Visible-light-mediated radical aryltrichloromethylation of N-arylacrylamides for the synthesis of trichloromethyl-containing oxindoles
-
Mei Zhu


Abstract
A practical and unified strategy has been described for the construction of trichloromethylated oxindoles via a visible light-promoted aryltrichloromethylation reaction of N-arylacrylamides with tetrachloromethane. These reactions are carried out at room temperature in good to excellent chemical yields with good functional group tolerance.
Introduction
Trichloromethyl functionality is widely found in organic chemistry and natural products [1–3]. The introduction of trichloromethyl group into organic framework strongly attracts synthetic pursuit of chemists because of its diverse synthetic applications and important biological properties of the substituted compounds [4, 5]. Nevertheless, up to date, methods for the synthesis of trichloromethylated oxindoles are quite rare [6–8]. Recently, Loh and co-workers reported the Fe-catalyzed coupling of N-arylacrylamides with dichloro- and tetrachloromethane using diaryliodonium salt as oxidant [7]. However, the drawbacks are the use of an oxidant diaryliodonium salt (2.0 equiv.) as well as the requirement of high temperature. A more concise synthesis is highly desirable.
Recently, visible-light photoredox catalysis has attracted substantial attention because of its environmental compatibility and versatility in promoting a large number of synthetically important reactions [9–12]. The visible-light-induced radical reaction of organohalides is an environmentally friendly alternative to traditional radical transformation by avoiding the use of hazardous radical initiators, and substantial achievements have been made [13–18]. Polyhalogenated alkanes, such as tetrachloromethane and bromotrichloromethane have been used as common radical precursors in various transformations leading to trichloromethyl functionalized compounds. For example, Stephenson and co-workers reported a radical addition reaction between tetrachloromethane and unfunctionalized alkenes [18]. Inspired by these results and in connection with our interest in radical cyclizations [19–22], we envisioned the synthesis of trichloromethylated oxindoles by photocatalyzed radical reaction of tetrachloromethane with N-arylacrylamides. This transformation allows the direct formation of a C-CCl3 bond and the construction of a oxindole ring in one reaction.
Results and discussion
At the outset of this investigation, N-arylacrylamide 1a and tetrachloromethane were chosen as the model substrates to optimize the reaction conditions. The complex [fac-Ir(III)(ppy)3] (ppy = 2-phenylpyridine) was chosen as the photocatalyst because of its superior reduction capacity in the excited state. Irradiation of the solution of 1a and CCl4 in DMF with a 5 W blue LED bulb in the presence of [fac-Ir(III)(ppy)3] and Na2HPO4 for 48 h at room temperature afforded the desired product 2a in 75% yield. Using CCl3Br as ·CCl3 radical precursor, oxindole 2a was isolated in 63% yield with some unidentifiable products. Other photocatalysts, such as, [Ir(ppy)2(dtbbpy)]PF6, Ru(bpy)3Cl2, eosin Y were also tested, but none of them gave better results than [fac-Ir(ppy)3]. A brief screen of the bases revealed that K2CO3 was the best choice for the reaction. Other inorganic bases, such as Na2CO3, NaOAc, and NaHCO3 also gave good yields of the isolated products. Indeed, the replacement of inorganic bases by an organic base (triethylamine) had a marked effect on the reaction, affording product 2a in only 52% yield. The reaction proceeds well in DMF and other solvents such as DMSO, CH3CN. In the control experiments, the reaction was found to be completely restrained without the catalyst or light irradiation.
With the optimized reaction conditions in hand, we further explored the scope of trichloromethylation with a variety of N-arylacrylamides (Scheme 1). It was found that various N-protected substrates, such as ethyl, isopropyl, butyl, benzyl or phenyl could be used as effective substituent group for this transformation (2a–f). Subsequently, the effect of substituents at the N-aryl moiety was examined. Both electron-donating and electron-withdrawing groups located in the para- or ortho position of the aromatic rings were found to be tolerated in this reaction, furnishing the corresponding oxindoles 2g–o in moderate to good yields. N-arylacrylamides 2g–h possessing electron-donating substituents gave the desired products in higher yields than the substrates 2i–l with electron-withdrawing groups. Moreover, the procedure appears to be sensitive to steric effects. Generally, substitutents in the para-position on the benzene are well tolerated. By contrast, the presence of ortho-substituents on the benzene reduce the yields of 2m–n. 3,5-Disubstituted N-arylacrylamide 1o was also transformed into the desired product 2o in 64% yield. Cyclization of meta-chloro-substituted substrates 1p resulted in a mixture of two regioisomers in a 2:1 ratio. Additionally, good results were also obtained with tetrahydroquinoline derivative 1q as a substrate.
