
Microwave-assisted and conventional synthesis of novel antimicrobial 1,2,4-triazole derivatives containing nalidixic acid skeleton
-
Sule Ceylan
 ,
Hacer Bayrak
,
Hacer Bayrak

Abstract
Carbothioamides 4a,b, obtained from nalidixic acid, were converted to the corresponding 1,3-thiazolidine derivatives 5a,b by cyclocondensation with 2-bromo-1-(4-chlorophenyl)ethanone. Treatment of 4a,b with base afforded 1,2,4-triazoles 6a,b. The synthesis of 1,3-oxazolidine 7 was performed by the reaction of compound 4a with ethyl bromoacetate. Treatment of 4a with acid produced 1,3,4-thiadiazole 8. The reaction of compounds 6a and 6b with several heterocyclic amines in the presence of formaldehyde gave the corresponding Mannich bases 9–15 containing various pharmacophore groups. Conventional and microwave-assisted methods were used for the synthesis. The effect of an acid catalyst on Mannich reactions was investigated. The structures of the newly synthesized compounds were elucidated on the basis of 1H NMR, 13C NMR, FTIR, EIMS techniques, and elemental analysis. All compounds were screened for their antimicrobial activity.
Introduction
Nalidixic acid [1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid, 1] was the first synthetic quinolone derivative introduced for the treatment of urinary tract infections in 1963 [1]. It inhibits DNA synthesis by promoting cleavage of bacterial DNA in the DNA-enzyme complexes of DNA gyrase, resulting in rapid bacterial death [2]. It is particularly effective against Gram-negative bacteria [3], [4]. Since the introduction of nalidixic acid for the treatment of bacterial infections, a number of derivatives have been synthesized. Among them, fluoroquinolones such as norfloxacin, pefloxacin, enoxacin, ofloxacin, and ciprofloxacin play a major role in the treatment of bacterial infections by displaying excellent pharmacokinetic properties, high antimicrobial activity, and low side effects [5], [6], [7], [8], [9]. Quinolones have been reported to have wide-ranging biological activities including antitubercular [10], fungicidal [11], antimalarial [12], anticancer and anti-HIV [12], antiviral [13], and antibacterial [14] properties.
The considerable biological importance of triazoles has stimulated a lot of interest in their derivatives [15]. A large number of 1,2,4-triazoles exhibit antibacterial, antifungal, antitubercular, analgesic, antiinflammatory, anticancer, anticonvulsant, antiviral, insecticidal, antidepressant, and central nervous system-affecting activities. 1,2,4-Triazoles, by virtue of their ambident nucleophilic centers, are good starting materials for the synthesis of several interesting N- and S-bridged heterocycles. The triazoles can be converted to thiazolotriazoles, triazolothiadiazoles, triazolooxazolidines, and various Mannich bases [16], [17], [18], [19], [20]. In addition, a 1,3,4-thiadiazole system constitutes the active part of several biologically active compounds [21], [22], [23], [24]. A thiazolidinone ring is an attractive target for combinatorial synthesis as its structure-activity relationship belongs to an important class of N- and S-containing heterocycles, which are widely used in drug design and synthesis [25], [26]. Related oxazolidinones are also biologically active [27], [28], [29], [30].
In this paper, we report the synthesis of new hybrid compounds which are derivatives of the systems mentioned above and possess an interesting profile of antimicrobial activity. The synthetic work was enhanced by using Mannich reaction [31], homogeneous catalysis [32], and eco-friendly microwave irradiation [33].
Results and discussion
Chemistry
The main aim of this study was to synthesize antimicrobial hybrid molecules containing different heterocyclic moieties. The synthetic strategies adopted to obtain the target compounds are depicted in Schemes 1–3. The synthesis was achieved by using both microwave (eco-friendly) and conventional methods. The structures of all products were established on the basis of physicochemical, spectral, and elemental analysis data.
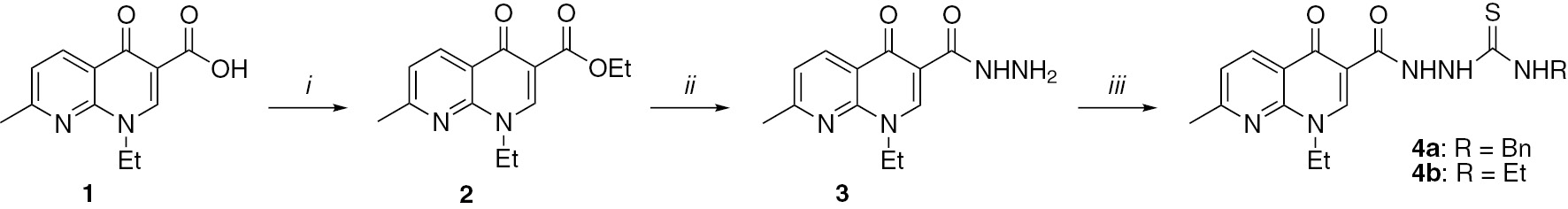
Reactions and conditions. i: H2SO4, EtOH, 22 h reflux or MW irradiation; ii: EtOH, H2NNH2, 6 h reflux or MW irradiation; iii: CH2Cl2, PhCH2NCS or EtNCS, 24 h room temprature or MW irradiation.
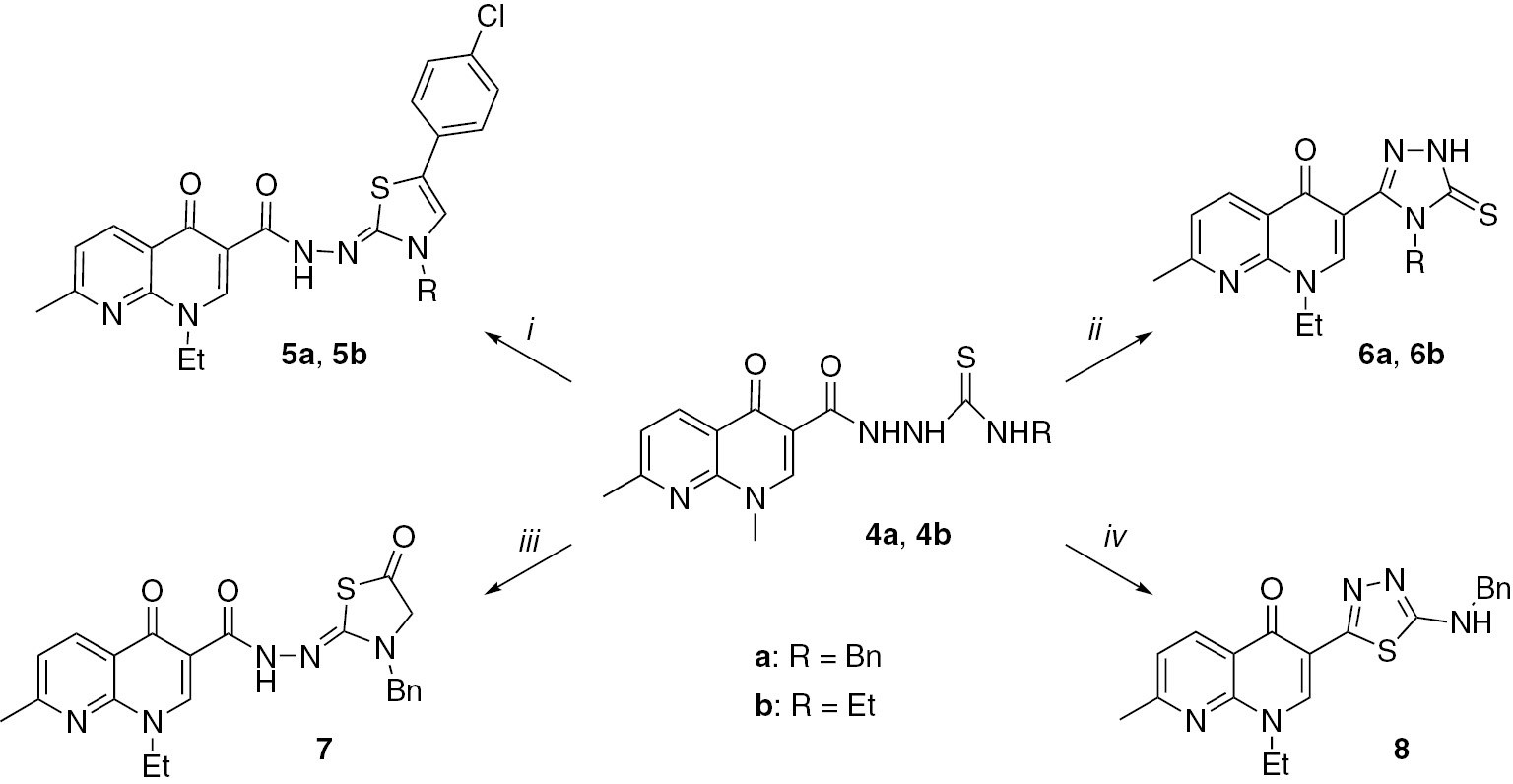
Reactions and conditions. i: EtOH, ClC6H4COCH2Br, CH3COONa, reflux for 54 h (5a) or EtOH, C2H5Br, CH3COONa, reflux for 45 h (5b) or MW irradiation; ii: 2 N NaOH in EtOH/H2O, 16 h (6a) or 14 h (6b) reflux or MW irradiation; iii: BrCH2COOEt in EtOH, CH3COONa, 24 h reflux or MW irradiation; iv: H2SO4, 3 h at room temperature or MW irradiation.
