Catalytic activity of the nanoporous MCM-41 surface for the Paal–Knorr pyrrole cyclocondensation
-
Kioumars Aghapoor
 ,
Mostafa M. Amini
,
Mostafa M. Amini

Abstract
The investigation of different oxide surfaces revealed that nanoporous silica (MCM-41) had the best catalytic activity for Paal–Knorr pyrrole synthesis. Despite the same composition, MCM-41 proved to be more effective than SiO2 itself, probably due to a significantly higher surface area of the SiO2 nanopores. The important features of this “clean” solvent-free protocol are the ease of recovery and the reuse of the catalyst for several cycles, operational simplicity, and easy product isolation and purification.
1 Introduction
The importance of heterogeneous catalysis has been well established for many processes in chemical industry [1, 2]. Indeed these heterogeneous catalysts play a key role in the development of environmentally benign processes and have several advantages such as easy handling and separation from reactants, improved recovery in order to be recycled, and cost-effectiveness in large scale. As the reaction occurs between the surface of these solid catalysts and reactants, scientific research aims to have a better understanding of these kinds of catalyzed chemical processes [3].
Pyrroles and their derivatives constitute an important class of heterocyclic compounds. The biological and medicinal properties of these compounds have been well established in the literature [4–6]. Several methodologies have already been reported to construct the pyrrole skeleton. The Paal–Knorr reaction is by far the simplest method employed for the condensation of 1,4-dicarbonyl compounds with primary amines in order to generate pyrroles [7–9].
Numerous catalysts and protocols have already been implemented to synthesize 2,5-disubstituted pyrroles, e.g., Lewis acids [10–14], heterogeneous solids having active acidic sites [15–21], ionic liquids [22, 23], deep eutectic solvents [24], an enzyme [25], and organocatalysts [26, 27]. Nevertheless, from the standpoint of synthesis, some of these procedures have restricted activity and suffer from operational and practical problems; they are costly to prepare and time consuming (causing high labor time). The sensitivity of some substituted substrates to the use of Lewis acid catalysts is also considered as a serious limitation. Therefore, the development of a mild and greener alternative remains essential in terms of sustainability for the Paal–Knorr synthesis of pyrroles.
Since the discovery of MCM-41 in 1992 [28, 29], a plethora of studies have been carried out on these mesoporous materials. These materials are well characterized and their physical properties are usually provided by X-ray diffraction (XRD) and nitrogen physisorption studies. The prominent features of MCM-41 reside in their large specific surface area and pore volume, uniform pore diameter, and high thermal stability. Due to these interesting characteristics, the scientific research in the preparation and use of MCM-41 (absorbent or catalyst) as a host for incorporation of different metals or organic moieties in chemical processes has increased exponentially [30–34]. However, the use of MCM-41 as a catalyst (by itself) has not received much attention.
Recently, we have observed the remarkable inherent catalytic activity of MCM-41 mesoporous materials for the preparation of aza heterocyclic compounds, e.g., polyhydroquinolines [35], benzoxazoles [36], and tetra-substituted imidazoles [37].
Taking into account the importance of MCM-41 mesoporous molecular sieves as potential solid catalysts for the synthesis of N-containing heterocycles and our interest in the development of greener protocols for N-heterocyclic condensation [38–40], we decided to explore the viability of using MCM-41 to promote the Paal–Knorr pyrrole synthesis (Scheme 1) that, to the best of our knowledge, has not been reported yet.
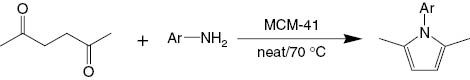
Condensation of hexane-2,5-dione with amines catalyzed by MCM-41.
2 Results and discussion
The mesoporous MCM-41 material was prepared through hydrothermal treatment following a methodology established by Gonçalves and co-workers [41]. The prepared material was characterized by XRD and nitrogen physisorption measurements.
2.1 Characterization of MCM-41
A small-angle powder XRD pattern of MCM-41 is presented in Fig. 1. The XRD pattern of MCM-41 shows four reflections: a very intense peak (1 0 0) and three additional high-order peaks (1 1 0), (2 0 0), and (2 1 0) with lower intensities. This pattern is characteristic of a hexagonal pore structure.
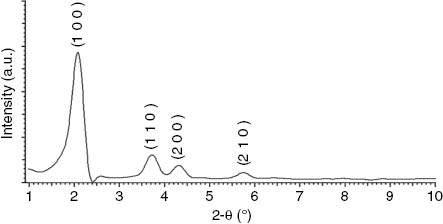
Small-angle powder XRD pattern of MCM-41.
