A Mn(II) coordination polymer from a polycarboxylate-containing ligand: synthesis, structural characterization, and properties
-
Xiao-Hong Zhu
 ,
Xiao-Chun Cheng
,
Xiao-Chun Cheng

Abstract
The neutral, four-fold protonated pyridine-3,5-dicarbox(3,5-dicarboxylatoanilide) (H4L) reacts with Mn(II) salts under hydrothermal conditions to yield a new complex: [Mn2(L)(H2O)2]·H2O (1), which has been characterized by single crystal X-ray diffraction, infrared spectroscopy, and elemental and thermogravimetric analyses. Complex 1 exhibits a binodal (4,8)-connected 3D framework with flu (412.612.84)(46)2 topology. The magnetic properties of 1 were investigated.
1 Introduction
In recent years, the crystal engineering of coordination polymers has received remarkable attention in view of their beautiful topologies and potential applications [1]. Consequently, a great number of polymers with interesting compositions and properties have been deliberately prepared via self-assembly using a variety of aromatic polycarboxylic ligands and discussed in some comprehensive reviews [2]. However, the self-assembly process is rather complicated and can be influenced by many factors such as the nature of metal ions, the flexibility or rigidity of the ligands, and acidic or basic media for the reaction systems [3]. Therefore, on one hand, it is still a challenge to exactly control the compositions and structures of resultant complexes, and on the other hand, the structures and properties of resultant complexes seem adjustable, which attracted us to do further study to understand the influence of the assembly process [4].
Recently, we have focused our attention on the reactions of metal salts with a polycarboxylic ligand: pyridine-3,5-dicarbox(3,5-dicarboxylatoanilide) (H4L in its four-fold protonated form) [5]. Our goals are to assembly coordination polymers and explore the influence of intrinsic features of ligands and experimental conditions on the structures of resultant polymers. Our selection is based on the following consideration: carboxylate group could adopt various coordination patterns such as μ1-η1:η0-monodentate, μ1-η1:η1-chelating, and μ2-η1:η1-bridging modes [6]. The four carboxylic acid groups of H4L could be partially or completely deprotonated by the alkaline reagents to generate L4−, HL3−, H2L2−, or H3L− anions, which could enrich the coordination patterns of H4L. Given its variable coordination modes, H4L can be regarded as a reliable candidate as a blocking linker. In this contribution, we report the preparation and characterization of a new coordination polymer [Mn2(L)(H2O)2]·H2O (1). The thermal stability and magnetic properties of 1 were examined.
2 Results and discussion
2.1 Preparation
MnCl2·4H2O reacts with the neutral, four-fold protonated pyridine-3,5-dicarbox(3,5-dicarboxylatoanilide) (H4L) in the presence of KOH under the hydrothermal condition at 120°C to produce the complex [Mn2(L)(H2O)2]·H2O (1) which is stable in air.
2.2 Structural description of [Mn2(L)(H2O)2]·H2O (1)
Complex 1 was obtained, which is a 3D framework based on Mn(II) and the four-fold deprotonated ligand L4−. It crystallizes in the monoclinic space group C2/c (Table 1). The asymmetric unit of 1 contains one Mn(II), half of L4−, one coordinated water, and 1/2 molecule of lattice water. Mn(II) is pentacoordinated (Fig. 1a) with distorted trigonal bipyramid coordination geometry, by four carboxylate O atoms and one water O atom. The bond distances around Mn(II) are from 2.0641(15) to 2.2106(19) Å; the coordination bond angles are in the range of 81.91(7) to 165.01(7)° (Table 2). In complex 1, the H4L was wholly deprotonated as a centrosymmetric L4− anion (Scheme 1). Two different carboxylate groups all exhibit μ2-η1:η1-bridging coordination mode, but the pyridyl N atom is free of coordination. So each L4− ligand links eight metal ions as a μ8-bridge, and each Mn(II) is coordinated by four L4−. This kind of interconnection extends infinitely to form a 3D framework (Fig. 1c). If only half of centrosymmetric L4− is considered, a 2D network can be viewed (Fig. 1b). Topological analysis shows that the architecture of 1 can be simplified as a binodal (4,8)-connected 3D framework with flu (412.612.84)(46)2 topology (Fig. 1d) [7].