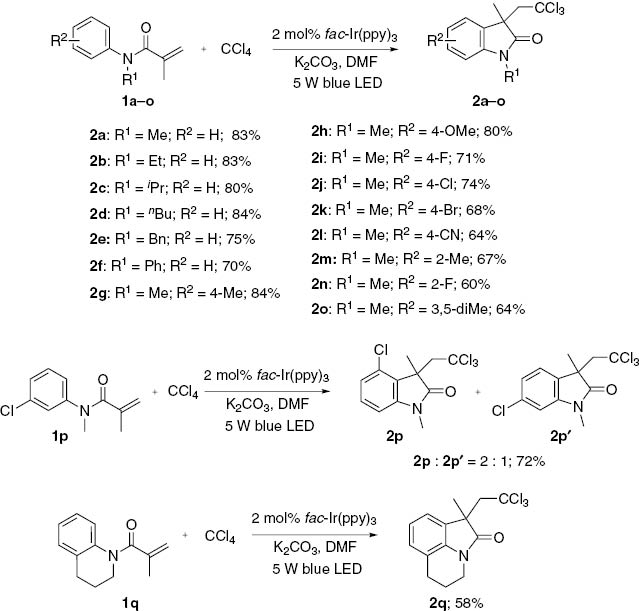
Synthesis of trichloromethylated oxindoles.
Conclusions
We report a facile assembly of 3,3-disubstituted oxindoles by visible-light-promoted aryltrichloromethylation of N-arylacrylamides with tetrachloromethane. The reaction tolerates a wide range of functional groups.
Experimental
All reactions were performed in a 20 mL tube equipped with a rubber septum at room temperature. Photo-irradiation was carried out with a 5 W blue LED. Solvents were purified or dried in a standard manner. 1H NMR spectra (400 MHz or 500 MHz), 13C NMR spectra (100 MHz or 125 MHz), and 19F NMR spectra were measured in CDCl3.·HR-MS analyses were recorded on Q-TOF Global mass spectrometer.
General procedure for the synthesis of trichloromethylated oxindoles 2a–q
To a mixture of N-arylacrylamide 1 (0.30 mmol), CCl4 (0.9 mmol), and K2CO3 (0.6 mmol) in 2.0 mL of DMF was added fac-Ir(ppy)3 (0.006 mmol, 2.0 mol%) under N2 atmosphere. The solution was stirred at room temperature and irradiated with 5 W blue LED for 48 h. Then the reaction mixture was treated with EtOAc and brine. The aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered and concentrated. The residue was purified by flash column chromatography on silica gel eluting with petroleum ether/ethyl acetate, 10:1, to give the oxindole 2a–q.
3-(2,2,2-Trichloroethyl)-1,3-dimethylindolin-2-one (2a) [7]
Yield 83%; yellow solid; mp 103–104°C; 1H NMR: δ 7.26–7.35 (m, 2H), 7.06 (t, J = 7.4 Hz, 1H), 6.88 (d, J = 7.6 Hz, 1H), 3.69 (d, J = 15.2 Hz, 1H), 3.34 (d, J = 15.2 Hz, 1H), 3.23 (s, 3H), 1.40 (s, 3H).
3-(2,2,2-Trichloroethyl)-1-ethyl-3-methylindolin-2-one (2b) [7]
Yield 83%; yellow solid; mp 95–97°C; 1H NMR: δ 7.36 (d, J = 7.6 Hz, 1H), 7.26–731 (m, 1H), 7.05 (t, J = 7.6 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 3.82–3.94 (m, 1H), 3.64–3.75 (m, 2H), 3.34 (d, J = 15.2 Hz, 1H), 1.38 (s, 3H), 1.25 (t, J = 7.2 Hz, 3H).