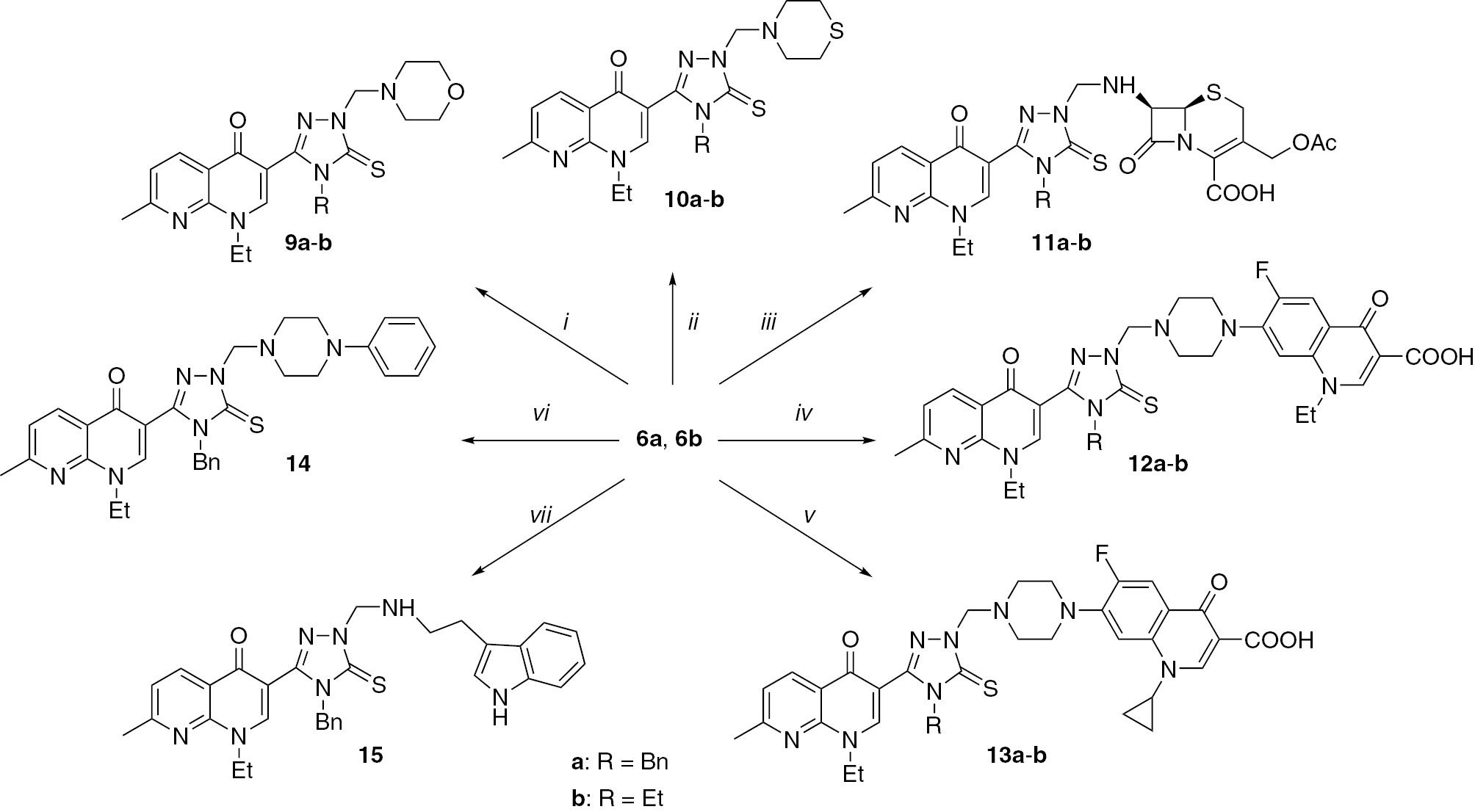
Conditions: amine, CH2O in DMSO (see Experimental). The amines: i, morpholine; ii, thiomorpholine; iii, 7-aminocephalosporanic acid; iv, norfloxacin; v, ciprofloxacin; vi, 1-phenylpiperazine; vii, tryptamine.
The esterification of nalidixic acid (1) with ethanol in the presence of a catalytic amount of sulfuric acid furnished ester 2. Then, compound 2 was converted to the acetohydrazide 3 by treatment with hydrazine hydrate. Synthesis of 3 required 20–22 h of heating under reflux conditions and the yield was 75–78%. Under microwave irradiation at 100°C for 15 min, the yield was 88–98%. Compound 3 was converted to carbothioamides 4a,b by treatment with benzylisothiocyanate and ethylisothiocyanate, respectively (Scheme 1). Again, the use of microwave irradiation was superior to the conventional synthesis. Condensation of compounds 4a,b with 4-chlorophenacyl bromide in ethanolic solution furnished the corresponding thiazoles 5a,b (Scheme 2). Again, the microwave method was preferred. The remaining preparations shown in Schemes 2 and 3 also benefited from the use of the microwave irradiation methodology. Triazoles 6a,b were obtained by intramolecular cyclization of compounds 4a,b. The aim was to introduce the 1,2,4-triazole nucleus to the nalidixic acid skeleton because it is known that more efficacious antibacterial compounds can be designed by joining two biologically active components together into a single molecular framework [34], [35], [36], [37], [38]. The synthesis of carbohydrazide 7 was performed by the reaction of compound 4a with ethyl bromoacetate in ethanolic solution, while the cyclization of 4a in an acidic medium yielded a 1,3,4-thiadiazole 8.
Mannich bases are physiologically reactive agents [39], [40], [41]. Mannich bases 9–15 were obtained by treatment of compounds 6a,b with formaldehyde and several biologically active amines (Scheme 3) [6], [8], [34], [42], [43]. Three different methods were used for the Mannich reactions. These were a conventional method in the absence of a catalyst, a conventional method in the presence of a catalyst, and microwave irradiation-assisted synthesis. The classical heating in the absence of any catalyst furnished the desired products in low-yield after 24 h. The same products were obtained in the yields of 53–97% at room temperature after 5 h in the presence of InCl3 or p-toluenesulfonic acid as a Brønsted-Lowry acid catalyst. In the microwave irradiation-assisted method the temperature was held constant at 50oC, the reaction time was 5 min and the Mannich bases were obtained in yields of 43–99%. Thus, microwave irradiation provided more efficient and green way to Mannich products and a relatively higher yield.
Antimicrobial activity
All synthesized compounds were screened for their antimicrobial activity. Compounds 2, 3, 4a,b, 5a,b, 6a,b, 7, and 8 are active against some of the test microorganisms with the minimal inhibition concentration (MIC) values in the range of 0.49–500 μg/mL (Table 1). Compounds 4a,b and 6a,b are exceptionally active against Escherichia coli (E. coli), Gram-negative bacteria. Mannich bases 9–15 show good-to-moderate activity against some of the test microorganisms. Among these, compounds 11a,b with a β-lactam nucleus attached to nalidixic acid skeleton display remarkable antimicrobial activities against two Gram-positive bacteria. Compound 11a is active against Mycobacterium smegmatis (M. smegmatis), a nonpigmented rapidly growing mycobacterium with the MIC value <1 μg/mL, which is better than that for standard drug streptomycin. Compound 11b is active against Enterococcus faecalis (E. faecalis), for which the MIC value of 6 μg/mL is better than that for the reference drug ampicillin. Exceptional activity can be observed for some other Mannich bases (Table 1).
Antimicrobial activity of the compounds.
| Comp. | Minimal inhibition concentration (μg/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ec | Yp | Pa | Sa | Ef | Bc | Ms | Ca | Sc | |
| 2 | 3.9 | 15.6 | – | – | – | 125 | – | 250 | 500 |
| 3 | 1.9 | 3.9 | 125 | 62.5 | 125 | 31.3 | – | 62.5 | 62.5 |
| 4a | 0.49 | 7.81 | 500 | 31.25 | – | 7.81 | 62.5 | 15.65 | 15.65 |
| 4b | 0.49 | 7.81 | – | 125 | 125 | 15.65 | 62.5 | 31.25 | 15.65 |
| 5a | 7.8 | 3.9 | – | – | – | 78 | – | – | – |
| 5b | 7.8 | 31.25 | – | – | – | 39 | – | – | – |
| 6a | 0.98 | 7.81 | – | 125 | – | 31.25 | – | 250 | – |
| 6b | 0.98 | 3.91 | 125 | 62.5 | – | 31.25 | 62.5 | – | – |
| 7 | 7.8 | 31.25 | – | – | 156 | 39 | – | – | – |
| 8 | 7.8 | 3.9 | – | – | 312.5 | >1000 | 125 | 78 | 39 |
| 9a | 7.8 | 31.25 | – | – | – | 39 | – | – | – |
| 9b | 3.9 | 62.5 | 156 | – | – | >1000 | 62.5 | – | – |
| 10a | 15.6 | 3.9 | – | – | – | 625 | – | – | 156 |
| 10b | 3.9 | 62.5 | 156 | – | 312.5 | >1000 | – | – | – |
| 11a | 125 | 7.8 | – | – | 156 | >1000 | <1 | – | 312.5 |
| 11b | 15.6 | 31.25 | 156 | – | 6 | 39 | – | – | – |
| 12a | <1 | 3.9 | 9.7 | 78 | 9.7 | 19 | 1.9 | – | 156 |
| 12b | <1 | 3.9 | 9.7 | – | 9.7 | 19 | 1.9 | – | – |
| 13a | <1 | <1 | 9.7 | 9.7 | – | 9.7 | <1 | – | 156 |
| 13b | <1 | <1 | 9.7 | – | 9.7 | 9.7 | <1 | – | – |
| 14 | 7.8 | 3.9 | – | – | – | 156 | – | – | – |
| 15 | 7.8 | 62.5 | – | – | – | 39 | – | – | 312.5 |
| Amp | 10 | 32 | >128 | 35 | 10 | 15 | |||
| Strep | 4 | ||||||||
| Flu | <8 | <8 | |||||||
Ec, E.coli ATCC 35218; Yp, Y. pseudotuberculosis ATCC 911; Pa, P. aeruginosa ATCC 10145; Sa, S. aureus ATCC 25923; Ef, E. faecalis ATCC 29212; Bc, B. cereus 709 Roma; Ms, M. smegmatis ATCC607; Ca, C. albicans ATCC 60193; Sc, S. cerevisiae RSKK 251; Amp, ampicillin; Strep, streptomycin; Flu, fluconazole; (–), no activity at test concentrations.