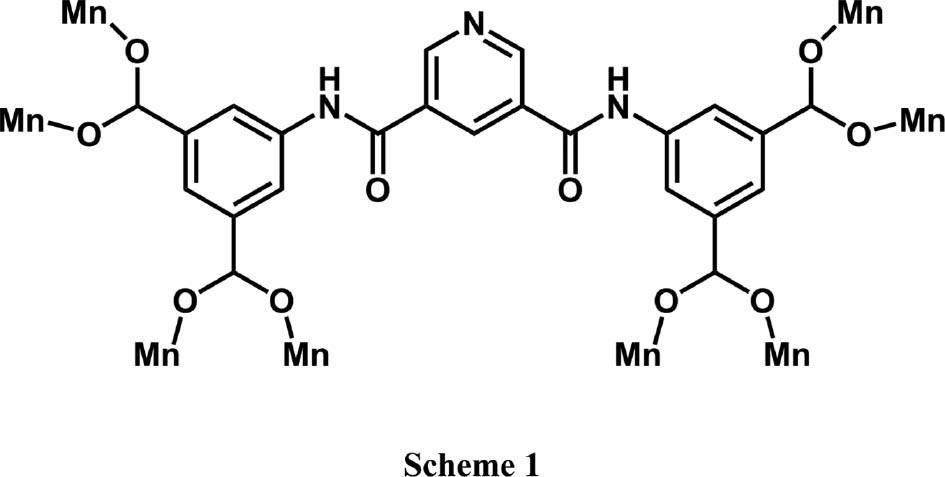
Coordination modes of H4L appearing in complex 1.
Crystal structure data for complex 1.
| [Mn2(L)(H2O)2]·H2O (1) | |
|---|---|
| Formula | C23H17N3O13Mn |
| Mr | 653.28 |
| Crystal size, mm3 | 0.10×0.10×0.10 |
| Crystal system | Monoclinic |
| Space group | C2/c |
| a, Å | 17.020(7) |
| b, Å | 16.874(6) |
| c, Å | 7.990(2) |
| β, deg | 96.393(15) |
| V, Å3 | 2280.3(13) |
| Z | 4 |
| Dcalcd., g cm−3 | 1.90 |
| μ(MoKα ), cm−1 | 1.2 |
| F(000), e | 1320 |
| hkl range | ±22, ±21, ±10 |
| θ range, deg | 3.19–27.48 |
| Refl. measured/unique/Rint | 10548/2600/0.0359 |
| Param. refined | 196 |
| R(F)a/wR(F2)b (all refls.) | 0.0394/0.0895 |
| GoF (F2)c | 1.050 |
| Δρfin (max/min), e Å−3 | 0.51/−0.34 |
aR(F)=Σ||Fo| – |Fc||/Σ|Fo|; bwR(F2)=[Σw(Fo2−Fc2)2/Σw(Fo2)2]1/2; w=[σ2(Fo2)+(AP)2+BP]−1, where P=(Max(Fo2, 0)+2Fc2)/3; cGoF=S=[Σw(Fo2−Fc2)2/(nobs−nparam)]1/2.