3-(2,2,2-Trichloroethyl)-1-isopropyl-3-methylindolin-2-one (2c) [7]
Yield 80%; yellow solid; mp 88–90°C; 1H NMR: δ 7.36 (d, J = 7.2 Hz, 1H), 7.25–7.30 (m, 2H), 6.98–7.04 (m, 2H), 4.59–4.69 (m, 1H), 3.69 (d, J = 15.2 Hz, 1H), 3.33 (d, J = 15.2 Hz, 1H), 1.48 (dd, J = 7.2, 4.4 Hz, 6H), 1.37 (s, 3H).
1-Butyl-3-(2,2,2-trichloroethyl)-3-methylindolin-2-one (2d)
Yield 84%; white solid; mp 94–95°C; 1H NMR: δ 7.35 (d, J = 8.0 Hz, 1H), 7.27–7.30 (m, 1H), 7.03–7.06 (m, 1H), 6.90 (d, J = 8.0 Hz, 1H), 3.63–3.79 (m, 3H), 3.35 (d, J = 15.6 Hz, 1H), 1.60–1.69 (m, 2H), 1.36–1.43 (m, 5H), 0.95 (t, J = 8.0 Hz, 3H); 13C NMR: δ 178.4, 142.8, 129.7, 128.4, 125.8, 121.7, 108.6, 96.3, 59.7, 48.0, 40.1, 29.2, 27.4, 20.3, 13.8. HR-MS. Calcd for [M+H]+, C15H19Cl3NO: m/z 334.0533. Found: m/z 334.0530.
1-Benzyl-3-(2,2,2-trichloroethyl)-3-methylindolin-2-one (2e) [7]
Yield 75%; white solid; mp 80–82°C; 1H NMR: δ = 7.19–7.37 (m, 7 H), 7.03 (t, J = 7.6 Hz, 1 H), 6.82 (d, J = 7.6 Hz, 1 H), 4.98 (d, J = 15.2 Hz, 1 H), 4.88 (d, J = 15.2 Hz, 1 H), 3.75 (d, J = 15.2 Hz, 1H), 3.38 (d, J = 15.2 Hz, 1H), 1.45 (s, 3 H).
3-(2,2,2-Trichloroethyl)-3-methyl-1-phenylindolin-2-one (2f)
Yield 70%; white solid; mp 99–101°C; 1H NMR: δ 7.55 (t, J = 8.0 Hz, 2H), 7.42–7.46 (m, 4H), 7.26 (t, J = 8.0 Hz, 1H), 7.13 (t, J = 8.0 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 3.82 (d, J = 15.2 Hz, 1H), 3.46 (d, J = 15.2 Hz, 1H), 1.56 (s, 3H); 13C NMR: δ 177.9, 143.2, 134.5, 129.7, 129.3, 128.4, 128.1, 126.3, 126.0, 122.5, 109.8, 96.3, 60.1, 48.2, 27.4. HR-MS. Calcd for [M+H]+, C17H15Cl3NO: m/z 354.0214. Found: m/z 354.0210.
3-(2,2,2-Trichloroethyl)-1,3,5-trimethylindolin-2-one (2g) [7]
Yield 84%; white solid; mp 108–109°C; 1H NMR: δ 7.16 (s, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 3.67 (d, J = 15.2 Hz, 1H), 3.30 (d, J = 15.2 Hz, 1H), 3.20 (s, 3H), 2.33 (s, 3H), 1.37 (s, 3H).
3-(2,2,2-Trichloroethyl)-5-methoxy-1,3-dimethylindolin-2-one (2h) [7]
Yield 80%; white solid; mp 103–105°C; 1H NMR: δ 6.96 (s, 1H), 6.85 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 3.79 (s, 3H), 3.68 (d, J = 15.2 Hz, 1H), 3.32 (d, J = 15.2 Hz, 1H), 3.21 (s, 3H), 1.38 (s, 3H).