Conclusions
Several biologically active hybrid compounds containing a nalidixic acid moiety were obtained. The antimicrobial screening showed that compounds containing norfloxacine, ciprofloxacine or 7-aminocephalosporanic acid system display excellent antimicrobial activity. Several compounds are more active against E. coli and M. smegmatis than the reference drug ampicillin and streptomycin.
Experimental
All chemicals were purchased from Fluka Chemie AG (Buchs, Switzerland) and used without further purification. Melting points were determined in open capillaries on a Büchi B-540 melting point apparatus and are uncorrected. Reactions were monitored by thin layer chromatography (TLC) on silica gel using 60 F254 aluminum sheets. The mobile phase was ethyl acetate/diethyl ether (1:1) and detection was under UV light. FT-IR spectra were recorded using a Perkin Elmer 1600 series FT-IR spectrometer. 1H NMR and 13C NMR spectra were registered in DMSO-d6 on a Bruker Avene II 400 MHz NMR spectrometer (400 MHz for 1H and 100 MHz for 13C). The elemental analysis was performed on a Costech Elemental Combustion System CHNS-O elemental analyzer.
Ethyl 1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylate (2)
Method 1
A solution of nalidixic acid (1, 2.31 g, 0.01 mol) and concentrated sulfuric acid (4 drops) in ethanol was heated under reflux for 22 h, then cooled to room temperature and concentrated under reduced pressure. The resultant white residue of 2 was crystallized from ethanol.
Method 2
A solution of compound 1 (1 mmol) and sulfuric acid (4 drops) in ethanol was microwave irradiated in a closed vessel at 100°C for 15 min and then poured into ice water. The resultant solid product 2 was collected by filtration and crystallized from ethanol.
Yield 76% (method 1); yield 88% (method 2); mp 207–208°C; IR (υmax, cm-1): 3072, 2980, 1720, 1683, 1616; 1H NMR: δ 1.3 (t, 3H J = 7.2 Hz), 1.4 (t, 3H, J = 6.9 Hz), 2.6 (s, 3H), 4.2 (q, 2H, J = 6.9 Hz,), 4.5 (q, 2H, J = 7.2 Hz), 7.4 (d, 1H, J = 8.1 Hz,), 8.4 (d, 1H, J = 8.1 Hz), 8.8 (s, 1H); 13C NMR: δ 14.3, 23.6, 27.9, 41.1, 44.5, 110.8, 118.2, 125.6, 136.2, 148.9, 154.6, 148.1, 162.2, 176.3. Anal. Calcd for C14H16N2O3: C, 64.60; H, 6.20; N, 10.76. Found: C, 64.25; H, 5.93; N, 10.41.
1-Ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carbohydrazide (3)
Method 1
A solution of compound 2 (2.60 g, 0.01 mol) and hydrazine hydrate (1.46 mL, 0.03 mol) in ethanol was heated under reflux for 20 h, then cooled and kept in the refrigerator for 12 h. The resultant solid was collected by filtration and crystallized from ethanol.
Method 2
A solution of compound 2 (1 mmol) and hydrazine hydrate (3 mmol) in ethanol was microwave-irradiated in a closed vessel at 100°C for 15 min. After the completion of the reaction, as monitored by TLC, the mixture was concentrated and the residue was crystallized from ethanol.
Yield 75% (method 1); yield 98% (method 2); mp 163–165°C; IR (υmax, cm-1): 3316, 3222, 3033, 2958, 1685, 1604; 1H NMR: δ 1.32 (s, 3H), 2.60 (s, 3H), 4.51 (s, 2H), 7.40 (s, 1H), 8.42 (s, 1H), 8.94 (s, 1H), 10.50 (s, 2H), 12.50 (s, 1H); 13C NMR: δ 14.3, 24.8, 40.1, 111.6, 119.4, 122.5, 135.8, 147.9, 153.6, 147.2, 163.1, 175.8. Anal. Calcd for C12H14N4O2: C, 58.53; H, 5.73; N, 22.75. Found: C, 58.40; H, 5.93; N, 22.48.
General synthesis of compounds 4a,b
Method 1
To a solution of compound 3 (2.46 g, 0.01 mol) in dichloromethane, benzyl isothiocyanate (for 4a, 2.46 g, 0.01 mol) or ethyl isothiocyanate (for 4b, 1.75 g, 0.02 mol), was added and the mixture was stirred at room temperature for 24 h and then concentrated under reduced pressure. The residue was crystallized from ethanol (4a) or from dimethyl sulfoxide/water (1:3) (4b).
Method 2
A mixture of compound 3 (1 mmol) and the corresponding isothiocyanate (1 mmol) in ethanol was irradiated in a closed vessel with the pressure control at 100°C for 10 min. After cooling the mixture was poured into ice water and the resultant solid was crystallized from an appropriate solvent.
N-Benzyl-2-[(1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-3-yl)carbonyl]hydrazinecarbothioamide (4a)
Yield 80% (method 1); yield 98% (method 2); mp 197–200°C; IR (υmax, cm-1): 3137, 3062, 2974, 1649, 1541, 1187; 1H NMR: δ 1.40 (t, 3H, J = 8.0 Hz), 2.71 (brs, 3H), 4.73 (d, 2H, J = 4.0 Hz), 4.94 (s, 2H), 7.29 (d, 1H, J = 8.0 Hz), 7.31–7.38 (m, 5H), 7.42 (d, 1H, J = 8.0 Hz), 8.58 (q, 1H, J = 8.0 Hz), 9.04 (s, 1H), 9.18 (s, 1H), 09.47 (s, 1H); 13C NMR: δ 15.5, 25.5, 46.6, 48.5, 119.1, 122.6, 123.4, 127.6, 127.7, 128.7, 129.3, 136.5, 148.6, 163.9, 165.1, 166.1, 150.0, 175.9, 178.5, 183.1. Anal. Calcd for C20H21N5O2S: C, 60.74; H, 5.35; N, 17.71. Found: C, 60.40; H, 5.83; N, 17.48.
N-Ethyl-2-[(1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-3-yl)carbonyl]hydrazinecarbothioamide (4b)
Yield 75% (method 1); yield 85% (method 2); mp 180–182°C; IR (υmax, cm-1): 3144, 3058, 2986, 1679, 1541, 1189; 1H NMR: δ 1.07 (t, 3H, J = 8.0 Hz), 1.39 (t, 3H, J = 8.0 Hz), 2.69 (brs, 3H), 3.41 (brs, 2H), 4.57 (q, 2H, J = 8.0 Hz), 7.44–7.47 (m, 4H), 7.56 (d, 1H, J = 8.0 Hz), 7.92 (brs, 2H), 8.53 (q, 1H, J = 8.0 Hz), 9.00 (s, 1H), 9.15 (s, 1H), 10.54 (s, 1H); 13C NMR: δ 14.8, 15.5, 25.5, 46.4, 47.2, 121.8, 123.0, 136.3, 148.5, 163.9, 165.1, 149.9, 175.8, 178.5, 182.1. Anal. Calcd for C15H19N5O2S: C, 54.04; H, 5.74; N, 21.01. Found: C, 54.40; H, 5.93; N, 21.10.
General synthesis of compounds 5a,b
Method 1
4-Chlorophenacyl bromide (2.33 g, 0.01 mol) and sodium acetate (4.1 g, 0.05 mol) were added to the solution of compound 4a (3.95 g, 0.01 mol, for 5a) or compound 4b (3.33 g, 0.01 mol, for 5b) in ethanol and the mixture was heated under reflux for 54 h (for 5a) or 45 h (for 5b), as monitored by TLC. Then, the mixture was cooled to room temperature and concentrated under reduced pressure. The resultant solid was filtered, washed with water and crystallized from dimethyl sulfoxide/water (1:3).
Method 2
A solution of 4-chlorophenacyl bromide (1 mmol), sodium acetate (5 mmol), compound 4a (1 mmol) or compound 4b (1 mmol) in ethanol was microwave-irradiated in a closed vessel at 150°C for 15 min. After completion of the reaction, as monitored by TLC, the mixture was poured into ice water. The crude product was filtered and crystallized from dimethyl sulfoxide/water (1:3).
N′-[3-Benzyl-5-(4-chlorophenyl)-1,3-thiazol-2(3H)-ylidene]-1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carbohydrazide (5a)
Yield 65% (method 1); yield 80% (method 2); mp 175–177°C; IR (υmax, cm-1): 3043, 3079, 2980, 1716, 1521, 1129; 1H NMR: δ 1.41 (brs, 3H), 2.67 (t, 3H, J = 8.0 Hz), 4.24 (brs, 2H, 4.41 (q, 2H, J = 8.0 Hz), 5.03 (s, 1H), 7.05 (d, 1H, J = 8.0 Hz), 7.17–7.63 (m, 9H), 7.62 (d, 1H, J = 8.0 Hz), 7.98 (brs, 1H), 9.17 (s, 1H); 13C NMR: δ 14.7, 25.5, 43.6, 47.3, 110.0, 117.2, 123.1, 127.8, 128.3, 129.5, 130.2, 130.3, 135.4, 136.1, 136.3, 140.3, 147.6, 148.1, 160.0, 166.2, 149.8, 166.8, 168.8, 176.1, 178.6. Anal. Calcd for C28H24ClN5O2S: C, 65.45; H, 4.56; N, 13.21. Found: C, 65.31; H, 4.78; N, 13.01.