Nitrogen sorption isotherms at 77 K for the prepared sample are presented in Fig. 2. The isotherm can be classified as type IV according to the IUPAC convention, and is typical of mesoporous materials [42].
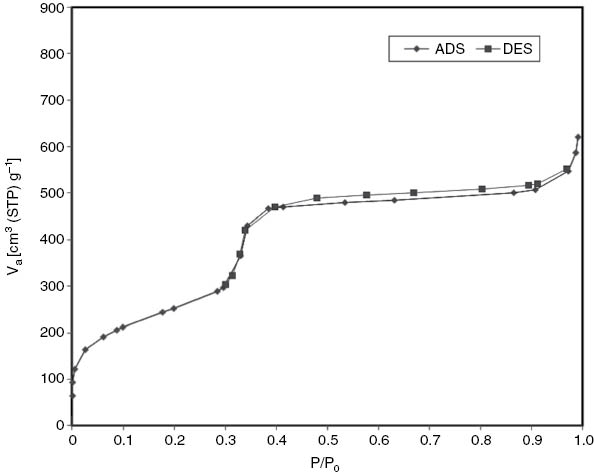
N2 adsorption–desorption isotherm of MCM-41.
2.2 Catalyst screening of various mineral oxides
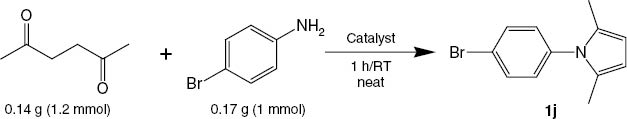
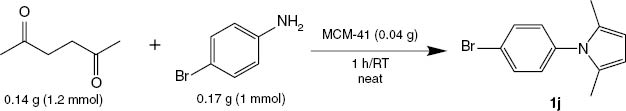
Initially, we examined the condensation of hexane-2,5-dione (1.2 mmol) with 4-bromoaniline (1 mmol) to evaluate the efficiency of various mineral oxides as catalysts (Table 1). The reaction was screened under solvent-free conditions at room temperature within 1 h.
Catalyst screening of various mineral oxides for the synthesis of 1j under neat conditions at room temperature within 1 h.
 | |||
|---|---|---|---|
| Entry | Catalyst, g | Time, min | Conversion, %a |
| 1 | – | 60 | 38 |
| 2 | MCM-41-HTb (0.02) | 60 | 60 |
| 3 | MCM-41-HT (0.04) | 60 | 65 |
| 4 | MCM-41-HT (0.06) | 60 | 61 |
| 5 | SiO2 (0.04) | 60 | 42 |
| 6 | TiO2 anatase (0.04) | 60 | 41 |
| 7 | Al2O3 neutral (0.04) | 60 | 15 |
| 8 | Al2O3 acidic (0.04) | 60 | 18 |
| 9 | Al2O3 basic (0.04) | 60 | 10 |
| 10 | ZnO (0.04) | 60 | 16 |
| 11 | Fe3O4 (0.04) | 60 | 22 |
aGas chromatography assay (%).
bMCM-41 prepared using a hydrothermal method.
Among these solid surfaces, Fe3O4, ZnO, and basic, neutral, and acidic Al2O3 showed poor catalytic activities (Table 1, entries 7–11). TiO2 and SiO2 gave moderate yields, whereas MCM-41 catalyzed the reaction to afford the desired product in good yield.
2.3 Catalyst screening of various MCM-41 materials
Various MCM-41 surfaces prepared using the conventional hydrothermal method and microwave heating were tested for the same reaction (Table 2). The reaction in the presence of various MCM-41 materials resulted in 57–65 % yield of product. However, a 10–18 % decrease was observed for the sample prepared using microwaves at 480 W for 20 min (Table 2, entry 8). Porosimetry data showed that the catalyst had a high surface area in the range of 1054–1275 m2 g−1. Nevertheless, it seems that the yield of product is a function of the surface area of the catalyst, and below 1100 m2 g−1, a notable decrease in the catalytic activity is observed (Table 2, entries 1–8).