3-(2,2,2-Trichloroethyl)-5-fluoro-1,3-dimethylindolin-2-one (2i) [7]
Yield 71%; white solid; mp 117–119°C; 1H NMR: δ 7.09–7.12 (m, 1H), 6.99–7.04 (m, 1H), 6.78–6.81 (m, 1 H), 3.69 (d, J = 15.2 Hz, 1H), 3.31 (d, J = 15.6 Hz, 1H), 3.22 (s, 3H), 1.39 (s, 3H).
5-Chloro-3-(2,2,2-trichloroethyl)-1,3-dimethylindolin-2-one (2j) [7]
Yield 74%; white solid; mp 108–110°C; 1H NMR: δ 7.45 (m, 2 H), 6.78 (d, J = 8.4 Hz, 1H), 3.69 (d, J = 15.6 Hz, 1H), 3.31 (d, J = 15.6 Hz, 1H), 3.22 (s, 3H),1.39 (s, 3H).
5-Bromo-3-(2,2,2-trichloroethyl)-1,3-dimethylindolin-2-one (2k) [7]
Yield 68%; white solid; mp 123–124°C; 1H NMR: δ 7.33 (d, J = 1.6 Hz, 1 H), 7.28 (dd, J = 8.4, 1.6 Hz, 1 H), 6.80 (d, J = 8.4 Hz, 1 H), 3.68 (d, J = 15.2 Hz, 1H), 3.30 (d, J = 15.2 Hz, 1H), 3.21 (s, 3H), 1.39 (s, 3H).
3-(2,2,2-Trichloroethyl)-1,3-dimethyl-2-oxoindoline-5-carbonitrile (2l) [7]
Yield 64%; white solid; mp 112–114°C; 1H NMR: δ 7.66–7.70 (m, 2H), 6.95 (d, J = 8.4 Hz, 1H), 3.70 (d, J = 15.2 Hz, 1H), 3.37 (d, J = 15.2 Hz, 1H), 3.26 (s, 3H), 1.43 (s, 3H).
3-(2,2,2-Trichloroethyl)-1,3,7-trimethylindolin-2-one (2m) [7]
Yield 67%; white solid; mp 90–92°C; 1H NMR: δ 6.97 (d, J = 7.6 Hz, 2H), 6.79 (t, J = 8.0 Hz, 1H), 3.79 (s, 3H), 3.69 (d, J = 15.2 Hz, 1H), 3.31 (d, J = 15.2 Hz, 1H), 3.21 (s, 3H), 1.39 (s, 3H).
3-(2,2,2-Trichloroethyl)-7-fluoro-1,3-dimethylindolin-2-one (2n) [7]
Yield 60%; white solid; mp 103–105°C; 1H NMR: δ 6.96–7.08 (m, 3H), 3.68 (d, J = 15.2 Hz, 1H), 3.44 (s, 3H), 3.32 (d, J = 15.2 Hz, 1H), 1.39 (s, 3H).
3-(2,2,2-Trichloroethyl)-1,3,4,6-tetramethylindolin-2-one (2o) [7]
Yield 64%; white solid; mp 109–111°C; 1H NMR: δ 6.63 (s, 1 H), 6.54 (s, 1 H), 3.61 (d, J = 15.2 Hz, 1H), 3.43 (d, J = 15.2 Hz, 1H), 3.19 (s, 3 H), 2.38 (s, 3 H), 2.35 (s, 3 H), 1.44 (s, 3 H).
4-Chloro-3-(2,2,2-trichloroethyl)-1,3-dimethylindolin-2-one (2p) and ethyl 6-chloro-3-(2,2,2-trichloroethyl)-1,3-dimethylindolin-2-one (2p′) (2:1)
Yield 72%; white solid; mp 88–90°C; 1H NMR: δ 7.24–7.29 (m, 1 H), 7.27 (s, 0.5 H), 7.04 (dd, J = 8.0, 2.0 Hz, 0.5 H), 6.98 (d, J = 8.0 Hz, 1 H), 6.89 (d, J = 2.0 Hz, 0.5 H), 6.80 (d, J = 8.0 Hz, 1 H), 3.75 (d, J = 15.2 Hz, 1H), 3.68 (d, J = 15.2 Hz, 0.5 H), 3.53 (d, J = 15.2 Hz, 1H), 3.33 (d, J = 15.2 Hz, 0.5 H), 3.23 (s, 3 H), 3.22 (s, 1.5 H), 1.54 (s, 3 H), 1.38 (s, 1.5 H); 13C NMR: δ 178.5, 177.0, 144.9, 144.4, 134.3, 132.4, 129.9, 127.8, 126.6, 126.5, 123.6, 121.9, 109.2, 107.0, 96.03, 96.0, 59.7, 57.5, 48.8, 47.7, 26.8, 26.78, 26.72, 22.9. HR-MS. Calcd for [M+H]+, C12H12Cl4NO: m/z 325.9668. Found: m/z 325.9662.