N′-[5-(4-Chlorophenyl)-3-ethyl-1,3-thiazol-2(3H)-ylidene]-1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carbohydrazide (5b)
Yield 70% (method 1); yield 87% (method 2); mp 192–193°C; IR (υmax, cm-1): 3068, 3068, 2971, 1702, 1519; 1H NMR: δ 1.05 (t, 3H, J = 8.0 Hz), 1.41 (t, 3H J = 4.0 Hz), 2.70 (brs, 3H), 3.67 (brs), 4.60 (q, 2H, J = 8.0 Hz), 5.03 (s, 1H), 7.48–7.58 (m, 6H), 7.58 (brs, 1H), 9.18 (s, 1H); 13C NMR: δ 13.3, 15.5, 25.5, 46.6, 47.3, 98.9, 109.1, 119.5, 120.0, 121.0, 123.1, 129.4, 129.5, 130.1, 131.0, 136.1, 148.8, 159.9, 150.1, 163.8, 166.0, 171.6, 178.6. Anal. Calcd for C23H22ClN5O2S: C, 59.03; H, 4.74; N, 14.97. Found: C, 59.33; H, 4.38; N, 14.69.
General synthesis of compounds 6a,b
Method 1
A solution of carbothioamide 4a (3.95 g, 0.01 mol, for 6a) or 4b (3.33 g, 0.01 mol, for 6b) in ethanol/water (1:1) was heated under reflux in the presence of 2 N NaOH for 16 h (for 6a) or 14 h (for 6b) with the progress of the reaction monitored by TLC. Then, the solution was cooled to room temperature and acidified to pH 7 with 37% HCl. The precipitate formed was filtered off, washed with water, and crystallized from methanol (for 6a) or ethanol /water (1:3) (for 6b).
Method 2
A mixture of compound 4a or 4b (1 mmol) and 2N NaOH (2 mmol) in ethanol (10 mL) was microwave-irradiated in a closed vessel at 150°C for 4 min with the progress of the reaction monitored by TLC. Then the resulting solution was cooled to room temperature and acidified to pH 5 with 37% HCl. The precipitate formed was filtered off, washed with water, and crystallized from an appropriate solvent indicated above.
3-(4-Benzyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)-1-ethyl-7-methyl-1,8-naphthyridin-4(1H)-one (6a)
Yield 63% (method 1); yield 86% (method 2); mp >270°C; IR (υmax, cm-1): 3061, 2985, 2959, 1717, 1517, 1128; 1H NMR: δ 1.17 (s, 3H), 2.50 (s, 3H), 4.33 (brs, 2H), 5.25 (s, 2H), 7.13–7.33 (m, 7H), 8.34 (s, 1H), 9.16 (s, 1H); 13C NMR: δ 15.4, 25.5, 45.6, 47.3, 109.2, 118.8, 121.6, 123.1, 127.2, 127.9, 128.8, 136.1, 136.9, 146.5, 148.8, 163.7, 150.0, 166.1, 174.7, 178.5. Anal. Calcd for C20H19N5OS: C, 63.64; H, 5.07; N, 18.55. Found: C, 63.28; H, 5.38; N, 18.89.
1-Ethyl-3-(4-ethyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)-7-methyl-1,8-naphthyridin-4(1H)-one (6b)
Yield 67% (method 1); yield 88% (method 2); mp 216-218°C; IR (υmax, cm-1): 3090, 2988, 2957, 1717 2, 1537, 1130; 1H NMR: δ 1.14 (t, 3H, J = 8.0 Hz), 1.41 (t, 3H, J = 8.0 Hz), 2.66 (s, 3H), 3.93 (q, 2H, J = 8.0 Hz), 4.49 (q, 2H, J = 8.0 Hz), 7.44–7.61 (m, 2H), 8.61 (d, 1H, J = 8.0 Hz), 9.19 (s, 1H); 13C NMR: δ 13.8, 15.5, 25.5, 45.9, 47.3, 108.8, 119.5, 121.9, 136.2, 149.1, 163.6, 148.5, 167.1, 174.9, 178.6. Anal. Calcd for C15H17N5OS: C, 57.12; H, 5.43; N, 22.21. Found: C, 57.28; H, 5.87; N, 22.60.
N′-[3-Benzyl-5-thioxo-1,3-oxazolidin-2-ylidene]- 1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carbohydrazide (7)
Method 1
Ethyl bromoacetate (1.28 mL, 0.01 mol) and sodium acetate (4.1 g, 0.05 mol) were added to the solution of compound 4a (3.95 g, 0.01 mol) in ethanol and the mixture was heated under reflux for 24 h. Then, the mixture was cooled to room temperature and concentrated under reduced pressure. The residue was filtered, washed with water, and crystallized from dimethyl sulfoxide/water (1:3).
Method 2
A mixture of compound 4a (1 mmol), ethyl bromoacetate (1 mmol) and sodium acetate (5 mmol) in ethanol was irradiated in a closed vessel at 150°C for 13 min with the progress of the reaction monitored by TLC. After the completion of the reaction, the mixture was poured into ice water and the resultant precipitate was filtered and crystallized from dimethyl sulfoxide/water (1:3).
Yield 55% (method 1); yield 76% (method 2); mp 230–232°C; IR (υmax, cm-1): 3031, 1698 2, 1495, 1127; 1H NMR: δ 1.42 (s, 3H), 4.23 (s, 2H), 4.63 (t, 3H, J = 8.0 Hz), 4.79 (s, 2H), 7.26-742 (m, 7H), 8.59 (t, 1H, J = 8.0 Hz), 9.03 (s, 1H); 13C NMR: δ 15.5, 25.4, 40.4, 44.1, 45.9, 109.1, 118.8, 123.1, 127.0, 127.3, 127.6, 127.7, 128.0, 136.1, 136.3, 148.8, 160.1, 150.1, 165.1, 166.1, 172.2, 178.6. Anal. Calcd for C22H21N5O3S: C, 60.67; H, 4.86; N, 16.08. Found: C, 60.32; H, 4.49 H; N, 16.28.
3-[5-(Benzylamino)-4,5-dihydro-1,3,4-thiadiazol-2-yl]-1-ethyl-7-methyl-1,8-naphthyridin-4(1H)-one (8)
Method 1
A solution of carbothioamide 4a (3.95 g, 0.01 mol) in concentrated sulfuric acid (11 mL, 0.20 mol) was stirred for 15 min at 0°C, then allowed to reach room temperature and stirred for an additional 3 h. The solution was poured into ice-cold water and made alkaline to pH 7 with ammonia. The precipitated product was filtered, washed with water, and crystallized from ethanol/water (1:3).
Method 2
A solution of compound 4a (1 mmol) in concentrated sulfuric acid (1.5 mL) was irradiated in a closed vessel at 50°C for 4 min with the progress of the reaction monitored by TLC. After completion of the reaction, the mixture was poured into ice water and the resultant crude product was collected by filtration and crystallized from ethanol/water (1:3).
Yield 74% (method 1); yield 83% (method 2); mp 193–195°C; IR (υmax, cm-1): 3197, 1715, 1518, 1080; 1H NMR: δ 1.41 (s, 3H), 2.67 (t, 3H, J = 8.0 Hz), 3.45 (brs, 2H), 4.63 (brs, 2H), 7.16–7.56 (m, 7H), 8.55 (brs, 1H), 9.16 (s, 1H); 13C NMR: δ 15.5, 25.5, 46.6, 47.9, 109.1, 119.9, 123.1, 127.3, 127.7, 128.6, 129.3, 136.0, 142.6, 148.9, 163.0, 165.1, 149.9, 166.0, 172.8, 178.5. Anal. Calcd for C20H19N5O2: C, 66.47; H, 5.30; N, 19.38. Found: C, 66.32; H, 4.89; N, 16.08.
General synthesis of compounds 9–13 (a,b)
Method 1
To a solution of compound 6a or 6b (0,01 mol) in dimethyl sulfoxide (10 mL), morpholine (for 9a and 9b, 0.87 mL, 0.01 mol) or thiomorpholine (for 10a and 10b, 0.94 mL, 0.01 mol) or 7-aminocephalosporanic acid (for 11a and 11b, 2.72 g, 0.01 mol) or norfloxacin (for 12a and 12b, 3.19 g, 0.01 mol) or ciprofloxacin (for 13a and 13b, 3.31 g, 0.01 mol) was added in the presence of formaldehyde (37%, 3.72 mL, 0.05 mol) and the mixture was stirred at room temperature for 22 h. The solution was poured into ice-cold water and the resultant solid was crystallized from ethanol/water (1:3) (for 9a,b, 10a,b), from ethyl acetate (for 11a,b) and from dimethyl sulfoxide/water (1:3) (for 12a,b, 13a,b).
Method 2
To a solution of compound 6a,b (1 mmol) in dimethyl sulfoxide (10 mL), morpholine (for 9a and 9b, 1 mmol) or thiomorpholine (for 10a and 10b, 1 mmol) or 7-aminocephalosporanic acid (for 11a and 11b, 1 mmol) or norfloxacin (for 12a and 12b, 1 mmol) or ciprofloxacin (for 13a and 13b, 1 mmol) was added in the presence of formaldehyde (5 mmol) and the mixture was stirred at room temperature for 15 min. Then, a catalytic amount of a suitable catalyst (InCl3 or p-TsOH) was added and the stirring was continued for an additional 5 h. The mixture was poured into ice water and a resultant solid was crystallized from an appropriate solvent indicated above.
