| 1s | VEGFR/PDGFR | O | H | 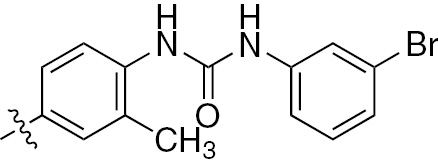 | – | 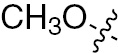 | 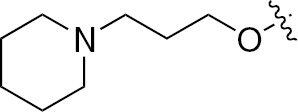 |
Quinazoline derivatives as multi-target EGFR and VEGFR family kinase inhibitors
VEGFR-2 plays a major role in tumor angiogenesis, which is regarded as the most important molecular target for inhibiting angiogenesis [19]. The drug targeting the EGFR and VEGFR-2 pathways can inhibit both tumor cells and tumor angiogenesis, hence a better therapeutic effect can be achieved. Vandetanib (ZD6474, 1i) is a potent tyrosine kinase inhibitor targeting the VEGFR tyrosine kinase (IC50=40 nm) and the EGFR tyrosine kinase (IC50=500 nm) [20]. Compound 1j has two individual reactive centers and was designed to target a cysteine residue located in the ATP binding pocket of both EGFR and VEGFR-2. The IC50 values against them are 4.1 nm and 113.4 nm, respectively [21]. The hydrogen bond donor lies at the para position of the aniline moiety of compound 1k and plays an important role in binding with amino acids of EGFR and VEGFR-2 [22].
In 2013, Zhang and co-workers [23] designed a hybrid approach that combines two privileged pharmacophores, namely 4-anilinoquinazoline and unsymmetrical diarylurea, to successfully deliver a novel series of multi-kinase inhibitors. Compound 1l exhibits profound activity against BRAF, BRAF V600E, VEGFR-2 and EGFR. The IC50 values against EGFR and VEGFR-2 are 165 nm and 95 nm, respectively. Compound 1m has a 1,3-dioxolane moiety fused to the quinazoline portion and a biphenylamino substituent as the aniline portion, which provide a significant anti-angiogenic effect in both in vitro and in vivo assays even at noncytotoxic concentrations [24]. Shi and co-workers [25] synthesized a series of EGFR/VEGFR-2 tyrosine kinase inhibitors. The inhibitory activities of the optimized compound 1n against EGFR and VEGFR-2 in vitro are 15 nm and 32 nm, respectively, which is better than that of the reference compound vandetanib. These findings may provide new insights for seeking new multi-targeting anticancer candidate compounds.
Quinazoline derivatives as multi-target VEGFR and PDGFR family inhibitors
VEGFR and platelet-derived growth factor receptor (PDGFR) mediate two important signaling pathways in tumor angiogenesis. Because of the compensation between signaling pathways, inhibition of either one of the signaling pathways can result in enhancing of the other signaling pathways, thus triggering drug resistance. Developing dual-target tyrosine kinase inhibitors for VEGFR and PDGFR can greatly improve the anti-angiogenesis and antitumor activities. Cediranib (AZD2171, 1o) is one of the most potent multi-target anti-angiogenesis drugs with the inhibitory activity of 5 nm for both VEGFR-2 and PDGFR-β [26], [27]. Compound 1p is a phenylurea derivative of 4-substituted quinazoline with inhibitory activities of 2.2 nm and 3.6 nm for VEGFR-2 and PDGFR, respectively [28].
An arylbenzothiazole derivative 1q demonstrates high inhibitory activities with the respective IC50 values of 60 nm, 170 nm and 540 nm for VEGFR2, PDGFR-β and TIE2 in vitro [29]. The pyrazole derivative 1r has a potent and balanced profile against PDGFR and VEGFR-2 with the respective IC50 values of 4 nm and 8 nm. It has the potential to become an anti-angiogenic drug in the clinic [30]. Urea derivatives, synthesized by Raveza and co-workers [31], allowed a significant increase in the inhibitory effects on the growth of three cancer cell lines PC3, HT29 and MCF7. Such compounds are highly potent inhibitors of VEGFR (1, 2 and 3), PDGFR-β and c-KIT with the IC50 values in the nanomolar range. Among them, the potent multi-kinase inhibitor 1s inhibits angiogenesis by preventing tube formation and inhibiting endothelial cell invasion.
Synthesis of quinazolinones and 4-anilino(or phenoxy)quinazolines
Methods based on intermediary of 4(3H)-quinazolinones for synthesis of 4-anilino(or phenoxy)quinazolines
4-Anilino(or phenoxy)quinazoline compounds demonstrate various useful biological and medicinal activities. The most common synthetic approach to these compounds involves the preparation of the intermediate 4-chloroquinazolines from 4(3H)-quinazolinones followed by treatment with anilines or phenols. Many synthetic methods for 4(3H)-quinazolinones have been reported [32]. The most common method for preparation of 4(3H)-quinazolinones 4 is based on the Niementowski reaction (Scheme 1). This reaction involves the fusion of anthranilic acid derivatives 1 with formamide 2 which generates an o-amidine intermediate 3. The procedure usually requires high temperatures and a tedious workup. To address these problems, Besson and co-workers [33] used microwave irradiation which improved the yield and decreased the reaction time.
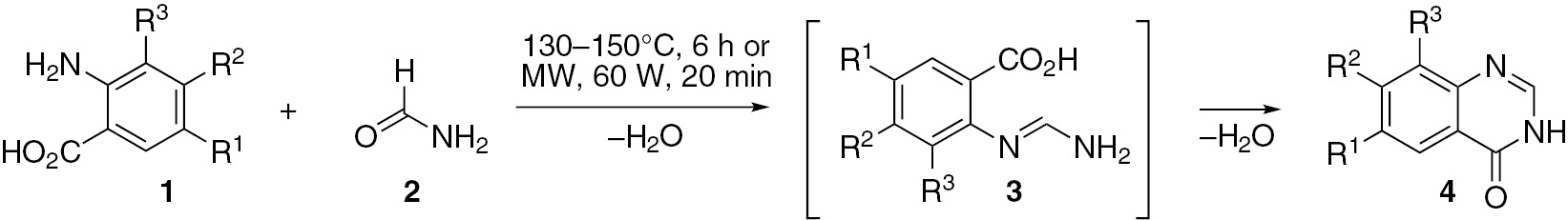
Compound 5 (Scheme 2) was prepared by Shao and co-workers [34] under microwave irradiation from m-trifluoromethylaniline (8) and 6,7-dimethoxy-4-chloroquinazoline (6). Compound 6 was obtained by treatment of 6-aminoveratric acid (7) with formamide in the presence of phosphorus oxychloride. This reaction apparently involves generation of a 4(3H)-quinazolinone as a transient intermediate product.