1-(2,2,2-Trichloroethyl)-5,6-dihydro-1-methyl-1H-pyrrolo[3,2,1-ij]quinolin-2(4H)-one (2q)
Yield 58%; white solid; mp 85–86°C; 1H NMR: δ 7.9 (d, J = 7.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.95 (t, J = 7.6 Hz, 1 H), 3.69 (d, J = 15.2 Hz, 1H), 3.33 (d, J = 15.2 Hz, 1H), 3.67–3.78 (m, 2H), 2.78–2.84 (m, 2H), 1.97–2.03 (m, 2H), 1.40 (s, 3 H); 13C NMR: δ 177.5, 139.0, 128.1, 127.2, 123.6, 121.5, 96.4, 59.8, 49.2, 39.2, 26.7, 24.7, 21.1. HR-MS. Calcd for [M+H]+, C14H25Cl3NO: m/z 318.0214. Found: m/z 318.0212.
Acknowledgments
We are grateful to the National Natural Science Foundation of China (project nos. 21202078 and U1204205) and the Program for Innovative Research Team (in Science and Technology) at the University of Henan Province (14IRTSTHN008).
References
[1] Gribble, G. W. Recently discovered naturally occurring heterocyclic organohalogen compounds. Heterocycles2012, 84, 157–207.10.3987/REV-11-SR(P)5Search in Google Scholar
[2] Xiao, Q.; Young, K.; Zakarian, A. An eficient synthesis of the fully elaborated isoindolinone unit of muironolide A. Org. Lett.2013, 15, 3314–3317.10.1021/ol401354aSearch in Google Scholar
[3] Flores, B.; Molinski, T. F. Assembly of the isoindolinone core of muironolide A by asymmetric intramolecular Diels–Alder cycloaddition. Org. Lett.2011, 13, 3932–3935.10.1021/ol201461nSearch in Google Scholar
[4] Beaumont, S.; Ilardi, E. A.; Monroe, L. R.; Zakarian, A. Valence tautomerism in titanium enolates: catalytic radical haloalkylation and application in the total synthesis of neodysidenin. J. Am. Chem. Soc.2010, 132, 1482–1483.10.1021/ja910154fSearch in Google Scholar
[5] Galonic, D. P.; Vaillancourt, F. H.; Walsh, C. T. Halogenation of unactivated carbon centers in natural product biosynthesis: trichlorination of leucine during barbamide biosynthesis. J. Am. Chem. Soc.2006, 128, 3900–3901.10.1021/ja060151nSearch in Google Scholar
[6] Chen, J.-R.; Yu, X.-Y.; Xiao, W.-J. Tandem radical cyclization of N-arylacrylamides: an emerging platform for the construction of 3,3-disubstituted oxindoles. Synthesis2015, 47, 604–629.10.1055/s-0034-1378944Search in Google Scholar
[7] Lu, M.-Z.; Loh, T.-P. Iron-catalyzed cascade carbochloromethylation of activated alkenes: highly efficient access to chloro-containing oxindoles. Org. Lett.2014, 16, 4698–4701.10.1021/ol502411cSearch in Google Scholar
[8] Liu, Y.; Zhang, J.-L.; Song, R.-J.; Li, J.-H. 1,2-Alkylarylation of activated alkenes with dual C–H bonds of arenes and alkyl halides toward polyhalo-substituted oxindoles. Org. Chem. Front.2014, 1, 1289–1294.10.1039/C4QO00251BSearch in Google Scholar
[9] Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Visible light photoredox patalysis with transition metal complexes: applications in organic synthesis. Chem. Rev.2013, 113, 5322–5363.10.1021/cr300503rSearch in Google Scholar
[10] Yoon, T. P. Visible light photocatalysis: the development of photocatalytic radical ion cycloadditions. ACS Catal.2013, 3, 895–902.10.1021/cs400088eSearch in Google Scholar
[11] Xuan, J.; Xiao, W.-J. Visible-light photoredox catalysis. Angew. Chem., Int. Ed.2012, 51, 6828–6838.10.1002/anie.201200223Search in Google Scholar