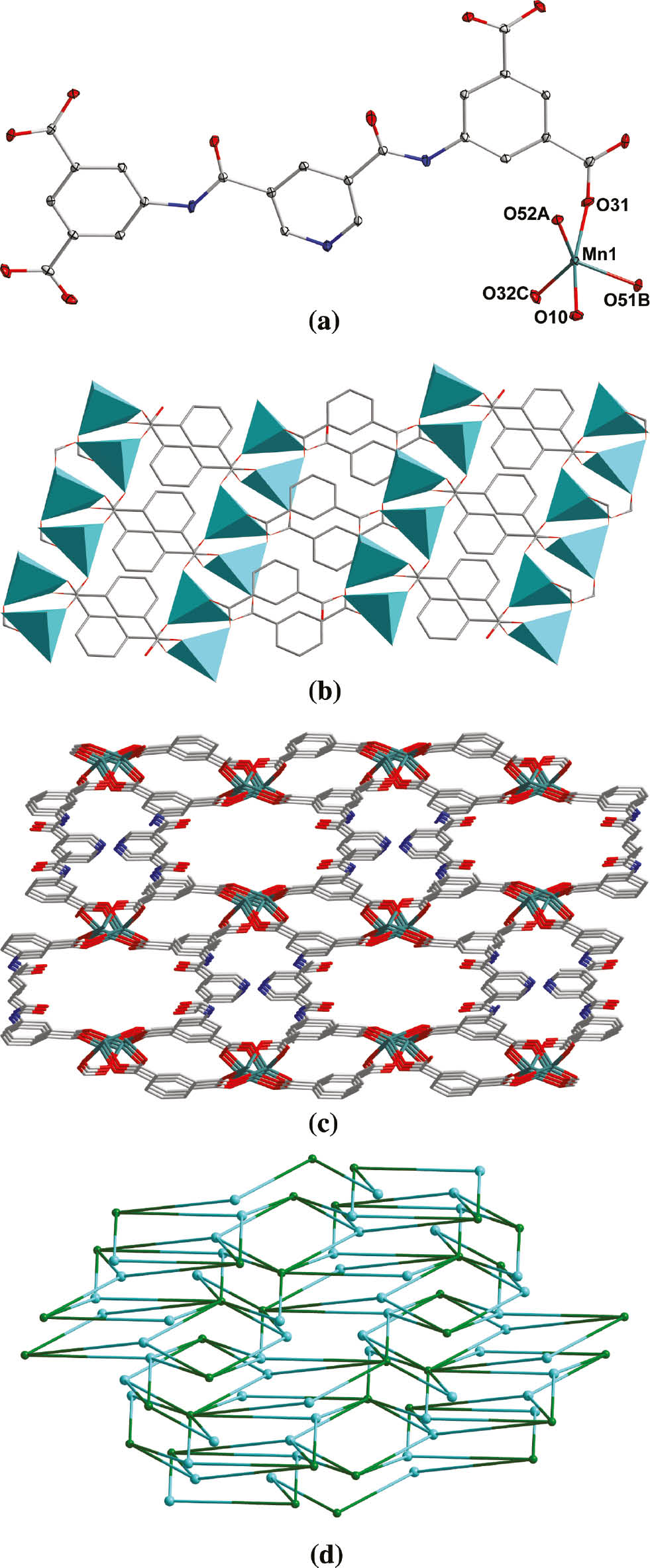
(a) The coordination environment of Mn(II) in complex 1 with the ellipsoids drawn at the 30% probability level. Hydrogen atoms and lattice water molecules are omitted for clarity. (b) View of the 2D network in 1. (c) View of the 3D architecture of 1. (d) Topological view of the 3D framework of 1.
Selected bond lengths (Å) and angles (deg) for complex 1a.
| [Mn2(L)(H2O)2]·H2O (1) | |||
|---|---|---|---|
| Mn(1)–O(10) | 2.211(2) | Mn(1)–O(31) | 2.1389(19) |
| Mn(1)–O(32)#1 | 2.0781(19) | Mn(1)–O(51)#2 | 2.0641(18) |
| Mn(1)–O(52)#3 | 2.0849(17) | ||
| O(10)–Mn(1)–O(31) | 165.00(7) | O(10)–Mn(1)–O(32)#1 | 94.54(7) |
| O(10)–Mn(1)–O(51)#2 | 81.91(7) | O(10)–Mn(1)–O(52)#3 | 84.91(6) |
| O(31)–Mn(1)–O(32)#1 | 99.71(7) | O(31)–Mn(1)–O(51)#2 | 90.62(6) |
| O(31)–Mn(1)–O(52)#3 | 89.07(6) | O(32)#1–Mn(1)–O(51)#2 | 101.58(7) |
| O(32)#1–Mn(1)–O(52)#3 | 131.64(6) | O(51)#2–Mn(1)–O(52)#3 | 125.97(6) |
aSymmetry transformations used to generate equivalent atoms: for 1: #1 x, 1−y, 1/2+z; #2 1/2−x, 1/2+y, 1/2−z; #3 1/2−x, 1/2−y, 1−z.