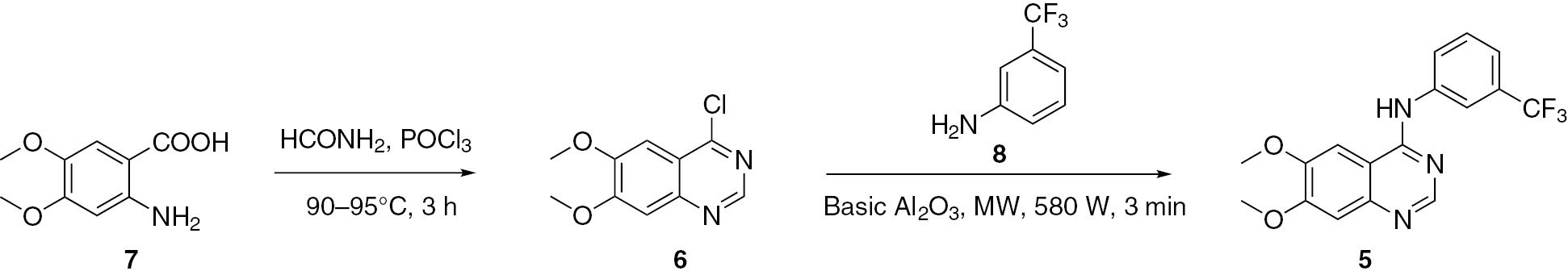
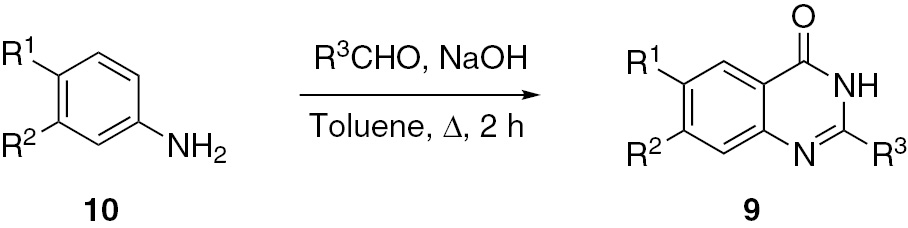
4(3H)-quinazolinones 9 were obtained by the reaction of substituted aromatic o-aminonitriles 10 with aldehydes in the presence of a base [35]. This transformation involves consecutive addition, intramolecular Pinner reaction, Dimroth rearrangement and oxidative dehydrogenation reactions. This synthesis has the advantages of readily available starting materials, mild reaction conditions and high yields of products (Scheme 3).
A microwave-assisted synthesis in an aqueous medium of quinazoline derivatives was developed [36]. Treatment of 2-aminobenzamide (11) with an excess of chloroacetyl chloride (12) under microwave irradiation furnished the intermediate 2-chloroacetamide 13. The crude product was isolated and directly cyclized in the presence of K2CO3 in water, under microwave irradiation, leading to the expected product 14 (Scheme 4).
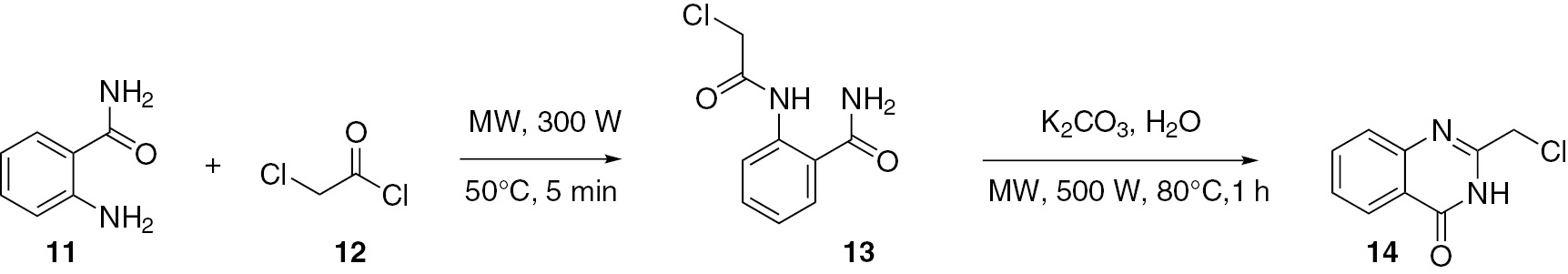
4-Anilino-7-nitroquinazoline (18) was synthesized [37] under microwave irradiation from aniline (17) and 7-nitro-4-chloroquinazoline (16). Using 4-nitroanthranilic acid (15) as the starting material, compound 16 was smoothly obtained through the Niementowski reaction followed by chlorination (Scheme 5).
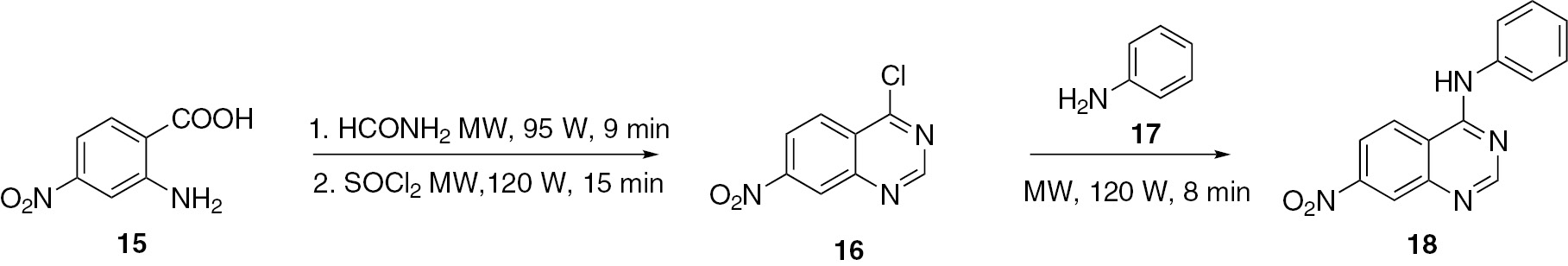
4-Chloroquinazoline (21) was prepared from 2-aminobenzonitrile (19). Compound 19 was first converted to quinazolin-4(3H)-one (20) by acid-catalyzed cyclization with formic acid under microwave irradiation. Treatment of 20 under microwave irradiation with an excess amount of POCl3 in N,N-diethylamine yielded the product 21 [38] (Scheme 6).