[12] Narayanam, J. M. R.; Stephenson, C. R. J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev.2011, 40, 102–113.10.1039/B913880NSearch in Google Scholar
[13] Shih, H.-W.; Vander Wal, M. N.; Grange, R. L.; MacMillan, D. W. C. Enantioselective α-benzylation of aldehydes via photoredox organocatalysis. J. Am. Chem. Soc.2010, 132, 13600–13603.10.1021/ja106593mSearch in Google Scholar
[14] Tucker, J. W.; Stephenson, C. R. J. Tandem visible light-mediated radical cyclization–divinylcyclopropane rearrangement to tricyclic pyrrolidinones. Org. Lett.2011, 13, 5468–5471.10.1021/ol202178tSearch in Google Scholar
[15] Jiang, H.; Cheng, Y.; Wang, R.; Zheng, M.; Zhang, Y.; Yu, Y. Synthesis of 6-alkylated phenanthridine derivatives using photoredox neutral somophilic isocyanide insertion. Angew. Chem., Int. Ed.2013, 52, 13289–13292.10.1002/anie.201308376Search in Google Scholar
[16] Kim, H.; Lee, C. Visible-light-induced photocatalytic reductive transformations of organohalide. Angew. Chem., Int. Ed.2012, 51, 12303–12306.10.1002/anie.201203599Search in Google Scholar
[17] Cheng, Y.; Gu, X.; Li, P. Visible-light photoredox in homolytic aromatic substitution: direct Arylation of arenes with aryl halides. Org. Lett.2013, 15, 2664–2667.10.1021/ol400946kSearch in Google Scholar
[18] Wallentin, C.-J.; Nguyen, J. D.; Finkbeiner, P.; Stephenson, C. R. J. Visible light-mediated atom transfer radical addition via oxidative and reductive quenching of photocatalysts. J. Am. Chem. Soc.2012, 134, 8875–8884.10.1021/ja300798kSearch in Google Scholar
[19] Fu, W.; Xu, F.; Fu, Y.; Xu, C.; Li, S.; Zou, D. A metal-free Route to CF3-containing oxindoles by PhI(OAc)2-mediated trifluoromethylation of N-arylacrylamides with TMSCF3. Eur. J. Org. Chem.2014, 2014, 709–712.10.1002/ejoc.201301512Search in Google Scholar
[20] Fu, W.; Zhu, M.; Zou, G. Synthesis of 3,3-disubstituted oxindoles by organoselenium-induced radical cyclizations of N-arylacrylamides. Heterocycl. Commun.2015, 21, 9–12.10.1515/hc-2014-0195Search in Google Scholar
[21] Fu, W.; Zhu, M.; Zou, G.; Xu, C.; Wang, Z. Visible-light-mediated trifluoroethylation of N-arylacrylamides with trifluoroethyl iodide: synthesis of CF3-containing oxindoles. Synlett2014, 25, 2513–2517.10.1055/s-0034-1379071Search in Google Scholar
[22] Fu, W.; Zhu, M.; Zou, G.; Xu, C.; Wang, Z. Visible-light-mediated radical aryldifluoroacetylation of N-arylacrylamides to give difluoroacetylated oxindoles. Asian J. Org. Chem.2014, 3, 1273–1276.10.1002/ajoc.201402199Search in Google Scholar
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Preliminary Communications
- Triphenylphosphine catalyzed domino reaction of dialkyl acetylenedicarboxylate with 3-aryl- 2-benzoylcyclopropane-1,1-dicarbonitrile
- Synthesis of colletotrichumine A
- Research Articles
- Synthesis of the spiroacetal fragments of spirofungins A and B, antibiotics isolated from Streptomyces violaceusniger Tü 4113
- Efficient synthesis and fungicidal activities of strobilurin analogues containing benzofuro [3,2-d]-1,2,4-triazolo[1,5-a]pyrimidinone side chains