Catalytic effect of various MCM-41 materials on the synthesis of 1j under neat conditions.
 | |||
|---|---|---|---|
| Entry | Catalyst | BET surface area, m2 g–1 | Conversion, %a |
| 1 | MCM-41-HTb | 1275 | 65 |
| 2 | MCM-41-360 W-20 minc | 1151 | 60 |
| 3 | MCM-41-360 W-40 minc | 1137 | 58 |
| 4 | MCM-41-360 W-60 minc | 1129 | 57 |
| 5 | MCM-41-360 W-90 minc | 1119 | 57 |
| 6 | MCM-41-300 W-20 minc | 1119 | 57 |
| 7 | MCM-41-420 W-20 minc | 1177 | 60 |
| 8 | MCM-41-480 W-20 minc | 1054 | 47 |
aGas chromatography assay (%).
bMCM-41 prepared using a hydrothermal method.
cMCM-41 prepared using microwave heating.
2.4 Optimization of the model reaction
To investigate the role of mesoporous MCM-41, the same reaction was carried out in the absence or presence of a catalyst at different temperatures. In the absence of MCM-41 under ambient (i.e., room temperature) and thermal conditions (i.e., 50 °C and 70 °C), the uncatalyzed reaction afforded the product 1j in 38 %, 70 %, and 82 % yield, respectively (Table 3, entries 1–3). When the same reaction was catalyzed by MCM-41, the reaction yield sharply increased under various conditions (Table 3, entries 4–6); e.g., in the presence of MCM-41 as a catalyst, the product 1j was obtained in 65 % yield (instead of 38 %) at room temperature. Accordingly, the MCM-41 catalyzed reaction at 70 °C after 1 h exhibited the highest conversion rate and afforded the product 1j quantitatively (Table 3, entry 6).
Temperature effect on the synthesis of 1j under neat conditions.
| Entry | Catalyst | Temperature, °C | Time, min | Conversion, %a |
|---|---|---|---|---|
| 1 | – | 25 | 60 | 38 |
| 2 | – | 50 | 60 | 70 |
| 3 | – | 70 | 60 | 82 |
| 4 | MCM-41-HTb (0.04) | 25 | 60 | 65 |
| 5 | MCM-41-HT (0.04) | 50 | 60 | 92 |
| 6 | MCM-41-HT (0.04) | 70 | 60 | 100 |
| 7 | MCM-41-HT (0.04) | 70 | 30 | 88 |
aGas chromatography assay (%).
bMCM-41 prepared using a hydrothermal method.
2.5 Evaluation of the reaction scope
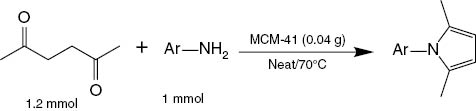
To expand the generality of this novel catalytic method, various aromatic primary amines were tested under the optimized conditions and the results are summarized in Table 4, indicating that both electron-donating (such as methoxy, methyl, and dimethyl) and electron-withdrawing (such as bromo, chloro, dichloro, and nitro) substituents on the aromatic ring of aniline underwent smooth reaction with hexane-2,5-dione giving excellent conversions in most cases. However, an extended reaction time is observed for nitro-substituted aniline derivatives (Table 4, entries 1g–1i) having a negative influence on the reaction rate.
Solvent-free synthesis of N-substituted pyrroles catalyzed by MCM-41 at 70 °C.
 | |||||
|---|---|---|---|---|---|
| Entry | Ar | Time, h | Conversion, %a | Yield, %b | M.p., °C |
| 1a | 4-Methoxyphenyl | 1 | 100 | 95 | 56–57 |
| 1b | Phenyl | 1 | 100 | 93 | 48–50 |
| 1c | 4-Methylphenyl | 1 | 100 | 94 | 44–45 |
| 1d | 3-Methylphenyl | 1 | 100 | 93 | Oil |
| 1e | 2-Methylphenyl | 1 | 97 | 91 | Oil |
| 1f | 2,5-Dimethylphenyl | 1 | 100 | 92 | Oil |
| 1g | 4-Nitrophenyl | 1/4 | 11/35 | 7/29 | 142–144 |
| 1h | 3-Nitrophenyl | 1/4 | 54/88 | 48/80 | 85 |
| 1i | 2-Nitrophenyl | 4 | 4 | – | – |
| 1j | 4-Bromophenyl | 1 | 100 | 94 | 75 |
| 1k | 4-Chlorophenyl | 1 | 100 | 93 | 55–57 |
| 1l | 3,4-Dichlorophenyl | 4 | 96 | 88 | Oil |
| 1m | Cyclohexyl | 1 | 100 | 73 | Oil |
| 1n | tert-Butyl | 4c | – | – | – |
| 1o | iso-Propyl | 4c | 100 | 72 | Oil |
aGas chromatography assay (%).
bYields refer to those of pure isolated products.
cThe reaction was carried out at 30 °C.