2.3 Thermal stability of complex 1
Thermogravimetric analysis (TGA) was carried out for complex 1, and the result is shown in Fig. 2. There is a weight loss of 3.0% in the temperature range of 90–120°C which corresponds to the liberation of lattice water (calcd 2.8%). A weight loss of 5.6% from 150 to 220°C is assigned to the release of coordinated water (calcd 5.5%), and the decomposition of the residue can be observed at 420°C.
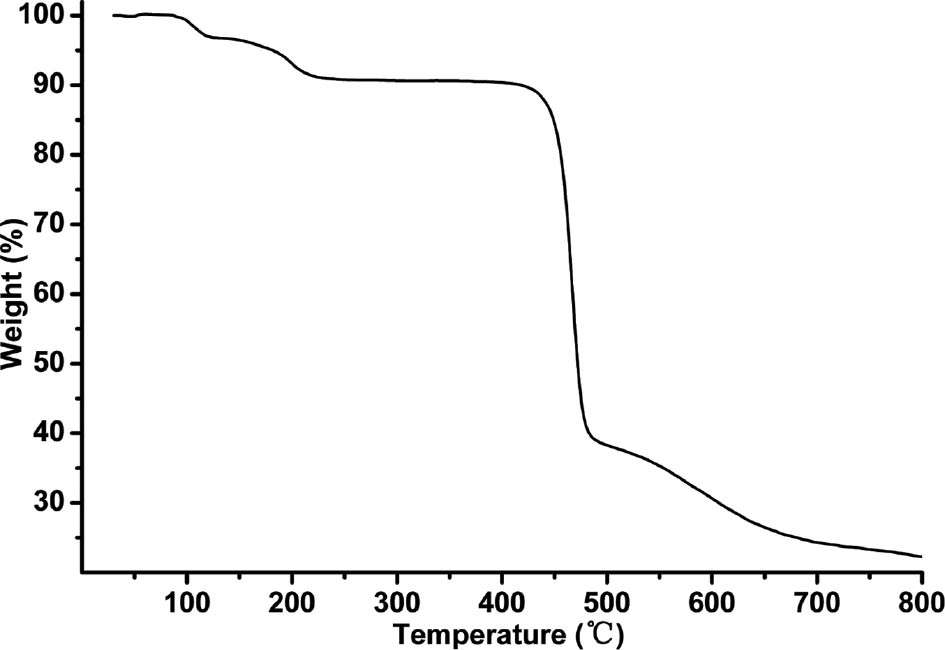
TGA curve of complex 1.
2.4 Magnetic properties of complex 1
The Mn(II) cations in 1 are bridged by carboxylate, which may mediate magnetic interactions [8]. Thus, the temperature dependence of the magnetic susceptibility of 1 was investigated from 300 to 1.8 K with an applied magnetic field of 2000 Oe. The χM, χM−1, and χMT vs. T curves for 1 are shown in Fig. 3.