Method 3
The mixture, obtained as described above for method 2, was irradiated at 50°C in a microwave oven for 5 min with the progress of the reaction monitored by TLC. After cooling the solution was poured into ice water and the resultant precipitate was filtered off and crystallized from an appropriate solvent indicated above.
3-[4-Benzyl-1-(morpholin-4-ylmethyl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl]-1-ethyl-7-methyl-1,8-naphthyridin-4(1H)-one (9a)
Yield 73% (method 1); yield 75% (method 2, InCl3); yield 77% (method 2, p-TsOH); yield 90% (method 3); mp 106–108°C; IR (υmax, cm-1): 1629, 1498, 1152, 1078; 1H NMR: δ 1.19 (t, 3H, J = 4.0 Hz), 2.56 (s, 4H), 2.73 (s, 3H), 3.36 (s, 4H), 4.37 (brs, 2H), 5.14 (s, 2H), 5.31 (s, 2H), 6.97 (brs, 2H), 7.15–7.30 (m, 5H), 7.46 (d, 2H J = 8.0 Hz), 8.52 (s, 1H); 13C NMR: δ 15.4, 25.3, 45.7, 48.6, 50.8, 66.6, 69.6, 108.6, 119.3, 121.8, 127.5, 127.8, 128.0, 128.8, 136.2, 136.3, 147.6, 148.8, 146.5, 163.7, 169.1, 174.7. Anal. Calcd for C25H28N6O2S: C, 63.00; H, 5.92; N, 17.63. Found: C, 63.28; H, 6.01; N, 17.32.
1-Ethyl-3-[4-ethyl-1-(morpholin-4-ylmethyl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl]-7-methyl-1,8-naphthyridin-4(1H)-one (9b)
Yield 65% (methods 1 and 2); yield 66% (method 2, p-TsOH); yield 85% (method 3); mp 209–210°C; IR (υmax, cm-1): 1710, 1519, 1162, 1081; 1H NMR: δ 1.17 (s, 3H), 1.40 (s, 3H), 2.68 (s, 3H), 3.35 (s, 4H), 3.57 (s, 4H), 3.95 (brs, 2H), 4.49 (s, 2H), 5.06 (s, 2H), 7.46 (d, 2H, J = 8.0 Hz), 8.46 (brs, 1H), 8.65 (s, 1H); 13C NMR: δ 13.6, 15.4, 25.4, 46.0, 50.8, 52.0, 66.5, 69.1, 108.2, 119.4, 121.6, 136.2, 147.2, 149.1, 146.8, 163.6, 168.1, 178.5. Anal. Calcd for C20H26N6O2S: C, 57.95; H, 6.32; N, 20.27. Found: C, 57.71; H, 6.01; N, 20.01.
3-[4-Benzyl-1-(thiomorpholin-4-ylmethyl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl]-1-ethyl-7-methyl-1,8-naphthyridin-4(1H)-one (10a)
Yield 40% (method 1); yield 53% (method 2, InCl3 or p-TsOH); yield 47% (method 3); mp 100–102°C; IR (υmax, cm-1): 3069, 2957, 1627, 1496, 1131; 1H NMR: δ 1.41 (t, 3H J = 8.0 Hz), 2.61 (s, 4H), 2.71 (s, 3H), 3.36 (s, 4H), 4.29 (q, 2H, J = 8.0 Hz), 4.64 (brs, 2H), 5.27 (s, 2H), 6.45 (brs, 1H), 7.00–7.95 (m, 7H), 8.51 (s, 1H); 13C NMR: δ 15.3, 25.3, 40.7, 45.7, 48.3, 52.3, 70.0, 108.7, 111.3, 117.8, 121.8, 127.2, 127.5, 127.7, 128.8, 132.9, 136.2, 136.2, 147.6, 146.5, 148.8, 163.7, 174.7. Anal. Calcd for C25H28N6OS2: C, 60.95; H, 5.73; N, 17.06. Found: C, 60.62; H, 5.91; N, 17.38.
1-Ethyl-3-[4-ethyl-1-(thiomorpholin-4-ylmethyl)-5-thioxo-4,5-dihydro-1H-1,2,4 -triazol-3-yl]-7-methyl-1,8-naphthyridin-4(1H)-one (10b)
Yield 35% (method 1); yield 55% (method 2, InCl3); yield 56% (method 2, p-TsOH); yield 43% (method 3); mp 192–193°C; IR (υmax, cm-1): 3087, 2980, 1710, 1518, 1128; 1H NMR: δ 1.17 (s, 3H), 1.40 (s, 3H), 2.67 (s, 3H), 2.99 (s, 4H), 3.34 (s, 4H), 3.95 (s, 2H), 4.49 (s, 2H), 5.08 (s, 2H), 7.45 (d, 2H, J = 8.0 Hz), 8.48 (brs, 1H), 8.63 (s, 1H); 13C NMR: δ 13.6, 15.5, 25.3, 46.0, 50.6, 52.8, 66.6, 70.4, 108.2, 119.4, 121.6, 136.2, 147.2, 149.1, 146.8, 163.6, 167.9, 178.5. Anal. Calcd for C20H26N6OS2: C, 55.79; H, 6.09; N, 19.52. Found: C, 55.61; H, 6.33; N, 19.28.
3-[(Acetoxy)methyl]-7-({[4-benzyl-3-(1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-3-yl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-1-yl]methyl}amino)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (11a)
Yield 55% (method 1); yield 64% (method 2, InCl3); yield 60% (method 2, p-TsOH); yield 74% (method 3); mp 148–150°C; IR (υmax, cm-1): 3305, 3057, 1771, 1715 4, 1556, 1130; 1H NMR: δ 1.41 (t, 3H, J = 8.0 Hz), 1.91 (s, 3H), 2.73 (brs, 3H), 3.43 (q, 2H, J = 4.0 Hz), 3.96 (s, 1H), 4.34 (brs, 2H), 5.02 (d, 2H, J = 8.0 Hz), 5.29 (brs, 2H), 5.45 (s, 2H), 5.54 (s, 1H), 7.04-7.95 (m, 7H), 8.58 (s, 1H), 9.17 (s, 1H), 14.04 (s, 1H); 13C NMR: δ 14.7, 22.01, 26.3, 27.0, 45.7, 47.3, 59.2, 63.2, 68.1, 71.3, 108.9, 109.1, 119.4, 123.0, 125.0, 127.3, 127.9, 128.8, 136.0, 136.2, 146.4, 148.8, 148.8, 120.5, 150.0, 163.5, 163.7, 167.3, 168.1, 174.7, 178.5. Anal. Calcd for C31H31N7O6S2: C, 62.26; H, 4.72; N, 14.82. Found: C, 62.01; H, 4.45; N, 14.51.
3-[(Acetoxy)methyl]-7-({[4-ethyl-3-(1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-3-yl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-1-yl]methyl}amino)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (11b)
Yield 58% (method 1); yield 69% (method 2, InCl3); yield 70% (method 2, p-TsOH); yield 81% (method 3); mp 140–142°C; IR (υmax, cm-1): 3280, 3047, 1768, 1710 4, 1562, 1130; 1H NMR: δ 1.15 (t, 3H, J = 8.0 Hz), 1.41 (t, 3H, J = 8.0 Hz), 1.91 (s, 3H), 2.71 (brs, 3H), 3.32 (brs, 2H), 3.92 (s, 1H), 4.47 (brs, 2H), 4.63 (brs, 2H), 5.01 (d, 2H, J = 8.0 Hz), 5.53 (s, 1H), 7.44 (d, 1H, J = 8.0 Hz), 7.59 (d, 1H, J = 8.0 Hz), 8.60 (s, 1H), 9.16 (s, 1H), 13.82 (s, 1H); 13C NMR: δ 13.8, 15.4, 21.5, 25.5, 27.1, 45.9, 47.3, 58.2, 64.3, 69.1), 72.0, 108.8, 109.8, 118.8, 123.1, 136.2, 148.5, 149.1, 119.4, 150.0, 163.5, 165.1, 166.0, 167.1, 174.9, 178.6. Anal. Calcd for C26H29N7O6S2: C, 52.07; H, 4.87; N, 16.35. Found: C, 52.31; H, 4.45; N, 16.51.
6-(4-{[4-Benzyl-3-(1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-3-yl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-1-yl]methyl}piperazin-1-yl)-1-ethyl-5-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (12a)
Yield 82% (method 1); yield 92% (method 2, InCl3); yield 91% (method 2, p-TsOH); yield 98% (method 3); mp 200–202°C; IR (υmax, cm-1): 3451, 1720, 1562, 1130; 1H NMR: δ 1.39 (t, 3H, J = 8.0 Hz), 1.96 (s, 3H), 2.64 (brs, 3H), 2.98 (s, 4H), 3.39 (brs, 4H), 4.34 (brs, 2H), 4.60 (brs, 2H), 5.25 (d, 4H, J = 8.0 Hz), 6.53 (s, 1H), 7.15–7.57 (m, 7H), 7.83 (brs, 1H), 8.47 (s, 1H), 8.92 (s, 1H), 14.89 (s, 1H); 13C NMR: δ 14.8 (CH3), 15.3 (CH3), 25.5 (CH3), 44.8 (N-2CH2), 46.4 (CH2), 47.2 (N-2CH2), 49.5, 49.9, 50.2, 106.0, 110.4, 112.3, 118.7, 119.6 and 119.7 (d, CH, J = 7.0 Hz), 121.8, 123.0, 126.1, 127.2 and 127.6 (d, CH, J = 47.0 Hz), 128.6 and 128.8 (d, CH, J = 17.0 Hz), 135.0 (CH), 135.9 and 136.2 (d, C, J = 26.0 Hz), 142.3 (C), 145.9 (CH), 146.5 and 147.7 (d, C, J = 122.0 Hz), 151.9 and 153.6 (d, C, J = 161.0 Hz), 165.1, 107.5, 108.4, 149.1, 149.9, 166.6, 169.1, 174.7, 176.5, 178.5. Anal. Calcd for C37H37FN8O4S: C, 62.70; H, 5.26; N, 15.81. Found: C, 62.31; H, 5.06; N, 16.03.