For comparison, the reactions with sterically more demanding primary aliphatic amines were also studied (Table 4, entries 1m–1o). In particular, with Ar = R = tert-butyl, no reaction was observed under these conditions.
2.6 Reusability of the catalyst
The feasibility of repeated use of MCM-41 was also examined. The recovery of the catalyst was very easy. After completion of the reaction, ethanol was added to the reaction mixture. The catalyst was simply filtered from the resulting mixture, and dried at 120 °C under reduced pressure for 2 h. The reused catalyst was stable under the reaction conditions, and its activity in terms of yields slightly decreased with increasing number of cycles of the reaction (conversion for six subsequent runs: 100 %– 100 %–100 %–99 %–95 %–92 %).
3 Conclusion
The direct condensation of 4-bromoaniline and hexane-2,5-dione as a model reaction in the presence of a few mineral oxides was investigated. Among these surfaces, nanoporous silica MCM-41 prepared through hydrothermal treatment (MCM-41-HT) exhibited the highest catalytic activity with regard to the transformation at 70 °C within 1 h.
Among various nanoporous MCM-41 materials (prepared under different conditions), it was observed that MCM-41-HT had the best performance in terms of conversion and yield. It is noteworthy to mention that MCM-41-HT had the highest surface area in comparison to other MCM-41 materials and much higher surface area than silica gel itself [43] (MCM-41 ≈ 1500 m2 g–1; SiO2 ≈ 400 m2 g–1); it may be deducted that the increased catalytic activity may be due to a significantly higher surface area.
Following these results, various aromatic primary amines were tested with hexane-2,5-dione under the optimized conditions. In most cases, MCM-41 showed high to excellent catalytic activity for the reaction. The ease of MCM-41 isolation from substrates and products and its reuse as a catalyst for several cycles are very beneficial from the economic and industrial point of view and can be further expanded into other catalysis areas.
4 Experimental section
4.1 Materials and methods
Sodium silicate solution (7.5–8.5 % Na2O, 25.5–28.5 % SiO2, Merck-105621) and cetyltrimethylammonium bromide (96 %, Fluka-52370) were used for the preparation of MCM-41 without additional purification. All mineral oxides were available commercially. All other chemicals (H2SO4, HCl, and organic solvents) were of analytical quality, and water was purified in an SG Water purification system (Barsbüttel, Germany).
All of the products were characterized by a comparison of their spectroscopic data with those of authentic samples [38–40, 44].
The powder XRD pattern was recorded at room temperature from 1° to 10° in 2θ by using a D8 Advance instrument (Bruker AXS GmbH, Karlsruhe, Germany) with CuKα radiation (λ = 0.15406 nm).
The N2 adsorption/desorption analyses were performed on a BELSORP-miniII instrument at 77 K (BEL Japan). MCM-41 was degassed at 120 °C for 1.5 h under inert gas flow prior to analysis. Specific surface area, total pore volume, and pore diameter of samples were obtained by the Brunauer–Emmett–Teller (BET) method using BELSORP analysis software.
A laboratory microwave oven MW 3100 (Landgraf Laborsysteme HLL GmbH, Langenhagen, Germany) equipped with a magnetic stirrer operating at 2450 MHz was used for syntheses of MCM-41 samples. In order to prepare MCM-41 under microwave conditions, the top of the reaction vessel (placed inside the microwave chamber) was attached to a water-cooled reflux condenser, located outside of the microwave chamber, for reflux purpose. The temperature inside the vessel was monitored using a Precision Temperature Logger EBI-2 T Type 311 (possessing an external probe) from ebro® Electronic GmbH and Co KG, Ingolstadt, Germany.
4.2 Preparation of MCM-41 using a hydrothermal method (MCM-41-HT)
MCM-41 was prepared through hydrothermal treatment by following a similar methodology according to Gonçalves and co-workers [41]. The following solutions were used: (A) a solution of 9.9 g sodium silicate in 30 mL deionized water; (B) a suspension of 8.12 g of cetyltrimethylammonium bromide in 80 mL deionized water.
In a typical experiment, solution A was added dropwise to a rapidly stirred suspension B. After 1-h stirring at ambient temperature, the obtained pH was 12.0. The pH was adjusted to 10.0 using dilute sulfuric acid (2 m) within 1 h.