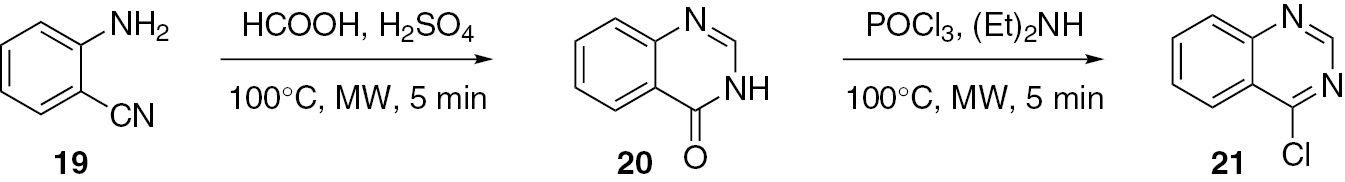
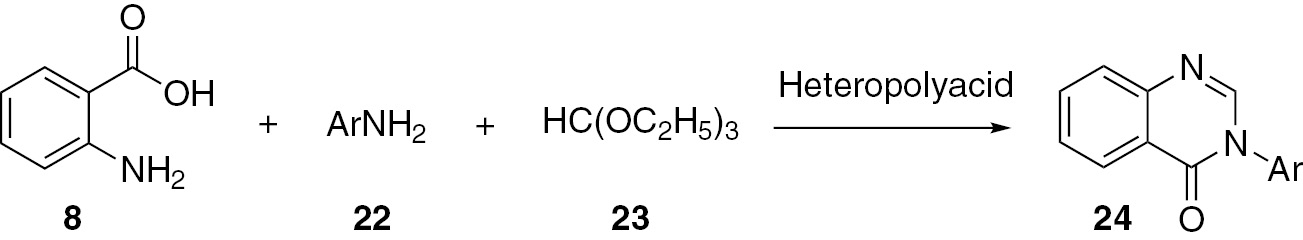
Ighilahriz and co-workers [39] developed a hetero-polyacid-catalyzed method for the synthesis of 4(3H)-quinazolinones 24 by cyclocondensation of anthranilic acid (8), ortho ester 23 and substituted anilines 22. This is a microwave-assisted, solvent-free, rapid and high yield reaction. The Keggin-type heteropolyacids are effective catalysts for this acid-catalyzed reaction (Scheme 7).
A zinc-catalyzed oxidation of 2-aminobenzamide (25) with a benzyl alcohol 26 was developed by Sharif et al. [40]. Various quinazolinones 27 were obtained in moderate to good yields. Treatment of aromatic aldehydes 28 with 2-aminobenzamide 25 under catalyst-free conditions afford the same products 27. Surprisingly, water is also a good medium for this reaction (Scheme 8).
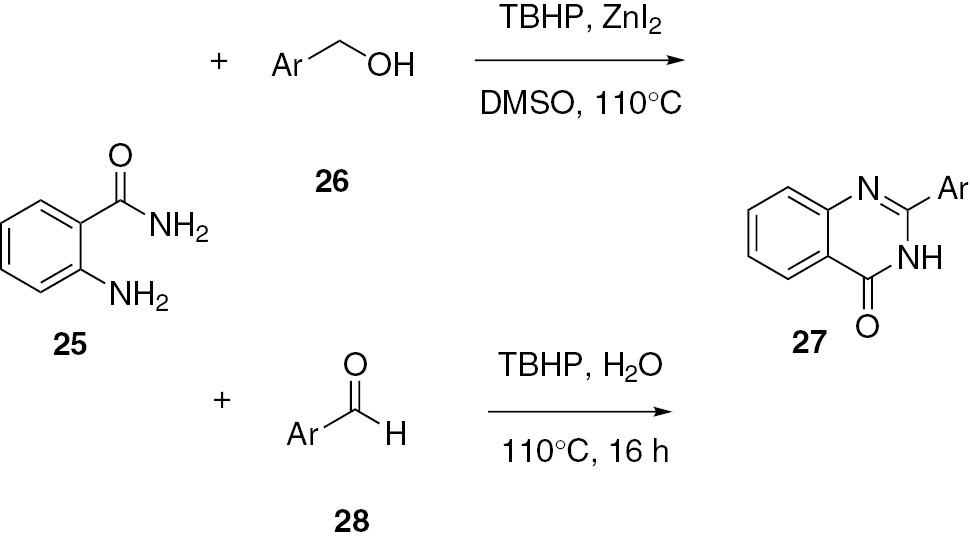
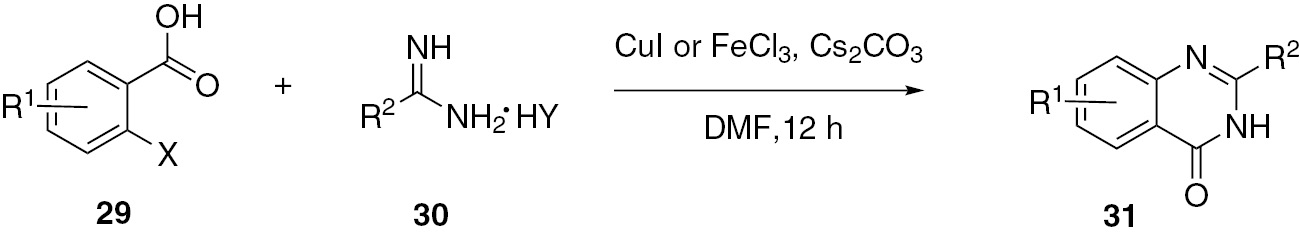
Metal-catalyzed Ullmann N-arylation can be used to construct N-heterocycles. Liu and co-workers [41] reported a simple, practical and efficient procedure for the preparation of quinazolinone derivatives under mild copper-catalyzed conditions. The target products 31 were obtained from 2-halobenzoic acids 29 and acetamidine hydrochlorides 30. Subsequently, they developed an iron-catalyzed method for the reaction. This reaction provides a new method for the construction of diverse and practical molecules of potential biological and medicinal activities (Scheme 9).
Other synthetic methods
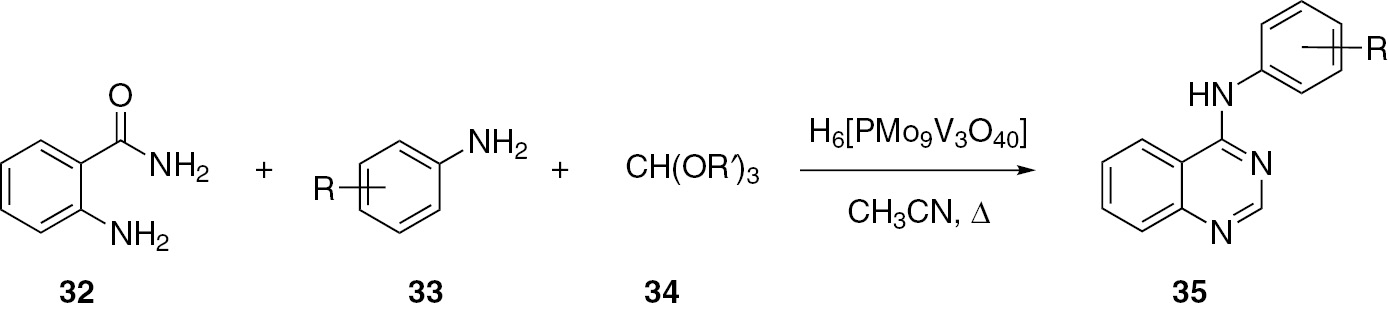
Heravi and co-workers [42] reported a one-pot method for the synthesis of 4-arylaminoquinazolines 35 by the reaction of anthranilamide (32), substituted aromatic amines 33 and ortho esters 34 catalyzed by heteropolyacid. The yield of the product 35 ranges from 80% to 95%. This method uses a simple workup and the catalyst is recyclable (Scheme 10).