1-Ethyl-6-(4-{[4-ethyl-3-(1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-3-yl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-1-yl]methyl}piperazin-1-yl)-5-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (12b)
Yield 85% (method 1); yield 98% (method 2, InCl3); yield 92% (method 2, p-TsOH); yield 96% (method 3); mp 220–222°C; IR (υmax, cm-1): 3372, 1710, 1517, 1130; 1H NMR: δ 1.17 (s, 3H), 1.39 (t, 3H J = 8.0 Hz), 1.97 (s, 3H), 2.66 (brs, 3H), 2.95 (s, 4H), 3.36 (brs, 4H), 3.96 (s, 2H), 4.48 (brs, 2H), 4.56 (brs, 2H), 5.18 (s, 2H), 7.13 (s, 1H), 7.42 (brs, 1H), 7.54 (brs, 1H), 7.80 (brs, 1H), 8.88 (s, 1H), 9.03 (s, 1H), 15.27 (s, 1H); 13C NMR: δ 13.6, 14.8, 15.4, 25.5, 43.3, 45.8, 48.0, 49.5, 49.9, 50.2, 106.2, 109.8, 110.8, 119.4 and 119.5 (d, C, J = 15.0 Hz), 121.6 and 122.9 (d, CH, J = 136.0 Hz), 135.9 and 136.1 (d, CH, J = 17.0 Hz), 137.5, 145.8, 149.1 and 149.9 (d, C, J = 84.0 Hz), 154.1 and 156.1 (d, C, J = 200.0 Hz), 163.6 and 165.1 (d, C, J = 153.0 Hz), 107.5, 108.2, 149.2, 149.9, 166.5, 168.1, 174.9, 176.5, 178.4. Anal. Calcd for C32H35FN8O4S: C, 59.43; H, 5.45; N, 17.33. Found: C, 59.31; H, 5.15; N, 17.03.
6-(4-{[4-Benzyl-3-(1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-3-yl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-1-yl]methyl}piperazin-1-yl)-1-cyclopropyl-5-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (13a)
Yield 82% (method 1); yield 90% (method 2, InCl3); yield 97% (method 2, p-TsOH); yield 97% (method 3); mp >270°C; IR (υmax, cm-1): 3300, 1716 3, 1481, 1130; 1H NMR: δ 1.18 (s, 2H), 1.32 (s, 2H), 1.41 (t, 3H, J = 8.0 Hz), 2.69 (brs, 3H), 2.99 (s, 4H), 3.38 (brs, 4H), 3.83 (brs, 2H), 4.33 (d, 4H, J = 8.0 Hz), 4.66 (brs, 1H), 5.25 (s, 2H), 6.98 (s, 1H), 7.13–7.84 (m, 7H), 7.95 (s, 1H), 8.61 (s, 1H), 9.01 (s, 1H), 15.15 (s, 1H); 13C NMR: δ 8.0 (2CH2), 15.3, 25.5, 36.3, 44.3 (N-2CH2), 47.3 (N-2CH2), 48.4, 49.9, 50.2, 106.9, 118.8, 119.0 and 120.3 (d, C, J = 129.0 Hz), 122.7, 123.9, 127.2, 127.6, 127.9 and 127.9 (d, CH, J = 7.0 Hz), 128.7 and 128.8 (d, CH, J = 6.0 Hz), 136.0, 136.1 (2C), 138.5 and 139.5 (d, C, J = 100.0 Hz), 144.5, 146.1 and 147.7 (d, C, J = 162.0 Hz), 148.6, 162.8 and 163.7 (d, C, J = 88.0 Hz)], 107.2, 118.6, 148.3, 148.8, 166.4, 169.1, 174.7, 176.7, 178.5. Anal. Calcd for C37H37FN8O4S: C, 63.32; H, 5.17; N, 15.55. Found: C, 63.28; H, 5.06; N, 15.21.
1-Cyclopropyl-6-(4-{[4-ethyl-3-(1-ethyl-7-methyl-4-oxo-1,4-dihydro-1,8-naphthyridin-3-yl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-1-yl]methyl}piperazin-1-yl)-5-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (13b)
Yield 79% (method 1); yield 85% (method 2, InCl3); yield 88% (method 2, p-TsOH);. yield 95% (method 3); mp 228–230°C; IR (υmax, cm-1): 3456, 1710 3, 1558, 1159; 1H NMR: δ 1.18 (s, 2H), 1.32 (s, 2H), 1.40 (t, 3H, J = 8.0 Hz), 1.96 (s, 3H), 2.69 (brs, 3H), 2.97 (s, 4H), 3.33 (brs, 4H), 3.82 (brs, 2H), 4.49 (d, 4H, J = 8.0 Hz), 4.64 (brs, 1H), 5.25 (s, 2H), 7.44–7.79 (m, 4H), 8.61 (s, 1H), 9.05 (s, 1H), 15.08 (s, 1H); 13C NMR: δ 8.0, 15.3, 25.4, 27.7, 36.4, 43.8, 45.9, 47.2, 48.8, 49.7, 51.2, 105.9, 109.8, 119.0 and 119.6 (d, C, J = 50.0 Hz), 121.7, 123.9, 125.9 and 125.9 (d, CH, J = 7.0 Hz), 128.1 and 128.8 (d, CH, J = 76.0 Hz), 135.0, 139.5 and 141.5 (d, C, J = 200.0 Hz), 146.1 and 148.6 (d, C, J = 253.0 Hz), 161.9 and 162.6 (d, C, J = 72.0 Hz), 107.7, 108.6, 143.6, 149.1, 166.2, 169.1, 173.9, 175.8, 178.3. Anal. Calcd for C33H35FN8O4S: C, 60.17; H, 5.36; N, 17.01. Found: C, 60.31; H, 5.15; N, 17.37.
General synthesis of compounds 14 and 15
Method 1
To a solution of compound 6a (0.01 mol) in dimethyl sulfoxide, 1-phenylpiperazine (for 14, 1.52 mL, 0.01 mol) or tryptamine (for 15, 1.60 g, 0.01 mol) was added in the presence of formaldehyde (37%, 3.72 mL, 0.05 mol) and the mixture was stirred at room temperature for 24 h, then poured into ice-cold water. The resultant precipitate was crystallized from ethanol/water (1:3).
Method 2
To a solution of compound 6a (1 mmol) in dimethyl sulfoxide, 1-phenylpiperazine (for 14, 1 mmol) or tryptamine (for 15, 1 mmol) was added in the presence of formaldehyde (5 mmol) and the mixture was stirred at room temperature for 15 min. Then, a catalytic amount of a catalyst (InCl3 or p-TsOH) was added and the stirring was continued for an additional 5 h. The mixture was poured into ice water and the resultant precipitate was crystallized from ethanol/water (1:3).
Method 3
A solution, prepared as described above, was irradiated at 50oC in the microwave oven for 5 min, then cooled to room temperature and poured into ice water. The resultant precipitate was filtered off and crystallized from ethanol/water (1:3).
3-{4-Benzyl-1-[(4-phenylpiperazin-1-yl)methyl]-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl}-1-ethyl-7-methyl-1,8-naphthyridin-4(1H)-one (14)
Yield 74% (method 1); yield 78% (method 2, InCl3); yield 80% (method 2, p-TsOH); yield 93% (method 3); mp 106–108°C; IR (υmax, cm-1): 1726, 1495, 1155; 1H NMR: δ 1.37 (t, 3H, J = 8.0 Hz), 2.59 (s, 3H), 2.92 (s, 4H), 3.13 (brs, 4H), 4.35 (brs, 2H), 5.23 (s, 2H), 5.32 (s, 2H), 6.77 (brs, 1H), 6.95 (d, 3H, J = 8.0 Hz), 7.15–7.29 (m, 7H), 7.45 (brs, 1H), 7.95 (s, 1H); 13C NMR: δ 15.4, 25.3, 47.3, 48.3, 48.8, 50.4, 69.4, 108.6, 116.1, 119.3, 119.4, 121.8, 127.4, 127.5, 127.8, 127.9, 128.6, 128.7, 128.8, 129.3, 136.2, 136.3, 147.6, 148.8, 151.5, 146.5, 163.7, 169.1, 174.7. Anal. Calcd for C31H33N7OS: C, 67.49; H, 6.03; N, 17.77. Found: C, 67.28; H, 6.06; N, 17.98.
3-[4-Benzyl-1-({[2-(1H-indol-3-yl)ethyl]amino}methyl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl]-1-ethyl-7-methyl-1,8-naphthyridin-4(1H)-one (15)
Yield 71% (method 1); yield 80% (method 2, InCl3), yield 82% (method 2, p-TsOH); yield 99% (method 3); mp 163–165°C; IR (υmax, cm-1): 3029 2, 1710, 1503, 1155; 1H NMR: δ 1.20 (t, 3H, J = 8.0 Hz), 2.66 (s, 3H), 4.36 (t, 2H, J = 8.0 Hz), 4.64 (t, 2H, J = 8.0 Hz), 5.12 (s, 2H), 5.30 (s, 2H), 5.54 (s, 2H), 6.99 (brs, 3H), 7.14–7.31 (m, 7H), 7.48 (d, 2H, J = 8.0 Hz), 8.54 (s, 1H), 9.01 (s, 1H), 9.21 (s, 1H), 14.93 (s, 1H); 13C NMR: δ 15.3, 25.3, 25.5, 45.6, 47.3, 48.2, 71.3, 108.7, 119.3, 121.8, 123.1, 127.6 (2CH), 128.0 (2CH), 128.8 (2CH), 136.1, 136.2 (2CH), 146.4 (2CH), 147.9, 148.8, 152.2, 163.7, 150.1, 164.9, 167.9, 174.7. Anal. Calcd for C31H31N7OS: C, 67.73; H, 5.68; N, 17.84. Found: C, 67.49; H, 6.05; N, 17.97.