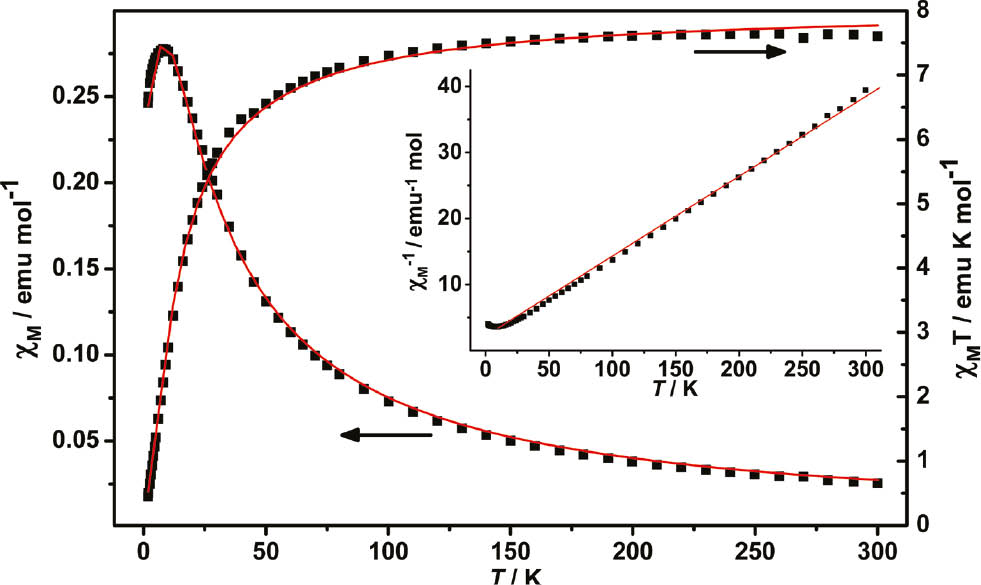
Temperature dependence of magnetic susceptibility χM, χM−1, and χMT for 1. The solid lines represent the fitted curve.
The value of χMT at 300 K is 7.66 emu K mol−1 which is larger than the value expected for a magnetically isolated Mn(II) cation (4.38 emu K mol−1, g=2.0) due to spin–orbital coupling, indicating a significant orbital contribution [8]. The temperature dependence of χM−1 above 50 K obeys the Curie–Weiss equation of χM−1=(T–θ)/C with the constants C=8.27 cm3 mol−1 K and θ=−18.4 K.
The negative value of θ and the shape of the χMT vs. T curve suggest that there may exist antiferromagnetic interactions between the neighboring Mn(II) centers [9]. In order to estimate the strength of the magnetic interactions in 1, the following equation was used [10]:
Here, A+B equals the Curie constant (C), and E1, E2 represent the ‘activation energies’ corresponding to the spin–orbit coupling and the magnetic exchange interaction, respectively. The obtained values of A+B=8.34 cm3 mol−1 K and E1/k=10.6 K agree with those given in a previous report [10]. The value of −E2/k=−1.68 K corresponding to J=−3.76 K further proves that antiferromagnetic interactions exist between neighboring Mn(II) ions in 1 [11].
3 Experimental section
All commercially available chemicals were of reagent grade and used as received without further purification. The H4L ligand was synthesized via the experimental procedure reported in the literature [5]. Elemental analysis of C, H, and N was taken on a Perkin-Elmer 240C elemental analyzer. Infrared spectra (IR) were recorded on a Bruker Vector22 FT-IR spectrophotometer by using KBr pellets. TGA was performed on a simultaneous SDT 2960 thermal analyzer under nitrogen atmosphere with a heating rate of 10°C min−1. The magnetic measurement in the temperature range of 1.8–300 K was carried out on a Quantum Design MPMS7 SQUID magnetometer in a field of 2 kOe (1 kOe=7.96×104 A m−1). Diamagnetic corrections were made with Pascal’s constants.
3.1 Preparation of [Mn2(L)(H2O)2]·H2O (1)
The reaction mixture of H4L (0.05 mmol, 24.7 mg), MnCl2·4H2O (0.10 mmol, 14.3 mg), and KOH (11.2 mg, 0.2 mmol) in 10 mL H2O was sealed in a 16 mL Teflon-lined stainless steel container and heated at 120°C for 3 days. After cooling to room temperature, colorless block crystals of 1 were collected by filtration and washed with water and ethanol several times (yield 50% based on H4L). – C23H17N3O13Mn2 (653.28): calcd. C 42.29, H 2.62, N 6.43; found C 42.50, H 2.42, N 6.26%. – IR (KBr pellet, cm−1): ν=3459 (m), 1621 (s), 1570 (s), 1468 (s), 1440 (s), 1417 (s), 1359 (s), 1238 (m), 1139 (m), 1061 (s), 879 (m), 762 (s), 733 (m), 647 (m), 586 (m).