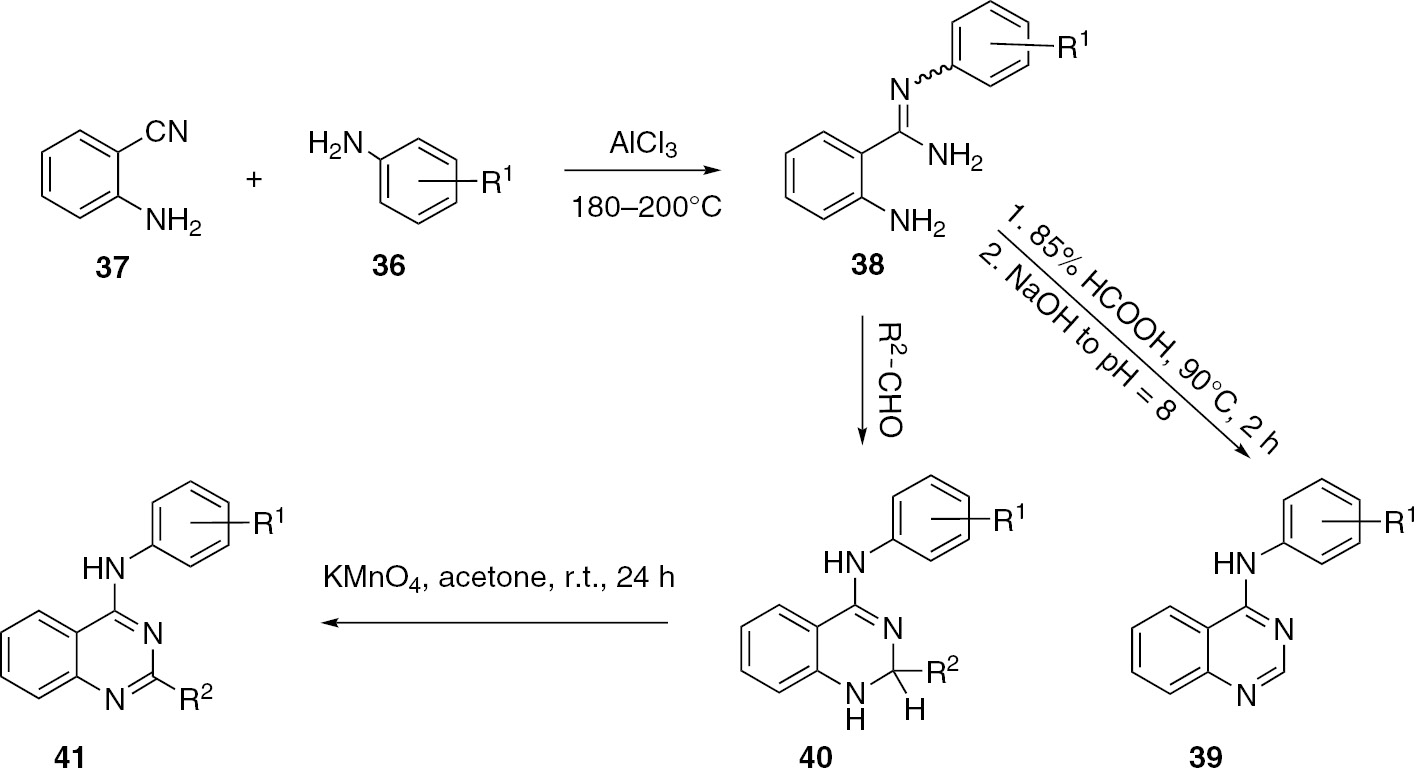
Szczepankiewicz and co-workers [43] described the synthesis of a series of 2-substituted 41 and 2-unsubstituted 4-arylaminoquinazolines 39 (Scheme 11). Thus, the treatment of 2-anthranilonitrile (37) with substituted anilines 36 in the presence of aluminum chloride gave the corresponding 2-amino-N-arylbenzamidines 38. Products 39 were obtained directly by heating compounds 38 in formic acid. The treatment of amidines 38 with aldehydes afforded substituted 1H-2,3-dihydroquinazolines 40 readily oxidizable by treatment with potassium permanganate to quinazolines 41.
Chandregowda and co-workers [44] reported an approach for the construction of a quinazoline starting from reduction of o-nitrobenzonitrile 42 to anthranilonitrile 43. Compound 43 was treated with a mixture of dimethylformamide (DMF) and dimethylacetamide (DMA) in toluene to give N,N-dimethylformamidine derivative 44. The tyrosine kinase inhibitor gefitinib (46) was prepared by treatment of 44 with aniline 45 (Scheme 12).