Antimicrobial activity
The following test microorganisms were obtained from the Hifzissihha Institute of Refik Saydam (Ankara, Turkey): E. coli ATCC35218, Yersinia pseudotuberculosis (Y. pseudotuberculosis) ATCC911, Pseudomonas aeruginosa (P. aeruginosa) ATCC43288, E. faecalis ATCC29212, Staphylococcus aureus (S. aureus) ATCC25923, Bacillus cereus (B. cereus) 709 Roma, M. smegmatis ATCC607, Candida albicans (C. albicans) ATCC60193, and Saccharomyces cerevisiae (S. cerevisia) RSKK 251. All synthesized compounds were weighed and dissolved in hexane to prepare a stock solution of 20.0 μg/mL. The antimicrobial effects of the substances were tested quantitatively by using double microdilution; the MIC values (μg/mL) were determined. The antibacterial and antifungal assays were performed in Mueller-Hinton broth (MH) (Difco, Detroit, MI, USA) at pH 7.3 and buffered yeast nitrogen base (Difco, Detroit, MI, USA) at pH 7.0, respectively. The microdilution test plates were incubated for 18–24 h at 35°C. Brain heart infusion broth (BHI) (Difco, Detriot, MI, USA) was used for M. smegmatis; incubation was for 48–72 h at 35°C [44]. Ampicillin, streptomycin, and fluconazole were used as standard antibacterial and antifungal drugs. Dimethyl sulfoxide with a dilution of 1:10 was used as a solvent control.
Acknowledgments
The financial support was provided by Scientific and Technological Research Council of Turkey (TUBITAK, Project no: 113Z181).
References
[1] Leisher, G. Y.; Froelich, E. J.; Gruett, M. D.; Bailey, J. H.; Brundage, P. R. 1,8-naphthyridine derivatives a new class of chemotherapeutic agents. J. Med. Pharm. Chem.1962, 5, 1063–1065.10.1021/jm01240a021Suche in Google Scholar PubMed
[2] Pedrini, A. M.; Geroldi, D.; Siccardi, A.; Balaschi, A. Studies on the mode of action of nalidixic acid. Eur. J. Biochem.1972, 25, 359–365.10.1111/j.1432-1033.1972.tb01704.xSuche in Google Scholar PubMed
[3] Crumplin, G. C.; Smith, J. T. Nalidixic acid: an antibacterial paradox. Antimicrob. Agents Chemother.1975, 8, 251–261.10.1128/AAC.8.3.251Suche in Google Scholar PubMed PubMed Central
[4] Aggarwal, N.; Kumar, R.; Srivastava, C.; Dureja, P.; Khurana, J. M.; Agric, J. Synthesis of nalidixic acid based hydrazones as novel pesticides. J.Agric. Food Chem.2010, 58, 3056–3061.10.1021/jf904144eSuche in Google Scholar PubMed
[5] Fang, K. C.; Chen, Y. L.; Sheu, J. Y.; Wang, T. C.; Tzeng, C. C. Synthesis, antibacterial, and cytotoxic evaluation of certain 7-substituted norfloxacin derivatives. J. Med. Chem.2000, 43, 3809–3812.10.1021/jm000153xSuche in Google Scholar PubMed
[6] Foroumadi, A.; Oboudiat, M.; Emami, S.; Karimollah, A.; Saghaee, L.; Moshafid, M. H.; Shafiee, A. Synthesis and antibacterial activity of N-[2-[5-(methylthio)thiophen-2-yl]-2-oxoethyl] and N-[2-[5-(methylthio)thiophen- 2-yl]-2-(oxyimino) ethyl]piperazinylquinolone derivatives. Bioorg. Med. Chem.2006, 14, 3421–3427.10.1016/j.bmc.2005.12.058Suche in Google Scholar PubMed
[7] Leyva, S.; Hernandez, H. Synthesis of norfloxacin analogues catalyzed by Lewis and Bronsted acids: an alternative pathway. J. Fluor. Chem.2010, 131, 982–988.10.1016/j.jfluchem.2010.07.002Suche in Google Scholar
[8] Foroumadi, A.; Emami, S.; Hassanzadeh, A.; Rajaee, M.; Sokhanvar, K.; Moshafib, M. H.; Shafiee, A. Synthesis and antibacterial activity of N-(5- benzylthio-1,3,4-thiadiazol-2-yl) and N-(5-benzylsulfonyl-1,3,4-thiadiazol-2- yl)piperazinyl quinolone derivatives. Bioorg. Med. Chem. Lett.2005, 15, 4488–4492.10.1016/j.bmcl.2005.07.016Suche in Google Scholar PubMed
[9] Abuo-Rahma, G. A. A.; Sarhan, H. A.; Gad, G. F. M. Design, synthesis, antibacterial activity and physicochemical parameters of novel N-4-piperazinyl derivatives of norfloxacin. Bioorg. Med. Chem.2009, 17, 3879–3886.10.1016/j.bmc.2009.04.027Suche in Google Scholar PubMed
[10] Shirodkar, P. Y.; Vartak Meghna, M. Synthesis, biological evaluation and QSAR evaluation of Mannich bases of 6-nitroquinazolones. Indian J. Heterocycl. Chem.2000, 9, 239–240.Suche in Google Scholar
[11] Pandey, V. K.; Misra, D.; Shukla, A. Synthesis and antiviral activity if 2- aryl-5-[3′-2′-methyl-6:8 substituted-quinazolyl)-phenyl]pyrazoles. Indian Drugs1994, 31, 532–536.Suche in Google Scholar
[12] Desai, N. C.; Shah, B. R.; Bhatt, J. J.; Patel, H. H.; Undavia, N. K.; Trivedi, P. B.; Narayanan, V. Synthesis of 2,3-disubstituted-3,1-quinazolin-4(4H)- ones as potential anticancer and anti-HIV agents. Indian J. Chem.1995, 34B, 201–208.Suche in Google Scholar
[13] Werbel, L. M.; Elslager, E. F.; Newton, L. S. Synthesis and antimalarial activity of a series of 2,4-diamino-6-[(N-alkylanilino)methyl]quinazolines. J. Heterocycl. Chem.1987, 24, 345–349.10.1002/jhet.5570240210Suche in Google Scholar
[14] Patel, N. B.; Lilakar, J. D. Synthesis of new substituted-4-(3H)-quinazolinones and their antibacterial activity. Indian J. Heterocyclic Chem.2001, 11, 85–86.Suche in Google Scholar
[15] Aggarwal, N.; Kumar, R.; Dureja, P.; Khurana, J. M. Synthesis, antimicrobial evaluation and QSAR analysis of novel nalidixic acid based 1,2,4-triazole derivatives. Eur. J. Med. Chem.2011, 46, 4089–4099.10.1016/j.ejmech.2011.06.009Suche in Google Scholar
[16] Holla, B. S.; Shivananda, M. K.; Shalini Shenoy, M.; Antony, G. Studies on arylfuran derivatives: part VII. Synthesis and characterization of some Mannich bases carrying halophenylfuryl moieties as promising antibacterial agents. Il Farmaco.1998, 53, 531–535.10.1002/chin.199922112Suche in Google Scholar
[17] Holla, B. S.; Rao, B. S.; Shridhara, K.; Akberali, P. M. Studies on arylfuran derivatives: Part XI. Synthesis, characterisation and biological studies on some Mannich bases carrying 2,4-dichlorophenylfurfural moiety. Il Farmaco. 2000, 55, 338–344.10.1016/S0014-827X(00)00033-1Suche in Google Scholar
[18] Holla B. S.; Poojary K. N.; Rao B. S.; Shivananda M. K. New bisamino mercaptotriazoles and bis-triazolothiadiazoles as possible anticancer agents. Eur. J. Med. Chem.2002, 37, 511–517.10.1016/S0223-5234(02)01358-2Suche in Google Scholar
[19] Holla, B. S.; Sooryanarayana Rao, B.; Gonsalves, R.; Sarojini, B. K.; Shridhara, K. Synthesis of some new biologically active thiadiazolotriazinones. Il Farmaco.2002, 57, 693–696.10.1016/S0014-827X(98)00036-6Suche in Google Scholar
[20] Demirbas, N.; Demirbas, A.; Karaoglu, S. A.; Celik, E. Synthesis and antimicrobial activities of some new [1,2,4]triazolo[3,4-b][1,3,4]thiadiazoles and [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Arkivoc2005, i, 75–91.10.3998/ark.5550190.0006.108Suche in Google Scholar
[21] Güzeldemirci, N. U.; Küçükbasmacı, Ö. Synthesis and antimicrobial activity evaluation of new 1,3,4-thiadiazoles bearing imidazo[2,1-b]thiazole moiety. Eur. J. Med. Chem.2010, 45, 63–68.10.1016/j.ejmech.2009.09.024Suche in Google Scholar PubMed
[22] Yi, H.; Xiaolong, L.; Jue, L.; Xiaocen, L., Guo, L.; Hai, L.; Yong, W. A novel and convenient route for the construction of 5-((1H-1,2,4-triazol-1-yl)methyl)-1H-indoles and their application in the synthesis of rizatriptan. Tetrahedron Lett. 2014, 55, 3938–3941.10.1016/j.tetlet.2014.05.063Suche in Google Scholar
[23] Li, P.; Shi, L.; Yang, L.; Chen, X. W.; Wu, F.; Shi, Q. C.; Xu, W. M.; He, M.; Hu, D. Y.; Song, B. A. Design, synthesis, and antibacterial activity against rice bacterial leaf blight and leaf streak of 2,5-disubstituted-1,3,4-oxadiazole/thiadiazole sulfone derivatives. Bioorg. Med. Chem. Lett.2014, 24, 1677–1680.10.1016/j.bmcl.2014.02.060Suche in Google Scholar PubMed