3.2 X-ray structure determination
The crystallographic data collection for complex 1 was carried out on a Bruker Smart ApexII CCD area-detector diffractometer using graphite-monochromatized MoKα radiation (λ=0.71073 Å) at T=293(2) K. The diffraction data were integrated by using the program Saint [12], which was also used for the intensity corrections for Lorentz and polarization effects. Semi-empirical absorption corrections were applied using the program Sadabs [13]. The structure of 1 was solved by Direct Methods, and all non-hydrogen atoms were refined anisotropically on F2 by the full-matrix least-squares techniques using the Shelxs/l-97 crystallographic software package [14], [15], [16], [17], [18]. In 1, all hydrogen atoms at C atoms were generated geometrically, while the ones of O1, O10, and N1 were found at reasonable positions in the difference Fourier maps and located there. The bond distance of N(1)–H(1) was restricted to 0.87±0.01 Å for a more reasonable length. The details of crystal parameters, data collection, and the graphite refinement are summarized in Table 1, and selected bond lengths and angles are listed in Table 2.
CCDC 1501754 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgments
The authors gratefully acknowledge Natural Science Foundation for Universities in Jiangsu Province (16KJB150005) and Huaiyin Institute of Technology (15HGZ006 and 491713325) for financial support of this work.
References
[1] T. R. Cook, Y. R. Zheng, P. J. Stang, Chem. Rev.2013, 113, 734.10.1021/cr3002824Search in Google Scholar
[2] L. Luo, K. Chen, Q. Liu, Y. Lu, T. A. Okamura, G. C. Lv, Y. Zhao, W. Y. Sun, Cryst. Growth Des.2013, 13, 2312.10.1021/cg301815wSearch in Google Scholar
[3] L. Luo, G. C. Lv, P. Wang, Q. Liu, K. Chen, W. Y. Sun, CrystEngComm2013, 15, 9537.10.1039/c3ce41056kSearch in Google Scholar
[4] F. L. Mao, J. Q. Tao, C. H. Dai, Z. Naturforsch.2015, 70b, 95.10.1515/znb-2014-0193Search in Google Scholar
[5] X. H. Zhu, J. J. Xia, H. Y. Sang, J. Chen, Z. Naturforsch.2017, 72b, 183.10.1515/znb-2016-0226Search in Google Scholar
[6] H. W. Kuai, X. C. Cheng, X. H. Zhu, J. Coord. Chem.2011, 64, 3323.10.1080/00958972.2011.608846Search in Google Scholar
[7] H. W. Kuai, X. C. Cheng, D. H. Li, T. Hu, X. H. Zhu, J. Solid State Chem.2015, 228, 65.10.1016/j.jssc.2015.04.021Search in Google Scholar
[8] J. W. Raebiger, J. L. Manson, R. D. Sommer, U. Geiser, A. L. Rheingold, J. S. Miller, Inorg. Chem.2001, 40, 2578.10.1021/ic001379tSearch in Google Scholar
[9] J. M. Herrera, A. Bleuzen, Y. Dromzée, M. Julve, F. Lloret, M. Verdaguer, Inorg. Chem.2003, 42, 7052.10.1021/ic034188+Search in Google Scholar
[10] J. M. Rueff, N. Masciocchi, P. Rabu, A. Sironi, A. Skoulios, Chem. Eur. J.2002, 8, 1813.10.1002/1521-3765(20020415)8:8<1813::AID-CHEM1813>3.0.CO;2-GSearch in Google Scholar
[11] J. M. Rueff, N. Masciocchi, P. Rabu, A. Sironi, A. Skoulios, Eur. J. Inorg. Chem.2001, 11, 2843.10.1002/1099-0682(200111)2001:11<2843::AID-EJIC2843>3.0.CO;2-TSearch in Google Scholar
[12] Saint, Program for Data Extraction and Reduction, Bruker AXS Inc., Madison, WI (USA) 2001.Search in Google Scholar
[13] G. M. Sheldrick, Sadabs, Program for Bruker Area Detector Absorption Correction, University of Göttingen, Göttingen (Germany) 1997.Search in Google Scholar