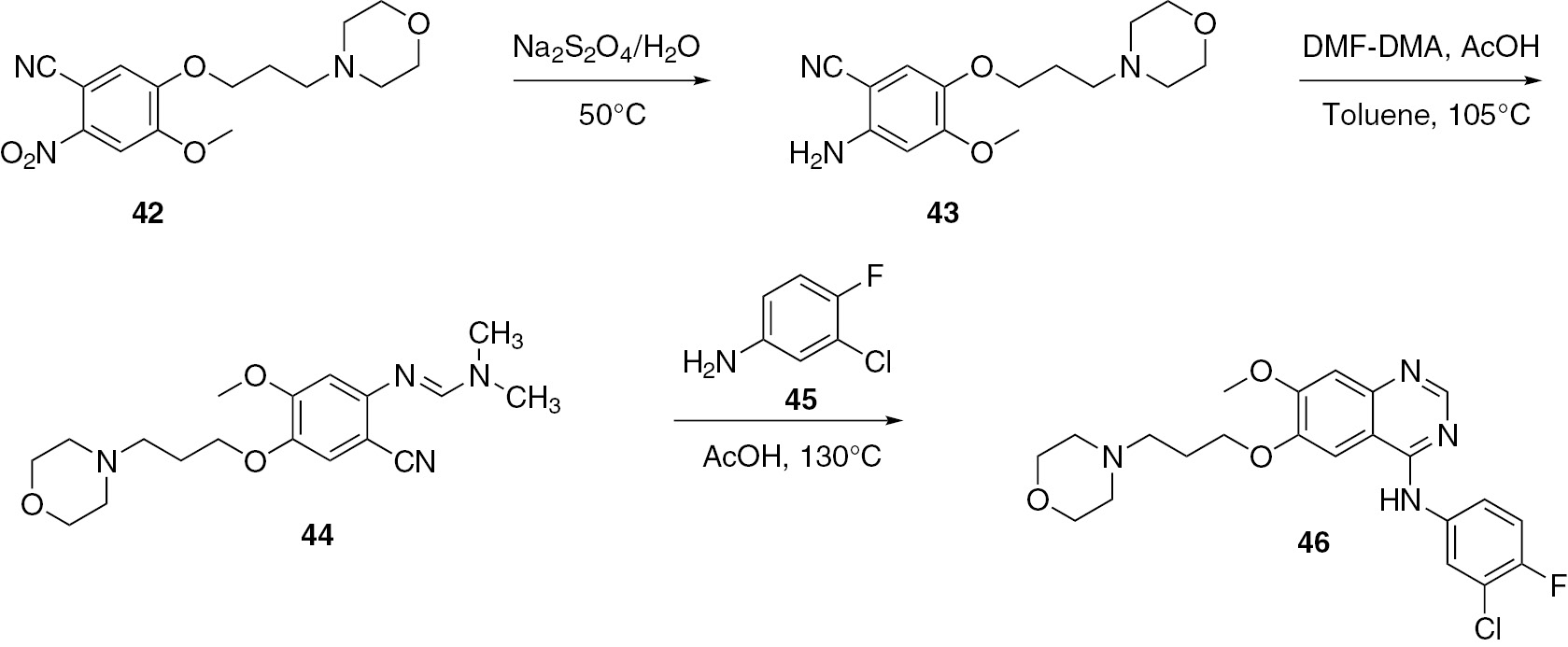
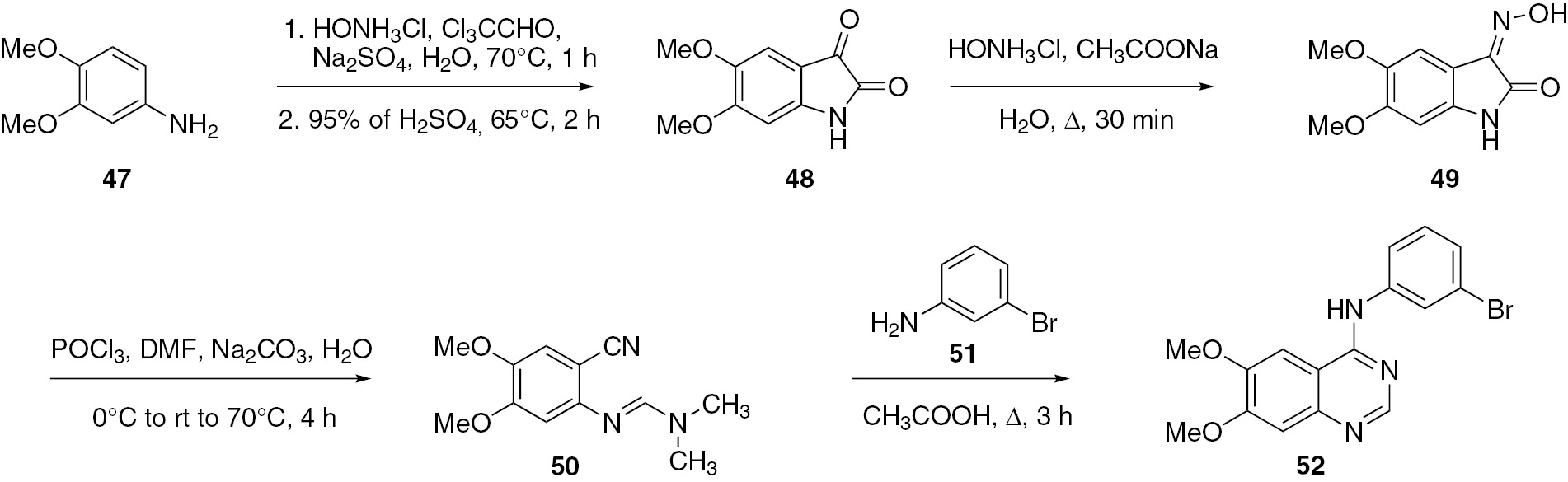
Wang and co-workers [45] used 3,4-dimethoxyaniline (47) as the starting material for the synthesis of PD153035 (52) in a total yield of 50%. The intermediates 48–50 were synthesized as shown in Scheme 13. The final intermediate product 50 was treated with aniline 51 to produce the target compound 52.
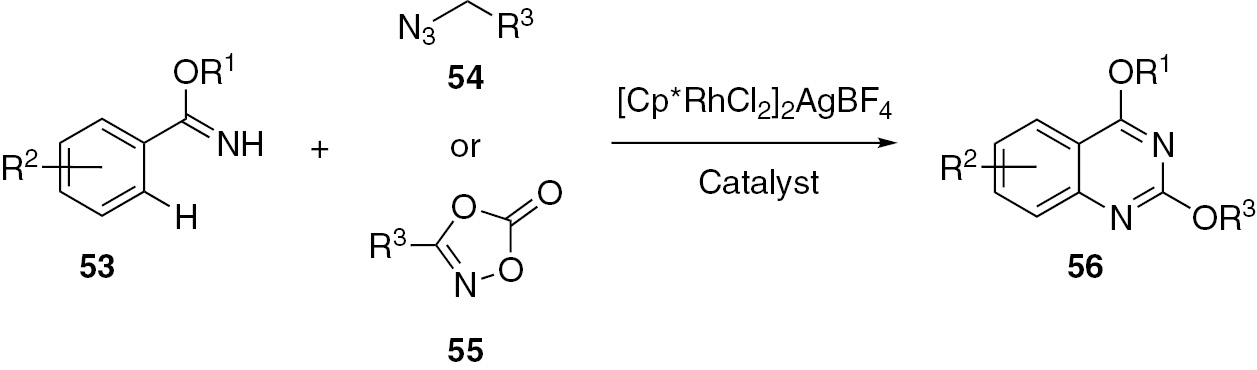
An annulation of benzimidates 53 with alkyl azides 54 or dioxazolones 55 for the preparation of quinazolines 56 was developed by Wang and co-workers [46], [47]. The operational simplicity, high atom efficiency and wide range of substrate scope make this method useful for the practical preparation of phenoxy-substituted quinazoline derivatives (Scheme 14).
Conclusions
In this paper, quinazoline inhibitors of tyrosine kinases including their synthesis are summarized. Modifications in the structure of quinazoline class of antitumor agents are mainly focused on introducing the side chains at the 6- and 7-positions of 4-anilino(or phenoxy)quinazoline. The use of bioisosterism, reactive splicing methods and computer-aided drug design are also important directions for optimizing the structure of the compounds.
Acknowledgments
The authors express their sincere gratitude to the Zhejiang University of Technology for supporting the access to databases needed for the literature survey. This work was supported by the Natural Science Foundation of Zhejiang Province (No. LY14B020004), Funder ID: 10.13039/501100004731.