[24] Zhang, M. Z.; Chen, Q.; Mulholland, N.; Beattie, D.; Irwin, D.; Gu, Y. C.; Yang, G. F.; Clough, J. Synthesis and antifungal activity of 3-(1,3,4-oxadiazol-5-yl)-indoles and 3-(1,3,4-oxadiazol-5-yl)methylindoles. Eur. J. Med. Chem.2013, 63, 22–32.10.1016/j.ejmech.2013.01.038Suche in Google Scholar PubMed
[25] Verma, A.; Saraf, S. K. 4-thiazolidinone – a biologically active scaffold. Eur. J. Med. Chem.2008, 43, 897–905.10.1016/j.ejmech.2007.07.017Suche in Google Scholar
[26] Arya, K.; Rawat, D. S.; Dandia, A.; Sasai, H. Bronsted acidic ionic liquids: green, efficient and reusable catalyst for synthesis of fluorinated spiro[indole-thiazinones/thiazolidinones] as antihistamic agents. J. Fluor. Chem.2012, 137, 117–122.10.1016/j.jfluchem.2012.03.003Suche in Google Scholar
[27] Anderegg, T. R.; Jones, R. N. Preliminary susceptibility testing guidelines for AZD2563, a long-acting oxazolidinone. Int. J. Antimicrob. Agents.2004, 23, 6–10.10.1016/j.ijantimicag.2003.05.007Suche in Google Scholar
[28] Brickner, S. J.; Hutchinson, D. K.; Barbachyn, M. R.; Manninen, P. R.; Ulanowicz, D. A.; Garmon, S. A. Synthesis and antibacterial activity of U-100592 and U-100766, two oxazolidinone antibacterial agents for the potential treatment of multidrug-resistant Gram-positive bacterial infections. J. Med. Chem.1996, 39, 673–679.10.1021/jm9509556Suche in Google Scholar
[29] Gemmell, C. G. Susceptibility of a variety of clinical isolates to linezolid: a European inter-country comparison. J. Antimicrob. Chemother.2001, 48, 47–52.10.1093/jac/48.1.47Suche in Google Scholar
[30] Diekema, D. J.; Jones, R. N. Oxazolidinone antibiotics. Lancet2001, 358, 1975–1982.10.1016/S0140-6736(01)06964-1Suche in Google Scholar
[31] Almajan, G. L.; Barbuceanu, S. F.; Almajan E. R.; Draghici, C.; Saramet, G. Synthesis, characterization and antibacterial activity of some triazole Mannich bases carrying diphenylsulfone moieties. Eur. J. Med. Chem.2009, 44, 3083–3089.10.1016/j.ejmech.2008.07.003Suche in Google Scholar PubMed
[32] Saadatjoo, N.; Golshekan, M.; Shariati, S.; Azizi, P.; Nemati, F. Ultrasound assisted synthesis of b-amino ketones via a Mannich reaction catalyzed by Fe3O4 magnetite nanoparticles as an efficient, recyclable and heterogeneous catalyst. Arabian J. Chem.2012, http://dx.doi.org/10.1016/j.arabjc.2012.11.018.10.1016/j.arabjc.2012.11.018Suche in Google Scholar
[33] Yun, M.; Yuehong, P.; Fei, L.; Hanqi, X.; Xiaofang, S. Microwave-assisted ultrafast synthesis of silver nanoparticles for detection of Hg2+. Spectrochimica Acta, Part A, Mol. Biomol. Spectrosc.2016, 153, 206–211.10.1016/j.saa.2015.08.004Suche in Google Scholar PubMed
[34] Mentese, M. Y.; Bayrak, H.; Uygun, Y.; Mermer, A.; Ulker, S.; Alpay Karaoglu, S.; Demirbas, N. Microwave assisted synthesis of some hybrid molecules derived from norfloxacin and investigation of their biological activities. Eur. J. Med. Chem. 2013, 67, 230–242.10.1016/j.ejmech.2013.06.045Suche in Google Scholar PubMed
[35] Kouznetsov, V. V.; Gomez-Barrio, A. Recent developments in the design and synthesis of hybrid molecules basedon aminoquinoline ring and their antiplasmodial evaluation. Eur. J. Med. Chem. 2009, 44, 3091–3113.10.1016/j.ejmech.2009.02.024Suche in Google Scholar
[36] Bayrak, H.; Demirbas, A.; Karaoglu, S. A.; Demirbas, N. Synthesis of some new 1, 2, 4-triazoles, their Mannich and Schiff bases and evaluation of their antimicrobial activities. Eur. J. Med. Chem. 2009, 44, 1057–1066.10.1016/j.ejmech.2008.06.019Suche in Google Scholar
[37] Bayrak, H.; Demirbas, A.; Demirbas, N.; Karaoglu, S. A. Synthesis of some new 1, 2, 4-triazoles starting from isonicotinic acid hydrazide and evaluation of their antimicrobial activities. Eur. J. Med. Chem. 2009, 44, 4362–4366.10.1016/j.ejmech.2009.05.022Suche in Google Scholar
[38] Basoglu, S.; Demirbas, A.; Ulker, S.; Karaoglu, S. A.; Demirbas, N. Design, synthesis and biological activities of some 7-aminocephalosporanic acid derivatives. Eur. J. Med. Chem. 2013, 69, 622–631.10.1016/j.ejmech.2013.07.040Suche in Google Scholar
[39] Zhao, Y. J.; Wei, W.; Su, Z. G.; Ma, G. H. Poly(ethylene glycol) prodrug for anthracyclines via N-Mannich base linker: design, synthesis and biological evaluation. Int. J. Pharm. 2009, 379, 90–99.10.1016/j.ijpharm.2009.06.013Suche in Google Scholar
[40] Roman, O. G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 89, 743–816.10.1016/j.ejmech.2014.10.076Suche in Google Scholar
[41] Ganiyat, K.; Willie, I. E.; Oluwakemi, O. Synthesis of Mannich bases: 2-(3- Phenylaminopropionyloxy)-benzoic acid and 3-phenylamino-1-(2, 4, 6-trimethoxy-phenyl)propan-1- one, their toxicity, ionization constant, antimicrobial and antioxidant activities. Food Chem.2014, 165, 515–521.10.1016/j.foodchem.2014.05.119Suche in Google Scholar
[42] Fekner, T.; Baldwin, J. E.; Adlington, R. M.; Fardeau, S.; Jones, T. W.; Prout, C. K.; Schofield, C. J. Synthesis of (6S)-cephalosporins from 6-aminopenicillanic acid. Tetrahedron2000, 56, 6053–6074.10.1016/S0040-4020(00)00486-5Suche in Google Scholar
[43] Dassonville-Klimpt, A.; Audic, N.; Sasaki, A.; Pillon, M.; Baudrin, E.; Mullié, C.; Sonnet, P. Synthesis and antibacterial activity of catecholate-ciprofloxacin conjugates. Bioorg. Med. Chem.2014, 22, 4049–4060.10.1016/j.bmc.2014.05.067Suche in Google Scholar PubMed
[44] Willanova, P. A. National Committee for Clinical Laboratory Standard. NCCLS Document M7-A3.1993, 13.Suche in Google Scholar
©2016 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Review
- Constituents from Chloranthaceae plants and their biological activities
- Preliminary Communications
- Design, synthesis and cytotoxicity evaluation of novel (E)-3-(3-aryl-1-phenyl-1H-pyrazol-4-yl)-1-(pyridin-3-yl)prop-2-en-1-ones as anticancer agents
- Synthesis of first selenodecalines: 2-aryl-4-phenyl octahydroselenochromenes
- Reasearch Articles
- Microwave-assisted and conventional synthesis of novel antimicrobial 1,2,4-triazole derivatives containing nalidixic acid skeleton
- One-pot synthesis of new triazole-sucrose derivatives via click chemistry and evaluation of their antitubercular activity
- One-pot synthesis of 2-hydrazonyl-4-phenylthiazoles via [PDBMDIm]Br-catalyzed reaction under solvent-free conditions
- Three-component one-pot synthesis of dihydrochromeno[4,3-b]pyrazolo[4,3-e]pyridines
Artikel in diesem Heft
- Frontmatter
- Review
- Constituents from Chloranthaceae plants and their biological activities
- Preliminary Communications
- Design, synthesis and cytotoxicity evaluation of novel (E)-3-(3-aryl-1-phenyl-1H-pyrazol-4-yl)-1-(pyridin-3-yl)prop-2-en-1-ones as anticancer agents
- Synthesis of first selenodecalines: 2-aryl-4-phenyl octahydroselenochromenes
- Reasearch Articles
- Microwave-assisted and conventional synthesis of novel antimicrobial 1,2,4-triazole derivatives containing nalidixic acid skeleton
- One-pot synthesis of new triazole-sucrose derivatives via click chemistry and evaluation of their antitubercular activity
- One-pot synthesis of 2-hydrazonyl-4-phenylthiazoles via [PDBMDIm]Br-catalyzed reaction under solvent-free conditions
- Three-component one-pot synthesis of dihydrochromeno[4,3-b]pyrazolo[4,3-e]pyridines