[14] G. M. Sheldrick, Shelxs/l-97, Programs for the Determination of Crystal Structure, University of Göttingen, Göttingen (Germany) 1997.Search in Google Scholar
[15] G. M. Sheldrick, Acta Crystallogr.1990, A46, 467.10.1107/S0108767390000277Search in Google Scholar
[16] G. M. Sheldrick, Acta Crystallogr.2008, A64, 112.10.1107/S0108767307043930Search in Google Scholar
[17] G. M. Sheldrick, Acta Crystallogr.2013, A69, s74.10.1107/S0108767313099364Search in Google Scholar
[18] G. M. Sheldrick, Acta Crystallogr.2015, C71, 3.Search in Google Scholar
©2017 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this Issue
- Improvement of the Van Leusen reaction in the presence of β-cyclodextrin: a green and efficient synthesis of oxazoles in water
- Nano-colloidal silica-tethered polyhedral oligomeric silsesquioxanes with eight branches of 3-aminopropyltriethoxysilane as high-performance catalyst for the preparation of bis-thiazolidinones under ultrasonic conditions
- A Mn(II) coordination polymer from a polycarboxylate-containing ligand: synthesis, structural characterization, and properties
- Syntheses, crystal structures and phosphorescence properties of cyclometalated iridium(III) bis(pyridylbenzaldehyde) complexes with dithiolate ligands
- A supramolecular tetranuclear zinc(II) complex constructed from an asymmetrical Salamo-type ligand: synthesis, structure and fluorescence properties
- Bioactive chemical constituents from the resin of Aloe vera
- Synthesis and structural characterization of Li3K3Eu7(BO3)9
- Ni6B22O39·H2O – extending the field of nickel borates
- High-pressure synthesis and crystal structure of α-Y2B4O9
- β-Y(BO2)3 – a new member of the β-Ln(BO2)3 (Ln=Nd, Sm, Gd–Lu) structure family
- Antiferromagnetic ordering in the plumbide EuPdPb
- Note
- Single-crystal structure refinement of YbF2 with a remark about YbH2
Articles in the same Issue
- Frontmatter
- In this Issue
- Improvement of the Van Leusen reaction in the presence of β-cyclodextrin: a green and efficient synthesis of oxazoles in water
- Nano-colloidal silica-tethered polyhedral oligomeric silsesquioxanes with eight branches of 3-aminopropyltriethoxysilane as high-performance catalyst for the preparation of bis-thiazolidinones under ultrasonic conditions
- A Mn(II) coordination polymer from a polycarboxylate-containing ligand: synthesis, structural characterization, and properties
- Syntheses, crystal structures and phosphorescence properties of cyclometalated iridium(III) bis(pyridylbenzaldehyde) complexes with dithiolate ligands
- A supramolecular tetranuclear zinc(II) complex constructed from an asymmetrical Salamo-type ligand: synthesis, structure and fluorescence properties
- Bioactive chemical constituents from the resin of Aloe vera
- Synthesis and structural characterization of Li3K3Eu7(BO3)9
- Ni6B22O39·H2O – extending the field of nickel borates
- High-pressure synthesis and crystal structure of α-Y2B4O9
- β-Y(BO2)3 – a new member of the β-Ln(BO2)3 (Ln=Nd, Sm, Gd–Lu) structure family
- Antiferromagnetic ordering in the plumbide EuPdPb
- Note
- Single-crystal structure refinement of YbF2 with a remark about YbH2