References
[1] Zhou, H.; Aguilar, A.; Chen, J.; Bai, L.; Liu, L.; Meagher, J. L.; Yang, C.-Y.; McEachern, D.; Cong, X.; Stuckey, J. A.; et al. Structure-based design of potent Bcl-2/Bcl-xL inhibitors with strong in vivo antitumor activity. J. Med. Chem.2012, 55, 6149–6161.10.1021/jm300608wSuche in Google Scholar PubMed PubMed Central
[2] Chandrika, P. M.; Ram Rao, A. R.; Narsaiah, B.; Raju, M. B. Quinazoline derivatives with potent anti-inflammatory and anti-allergic activities. Int. J. Chem. Sci.2008, 6, 1119–1146.Suche in Google Scholar
[3] Kung, P.-P.; Casper, M. D.; Cook, K. L.; Laura, W.-L.; Risen, L. M.; Vickers, T. A.; Ranken, R.; Blyn, L. B.; Wyatt, J. R.; Cook, P. D.; et al. Structure-activity relationships of novel 2-substituted quinazoline antibacterial agents. J. Med. Chem.1999, 42, 4705–4713.10.1021/jm9903500Suche in Google Scholar PubMed
[4] Malamas, M. S.; Millen, J. Quinazolineacetic acids and related analogs as aldose reductase inhibitors. J. Med. Chem.1991, 34, 1492–1503.10.1021/jm00108a038Suche in Google Scholar PubMed
[5] Madapa, S.; Tusi, Z.; Mishra, A.; Srivastava, K.; Pandey, S. K.; Tripathi, R.; Puri, S. K.; Batra, S. Search for new pharmacophores for antimalarial activity. Part II: Synthesis and antimalarial activity of new 6-ureido-4-anilinoquinazolines. Bioorg. Med. Chem.2009, 17, 222–234.10.1016/j.bmc.2008.11.005Suche in Google Scholar PubMed
[6] Lowe, J. A.; Archer, R. L.; Chapin, D. S.; Chen, J. B.; Helweg, D.; Johnson, J. L.; Koe, B. K.; Lebel, L. A.; Moore, P. F.; Nielsen, J. A. Structure-activity relationship of quinazolinedione inhibitors of calcium-independent phosphodiesterase. J. Med. Chem.1991, 34, 624–628.10.1021/jm00106a024Suche in Google Scholar PubMed
[7] Jiang, M.; Liu, D.; Lan, S.-P. Progressed in quinolines as protein tyrosine kinase inhibitors. Chem. Reagents2013, 35, 333–336.Suche in Google Scholar
[8] Liu, J.; Wang, L.; Yang, X.-M. Multi-targeted protein tyrosine kinase inhibitor: research advances. J. Int. Pharm. Res.2009, 36, 161–171.Suche in Google Scholar
[9] Gong, F.-H.; Sun, L.-P. Research progresses in antitumor activity of multiple target 4-substituted anilinoquinazoline derivatives. Prog. Pharm. Sci.2014, 38, 25–30.Suche in Google Scholar
[10] Moreira, C.; Kaklamani, V. Lapatinib and breast cancer: current indications and outlook for the future. Expert Rev. Anticancer Ther.2010, 10, 1171–1182.10.1586/era.10.113Suche in Google Scholar PubMed
[11] Rao, G. W.; Xu, G. J.; Wang, J.; Jiang, X. L.; Li, H. B. Synthesis, antitumor evaluation and docking study of novel 4-anilinoquinazoline derivatives as potential epidermal growth factor receptor (EGFR) inhibitors. Chem. Med. Chem.2013, 8, 928–933.10.1002/cmdc.201300120Suche in Google Scholar PubMed
[12] Eskens, F. A.; Mom, C. H.; Planting, A. S.; Gietema, J. A.; Amelsberg, A.; Huisman, H.; van Doorn, L.; Burger, H.; Stopfer, P.; Verweij, J.; et al. A phase I dose escalation study of BIBW 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br. J. Cancer2008, 98, 80–85.10.1038/sj.bjc.6604108Suche in Google Scholar PubMed PubMed Central
[13] Engelman, J. A.; Zejnullahu, K.; Gale, C. M.; Lifshits, E.; Gonzales, A. J.; Shimamura, T.; Zhao, F.; Vincent, P. W.; Naumov, G. N.; Bradner, J. E.; et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res.2007, 67, 11924–11932.10.1158/0008-5472.CAN-07-1885Suche in Google Scholar PubMed
[14] Suzuki, T.; Fujii, A.; Ohya, J.; Amano, Y.; Kitano, Y.; Abe, D.; Nakamura, H. Pharmacological characterization of MP-412 (AV-412), a dual epidermal growth factor receptor and ErbB2 tyrosine kinase inhibitor. Cancer Sci.2007, 98, 1977–1984.10.1111/j.1349-7006.2007.00613.xSuche in Google Scholar PubMed
[15] Klutchko, S. R.; Zhou, H.-R.; Winters, R. T.; Tran, T. P.; Bridges, A. G.; Althaus, I. W.; Amato, D. M.; Elliott, W. L.; Ellis, P. A.; Meade, M. A.; et al. Tyrosine kinase inhibitors. 19. 6-Alkynamides of 4-anilinoquinazolines and 4-anilinopyrido[3, 4-d]pyrimidines as irreversible inhibitors of the erbB family of tyrosine kinase receptors. J. Med. Chem.2006, 49, 1475–1485.10.1021/jm050936oSuche in Google Scholar
[16] Ballard, P.; Barlaam, B. C.; Bradbury, R. H.; Dishington, A.; Hennequin, L. F.; Hickinson, D. M.; Hollingsworth, I. M.; Kettle, J. G.; Klinowska, T.; Ogilvie, D. J.; et al. Neutral 5-substituted 4-anilinoquinazolines as potent, orally active inhibitors of erbB2 receptor tyrosine kinase. Bioorg. Med. Chem. Lett.2007, 17, 6326–6329.10.1016/j.bmcl.2007.08.073Suche in Google Scholar
[17] Barlaam, B.; Anderton, J.; Ballard, P.; Bradbury, R. H.; Hennequin, L. F.; Hickinson, D. M.; Kettle, J. G.; Kirk, G.; Klinowska, T.; Lambert-van der Brempt, C.; et al. Discovery of ADZ8931, an equipotent, reversible inhibitor of signaling by EGFR, HER2, and HER3 receptors. ACS Med. Chem. Lett.2013, 4, 742–746.10.1021/ml400146cSuche in Google Scholar
[18] Chen, X.; Du, Y.; Sun, H.; Wang, F.; Kong, L.; Sun, M. Synthesis and biological evaluation of novel tricyclic oxazine and oxazepine fused quinazolines. Part 1: erlotinib analogs. Bioorg. Med. Chem. Lett.2014, 24, 884–887.10.1016/j.bmcl.2013.12.079Suche in Google Scholar