The mixture was then autoclaved at 373 K for 2 days in Teflon-lined stainless steel reaction vessels. The solid product was recovered by filtration and washed with deionized water, until the filtrate had the final pH 7.0. The white precipitate was dried at 373 K under vacuum and calcined at 813 K for 6 h to remove the surfactant leading to MCM-41-HT.
4.3 Preparation of MCM-41 using microwave heating
In this procedure, after adjusting the pH of the suspension to 10.0 (as mentioned above), antifoam SILFAR® S184 (0.2 g) was added to the obtained suspension and stirred for a further 1 h. The resulting mixture was loaded into the microwave vessel. The vessel was then inserted into the microwave chamber (equipped with a water-cooled reflux condenser), and the reaction was carried out maintaining the power at 300, 360, 420, and 480 W (50 %, 60 %, 70 %, and 80 % of maximum power, respectively) for an appropriate time (recorded temperature during irradiation was 96.5 °C). The solid product was recovered by filtration and washed with deionized water, until the filtrate had the final pH 7.0. The white precipitate was dried at 373 K under vacuum and calcined at 813 K for 6 h to remove the surfactant leading to MCM-41 prepared by microwave irradiation.
4.4 General procedure for Paal–Knorr pyrrole cyclocondensation
In a typical reaction, primary aromatic amine (1 mmol), hexane-2,5-dione (1.2 mmol), and MCM-41 (0.04 g) were stirred at 70 °C for the appropriate reaction time. After completion of the reaction (monitored by TLC or GC), ethanol (2 mL) was added to the reaction mixture, and the catalyst was recovered by filtration. The organic medium was removed with a rotary evaporator under reduced pressure to afford the product 1. The crude product was passed through a short column of neutral alumina [eluted with ethyl acetate–hexane (3:7)] to give the pure product.
4.5 Selected spectroscopic data
N-(4′-Bromophenyl)-2,5-dimethylpyrrole (1j): M.p. 75 °C. – 1H NMR (500 MHz, CDCl3): δ = 7.60 (d, 2H), 7.15 (d, 2H), 5.90 (s, 2H), 2.05 (s, 6H). – 13C NMR (125 MHz, CDCl3): δ = 138.0, 132.2, 129.8, 128.6, 121.5, 106.0, 12.9. – MS (EI): m/z (rel. intensity, %) = 251 (80) [M]+, 250 (100), 249 (78), 169 (30), 154 (30), 129 (20).
N-(4′-Chlorophenyl)-2,5-dimethylpyrrole (1k): M.p. 55– 57 °C. – 1H NMR (500 MHz, CDCl3): δ = 7.42 (d, 2H), 7.14 (d, 2H), 5.90 (s, 1H), 2.02 (s, 6H). – 13C NMR (125 MHz, CDCl3): δ = 137.5, 133.5, 129.5, 129.4, 128.5, 106.1, 12.9. – MS (EI): m/z (rel. intensity, %) = 206 (80), 205 (100) [M]+, 191 (10), 170 (25), 155 (25), 130 (20), 111 (10).
References
[1] F. Zaera, Catal. Lett.2012, 142, 501.10.1007/s10562-012-0801-9Search in Google Scholar
[2] D. W. Lee, B. R. Yoo, J. Ind. Eng. Chem.2014, 20, 3947.10.1016/j.jiec.2014.08.004Search in Google Scholar
[3] M. A. Barteau, Chem. Rev.1996, 96, 1413.10.1021/cr950222tSearch in Google Scholar
[4] A. Fürstner, H. Szillat, B. Gabor, R. Mynott, J. Am. Chem. Soc.1998, 120, 8305.10.1021/ja981183gSearch in Google Scholar
[5] P. A. Jacobi, L. D. Coutts, J. Guo, S. I. Hauck, S. H. Leung, J. Org. Chem.2000, 65, 205.10.1021/jo991503uSearch in Google Scholar
[6] A. Fürstner, Angew. Chem. Int. Ed.2003, 42, 3582.10.1002/anie.200300582Search in Google Scholar