- Synthesis of 2-amino-6,7,8,9-tetrahydro-6-phenethyl-3H-pyrimido[4,5-e][1,4]diazepin-4(5H)-one: a model for a potential pyrimido[4,5-e][1,4]diazepine-based folate anti-tumor agent
- Cascade assembling of pyrazolin-5-ones and benzylidenemalononitriles: the facile and efficient approach to medicinally relevant spirocyclopropylpyrazolone scaffold
- Synthesis, characterization and bioactivity of novel 5,6-dihydropyrrolo[3,4-c]pyrazol-4- (1H)one derivatives
- Molecular modeling and synthesis of new 1,5-diphenylpyrazoles as breast cancer cell growth inhibitors
- An efficient synthesis of 11-aryl-10-oxo-7,8,10,11-tetrahydro-1H-[1,2,3]triazolo [4′,5′:3,4]benzo[1,2-b][1,6]naphthyridine derivatives under catalyst-free conditions
- Mechanochemical synthesis of 2,2-difluoro-4, 6-bis(β-styryl)-1,3,2-dioxaborines and their use in cyanide ion sensing
- Visible-light-mediated radical aryltrichloromethylation of N-arylacrylamides for the synthesis of trichloromethyl-containing oxindoles
- A stereolibrary of conformationally restricted amino acids based on pyrrolidinyl/piperidinyloxazole motifs
- Synthesis of [1,3]thiazolo[3,2-b][1,2,4]triazol-7-ium and [1,2,4]triazolo[5,1-b][1,3]thiazin-4-ium salts via regioselective electrophilic cyclization of 3-[(2-alken-1-yl)sulfanyl]-4H-1,2,4-triazoles
Articles in the same Issue
- Frontmatter
- Preliminary Communications
- Triphenylphosphine catalyzed domino reaction of dialkyl acetylenedicarboxylate with 3-aryl- 2-benzoylcyclopropane-1,1-dicarbonitrile
- Synthesis of colletotrichumine A
- Research Articles
- Synthesis of the spiroacetal fragments of spirofungins A and B, antibiotics isolated from Streptomyces violaceusniger Tü 4113
- Efficient synthesis and fungicidal activities of strobilurin analogues containing benzofuro [3,2-d]-1,2,4-triazolo[1,5-a]pyrimidinone side chains
- Synthesis of 2-amino-6,7,8,9-tetrahydro-6-phenethyl-3H-pyrimido[4,5-e][1,4]diazepin-4(5H)-one: a model for a potential pyrimido[4,5-e][1,4]diazepine-based folate anti-tumor agent
- Cascade assembling of pyrazolin-5-ones and benzylidenemalononitriles: the facile and efficient approach to medicinally relevant spirocyclopropylpyrazolone scaffold
- Synthesis, characterization and bioactivity of novel 5,6-dihydropyrrolo[3,4-c]pyrazol-4- (1H)one derivatives
- Molecular modeling and synthesis of new 1,5-diphenylpyrazoles as breast cancer cell growth inhibitors
- An efficient synthesis of 11-aryl-10-oxo-7,8,10,11-tetrahydro-1H-[1,2,3]triazolo [4′,5′:3,4]benzo[1,2-b][1,6]naphthyridine derivatives under catalyst-free conditions
- Mechanochemical synthesis of 2,2-difluoro-4, 6-bis(β-styryl)-1,3,2-dioxaborines and their use in cyanide ion sensing
- Visible-light-mediated radical aryltrichloromethylation of N-arylacrylamides for the synthesis of trichloromethyl-containing oxindoles
- A stereolibrary of conformationally restricted amino acids based on pyrrolidinyl/piperidinyloxazole motifs
- Synthesis of [1,3]thiazolo[3,2-b][1,2,4]triazol-7-ium and [1,2,4]triazolo[5,1-b][1,3]thiazin-4-ium salts via regioselective electrophilic cyclization of 3-[(2-alken-1-yl)sulfanyl]-4H-1,2,4-triazoles