[19] Ferrara, N. VEGF as a therapeutic target in cancer. Oncology2005, 69 Suppl 3, 11–16.10.1159/000088479Suche in Google Scholar
[20] Lee, D.; Natale, R. B.; Nadler, E.; Jain, V. K. Phase II data with ZD6474, a small-molecule kinase inhibitor of epidermal growth factor receptor and vascular endothelial growth factor receptor, in previously treated advanced non–small-cell lung cancer. Clin. Lung Cancer.2005, 7, 89–91.10.1016/S1525-7304(11)70394-1Suche in Google Scholar
[21] Wissner, A.; Fraser, H. L.; Ingalls, C. L.; Dushin, R. G.; Floyd, M. B.; Cheung, K.; Nittoli, T.; Ravi, M. R.; Tan, X.; Loganzo, F. Dual irreversible kinase inhibitors: quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2. Bioorg. Med. Chem.2007, 15, 3635–3648.10.1016/j.bmc.2007.03.055Suche in Google Scholar PubMed
[22] Barbosa, M. L.; Lima, L. M.; Tesch, R.; Sant’Anna, C. M.; Totzke, F.; Kubbutat, M. H.; Schachtele, C.; Laufer, S. A.; Barreiro, E. J. Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors. Eur. J. Med. Chem.2014, 71, 1–14.10.1016/j.ejmech.2013.10.058Suche in Google Scholar PubMed
[23] Zhang, Q.-W.; Diao, Y.-Y.; Wang, F.; Fu, Y.; Tang, F.; You, Q.-D.; Zhou, H.-Y. Design and discovery of 4-anilinoquinazoline ureas as multikinase inhibitors targeting BRAF, VEGFR-2 and EGFR. Chem. Med. Chem.2013, 4, 979.10.1039/c3md00096fSuche in Google Scholar
[24] Conconi, M. T.; Marzaro, G.; Urbani, L.; Zanusso, I.; Di Liddo, R.; Castagliuolo, I.; Brun, P.; Tonus, F.; Ferrarese, A.; Guiotto, A.; et al. Quinazoline-based multi-tyrosine kinase inhibitors: synthesis, modeling, antitumor and antiangiogenic properties. Eur. J. Med. Chem.2013, 67, 373–383.10.1016/j.ejmech.2013.06.057Suche in Google Scholar PubMed
[25] Shi, Y.; Hu, Q.-Q.; Yu, B.; Li, Y.-L.; Xiong, D.-S. Anti-tumor activity of novel 4-aminophenylquinazoline tyrosine kinase inhibitors. Drugs Clinic2014, 9, 969–973.Suche in Google Scholar
[26] Wedge, S. R.; Kendrew, J.; Hennequin, L. F.; Valentine, P. J.; Barry, S. T.; Brave, S. R.; Smith, N. R.; James, N. H.; Dukes, M.; Curwen, J. O.; et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res.2005, 65, 4389–4400.10.1158/0008-5472.CAN-04-4409Suche in Google Scholar PubMed
[27] Bender, D.; Sill, M. W.; Lankes, H. A.; Reyes, H. D.; Darus, C. J.; Delmore, J. E.; Rotmensch, J.; Gray, H. J.; Mannel, R. S.; Schilder, J. M.; et al. A phase II evaluation of cediranib in the treatment of recurrent or persistent endometrial cancer: an NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol.2015, 138, 507–512.10.1016/j.ygyno.2015.07.018Suche in Google Scholar PubMed PubMed Central
[28] Kubo, K.; Shimizu, T.; Ohyama, S.; Murooka, H.; Iwai, A.; Nakamura, K.; Hasegawa, K.; Kobayashi, Y.; Takahashi, N.; Takahashi, K.; et al. Novel potent orally active selective VEGFR-2 tyrosine kinase inhibitors: synthesis, structure-activity relationships, and antitumor activities of N-phenyl-N′-{4-(4-quinolyloxy)phenyl}ureas. J. Med. Chem.2005, 48, 1359–1366.10.1021/jm030427rSuche in Google Scholar PubMed
[29] Tasler, S.; Muller, O.; Wieber, T.; Herz, T.; Pegoraro, S.; Saeb, W.; Lang, M.; Krauss, R.; Totzke, F.; Zirrgiebel, U.; et al. Substituted 2-arylbenzothiazoles as kinase inhibitors: hit-to-lead optimization. Bioorg. Med. Chem.2009, 17, 6728–6737.10.1016/j.bmc.2009.07.047Suche in Google Scholar PubMed
[30] Ple, P. A.; Jung, F.; Ashton, S.; Hennequin, L.; Laine, R.; Morgentin, R.; Pasquet, G.; Taylor, S. Discovery of AZD2932, a new quinazoline ether inhibitor with high affinity for VEGFR-2 and PDGFR tyrosine kinases. Bioorg. Med. Chem. Lett.2012, 22, 262–266.10.1002/chin.201220167Suche in Google Scholar
[31] Ravez, S.; Barczyk, A.; Six, P.; Cagnon, A.; Garofalo, A.; Goossens, L.; Depreux, P. Inhibition of tumor cell growth and angiogenesis by 7-aminoalkoxy-4-aryloxyquinazoline ureas, a novel series of multi-tyrosine kinase inhibitors. Eur. J. Med. Chem.2014, 79, 369–381.10.1016/j.ejmech.2014.04.007Suche in Google Scholar PubMed
[32] Abdou, I. M.; Al-Neyadi, S. S. Synthesis of quinazolines and quinazolinones via palladium-mediated approach. Heterocycl. Commun.2015, 21, 115–132.10.1515/hc-2014-0181Suche in Google Scholar
[33] Alexandre, F. R.; Berecibar, A.; Besson, T. ChemInform abstract: microwave-assisted Niementowski reaction. Back to the roots. Tetrahedron Lett.2002, 33, 3911–3913.10.1002/chin.200236153Suche in Google Scholar
[34] Shao, H. Z.; Zhou, Y.; Xia, M. Synthesis and bioactivity of 6,7-dimethoxy- (3′trifluromethylphenyl)quinazoline. Chin. J. Synth. Chem.2006, 14, 66–68.Suche in Google Scholar
[35] Change, L.; Qiyao, Y.; Jianhong, T.; Jiarong, L. One-pot synthesis of 4(3H)- quinazolinone under basic conditions. Chin. J. Org. Chem.2012, 32, 532–534.10.6023/cjoc1108102Suche in Google Scholar
[36] Kabri, Y.; Gellis, A.; Vanelle. P. Microwave-assisted synthesis in aqueous medium of new quinazoline derivatives as anticancer agent precursors. Green. Chem.2009, 11, 201–208.10.1039/B816723KSuche in Google Scholar
[37] Zhu, Y.-X.; Ma, Y.-Z.; Zhang, Y.; Liu, Y.-X.; Zhi, H. Microwave-assisted synthesis of 4-anilino-7-aminoquinazoline. Fine Chem. Intermed.2008, 38, 35–36.Suche in Google Scholar
[38] Saari, R.; Torma, J. C.; Nevalainen, T. Microwave-assisted synthesis of quinoline, isoquinoline, quinoxaline and quinazoline derivatives as CB2 receptor agonists. Bioorg. Med. Chem.2011, 19, 939–950.10.1016/j.bmc.2010.11.059Suche in Google Scholar
[39] Ighilahriz, K.; Boutemeur, B. Chami, F.; Rabia, C.; Hamdi, M.; Hamdi, S. M. A microwave-assisted and heteropolyacids-catalysed cyclocondensation reaction for the synthesis of 4(3H)-quinazolinones. Molecules2008, 13, 779–789.10.3390/molecules13040779Suche in Google Scholar
[40] Sharif, M.; Opalach, J.; Langer, P; Beller, M.; Wu, X.-F. Oxidative synthesis of quinazolinones and benzothiadiazine 1,1-dioxides from 2-aminobenzamide and 2-aminobenzenesulfonamide with benzyl alcohols and aldehydes. RSC Advances2014, 4, 8–17.10.1039/C3RA45765FSuche in Google Scholar
[41] Liu, X.; Fu, H.; Jiang, Y.; Zhao, Y. A simple and efficient approach to quinazolinones under mild copper-catalyzed conditions. Angew. Chem. Int. Ed.2009, 121, 354–357.10.1002/ange.200804675Suche in Google Scholar
[42] Heravi, M. M.; Sadjadi, S.; Haj, N. M.; Oskooie, H. A.; Shoar, R. H.; Bamoharram, F. F. A novel multi-component synthesis of 4-arylaminoquinazolines. Tetrahedron Lett.2009, 50, 943–945.10.1016/j.tetlet.2008.12.044Suche in Google Scholar
[43] Szczepankiewicz, W.; Suwinski, J.; Bujok, R. Synthesis of 4-arylaminoquinazolines and 2-aryl-4-arylaminoquinazolines from 2-aminobenzonitrile, anilines and formic acid or benzaldehydes. Tetrahedron2001, 32, 9343–9349.10.1016/S0040-4020(00)00899-1Suche in Google Scholar
[44] Chandregowda, V.; Rao, G. V.; Reddy, G. C. Convergent approach for commercial synthesis of gefitinib and erlotinib. Org. Process Res. Dev.2007, 813–816.10.1021/op700054pSuche in Google Scholar
[45] Wang, Z.; Wang, C.; Sun, Y.; Zhang, N.; Liu, Z.; Liu, J. A novel strategy to the synthesis of 4-anilinoquinazoline derivatives. Tetrahedron2014, 70, 906–913.10.1016/j.tet.2013.12.028Suche in Google Scholar
[46] Wang, X.; Jiao, N. Rh- and Cu-cocatalyzed aerobic oxidative approach to quinazolines via [4 + 2] C-H annulation with alkyl azides. Org. Lett.2016, 18, 2150–2153.10.1021/acs.orglett.6b00774Suche in Google Scholar PubMed
[47] Wang, J.; Zha, S.; Chen, K.; Zhang, F.; Song, C.; Zhu, J. Quinazoline synthesis via Rh(III)-catalyzed intermolecular C-H functionalization of benzimidates with dioxazolones. Org. Lett.2016, 18, 2062–2065.10.1021/acs.orglett.6b00691Suche in Google Scholar PubMed
©2018 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- Review
- Research progress in quinazoline derivatives as multi-target tyrosine kinase inhibitors
- Research Articles
- A bio-inspired approach to proline-derived 2,4-disubstituted oxazoles
- Solvent-free preparation of 3-aryl-2-[(aryl)(arylamino)]methyl-4H-furo[3,2-c]chromen-4-one derivatives using ZnO-ZnAl2O4 nanocomposite as a heterogeneous catalyst
- Synthesis of 4-arylethyl-6-arylpyrimidine-2-thiols through aza-Michael addition/nucleophilic addition/aromatization tandem reactions
- 3-Carboxy-1-sulfopyridin-1-ium chloride ([CPySO3H]+Cl−): an efficient catalyst for one-pot synthesis of hexahydroquinoline-3-carboxamides
- Synthesis of new 3H-chromeno[2,3-d]pyrimidine-4,6(5H,7H)-diones via the tandem intramolecular Pinner/Dimroth rearrangement
- A new synthesis of pyrrolo[3,2-d]pyrimidine derivatives by a one-pot, three-component reaction in the presence of L-proline as an organocatalyst
- Diversity-oriented synthesis of amide derivatives of tricyclic thieno[2,3-d]pyrimidin-4(3H)-ones and evaluation of their influence on melanin synthesis in murine B16 cells
- Synthesis, crystal structure, molecular docking studies and bio-evaluation of some N4-benzyl-substituted isatin- 3-thiosemicarbazones as urease and glycation inhibitors
- Ultrasound-assisted synthesis and antimicrobial activity of tetrazole-based pyrazole and pyrimidine derivatives
Artikel in diesem Heft
- Frontmatter
- Review
- Research progress in quinazoline derivatives as multi-target tyrosine kinase inhibitors
- Research Articles
- A bio-inspired approach to proline-derived 2,4-disubstituted oxazoles
- Solvent-free preparation of 3-aryl-2-[(aryl)(arylamino)]methyl-4H-furo[3,2-c]chromen-4-one derivatives using ZnO-ZnAl2O4 nanocomposite as a heterogeneous catalyst
- Synthesis of 4-arylethyl-6-arylpyrimidine-2-thiols through aza-Michael addition/nucleophilic addition/aromatization tandem reactions
- 3-Carboxy-1-sulfopyridin-1-ium chloride ([CPySO3H]+Cl−): an efficient catalyst for one-pot synthesis of hexahydroquinoline-3-carboxamides
- Synthesis of new 3H-chromeno[2,3-d]pyrimidine-4,6(5H,7H)-diones via the tandem intramolecular Pinner/Dimroth rearrangement
- A new synthesis of pyrrolo[3,2-d]pyrimidine derivatives by a one-pot, three-component reaction in the presence of L-proline as an organocatalyst
- Diversity-oriented synthesis of amide derivatives of tricyclic thieno[2,3-d]pyrimidin-4(3H)-ones and evaluation of their influence on melanin synthesis in murine B16 cells
- Synthesis, crystal structure, molecular docking studies and bio-evaluation of some N4-benzyl-substituted isatin- 3-thiosemicarbazones as urease and glycation inhibitors
- Ultrasound-assisted synthesis and antimicrobial activity of tetrazole-based pyrazole and pyrimidine derivatives