[7] V. F. Ferreira, M. C. B. V. De Souza, A. C. Cunha, L. O. R. Pereira, M. L. G. Ferreira, Org. Prep. Proced. Int.2001, 33, 411.10.1080/00304940109356613Search in Google Scholar
[8] G. Minetto, L. F. Raveglia, M. Taddei, Org. Lett.2004, 6, 389.10.1021/ol0362820Search in Google Scholar
[9] U. Joshi, M. Pipelier, S. Naud, D. Dubreuil, Curr. Org. Chem.2005, 9, 261.10.2174/1385272053369132Search in Google Scholar
[10] B. K. Banik, I. Banik, M. Renteria, S. K. Dasgupta, Tetrahedron Lett.2005, 46, 2643.10.1016/j.tetlet.2005.02.103Search in Google Scholar
[11] J. Chen, H. Wu, Z. Zheng, C. Jin, X. Zhang, W. Su, Tetrahedron Lett.2006, 47, 5383.10.1016/j.tetlet.2006.05.085Search in Google Scholar
[12] Z.-H. Zhang, J.-J. Li, T.-S. Li, Ultrasonics Sonochem.2008, 15, 673.10.1016/j.ultsonch.2008.02.008Search in Google Scholar
[13] J.-X. Chen, M.-C. Liu, X.-L. Yang, J.-C. Ding, H.-Y. Wu, J. Braz. Chem. Soc.2008, 19, 877.10.1590/S0103-50532008000500011Search in Google Scholar
[14] A. A. Jafari, H. Mahmoudi, Environ. Chem. Lett.2013, 11, 157.10.1007/s10311-012-0391-1Search in Google Scholar
[15] M. Curini, F. Montanari, O. Rosati, E. Lioy, R. Margarita, Tetrahedron Lett.2003, 44, 3923.10.1016/S0040-4039(03)00810-4Search in Google Scholar
[16] B. K. Banik, S. Samajdar, I. Banik, J. Org. Chem.2004, 69, 213.10.1021/jo035200iSearch in Google Scholar
[17] M. Abid, A. Spaeth, B. Török, Adv. Synth. Catal.2006, 348, 2191.10.1002/adsc.200606200Search in Google Scholar
[18] A. Teimouri, A. N. Chermahini, Chin. J. Chem.2012, 30, 372.10.1002/cjoc.201100143Search in Google Scholar
[19] N. T. S. Phan, T. T. Nguyen, Q. H. Luu, L. T. L. Nguyen, J. Mol. Catal. A: Chem.2012, 363–364, 178.10.1016/j.molcata.2012.06.007Search in Google Scholar
[20] L. Gao, L. Bing, Z. Zhang, H. Kecheng, H. Xiaoyun, K. Deng, J. Organomet. Chem.2013, 735, 26.10.1016/j.jorganchem.2013.03.018Search in Google Scholar
[21] S. Cheraghi, D. Saberi, A. Heydari, Catal. Lett.2014, 144, 1339.10.1007/s10562-014-1197-5Search in Google Scholar
[22] B. Wang, Y. Gu, C. Luo, T. Yang, L. Yang, J. Suo, Tetrahedron Lett.2004, 45, 3417.10.1016/j.tetlet.2004.03.012Search in Google Scholar
[23] J. S. Yadav, B. V. S. Reddy, B. Eeshwaraiah, M. K. Gupta, Tetrahedron Lett.2004, 45, 5873.10.1016/j.tetlet.2004.05.152Search in Google Scholar
[24] S. Handy, K. Lavender, Tetrahedron Lett.2013, 54, 4377.10.1016/j.tetlet.2013.05.122Search in Google Scholar
[25] H. Zheng, Q. Shi, K. Du, Y. Mei, P. Zhang, Mol. Divers.2013, 17, 245.10.1007/s11030-013-9426-1Search in Google Scholar
[26] N. Azizi, A. Davoudpour, F. Eskandari, E. Batebi, Monatsh. Chem.2013, 144, 405.10.1007/s00706-012-0841-2Search in Google Scholar
[27] S. Menuel, J. Rousseau, C. Rousseau, E. Vaiciunaite, J. Dodonova, S. Tumkevicius, E. Monflier, Eur. J. Org. Chem.2014, 2014, 4356.10.1002/ejoc.201402327Search in Google Scholar
[28] C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli, J. S. Beck, Nature1992, 359, 710.10.1038/359710a0Search in Google Scholar
[29] J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins, J. L. Schlenker, J. Am. Chem. Soc.1992, 114, 10834.10.1021/ja00053a020Search in Google Scholar
[30] X. S. Zhao, G. Q. M. Lu, G. J. Millar, Ind. Eng. Chem. Res.1996, 35, 2075.10.1021/ie950702aSearch in Google Scholar
[31] A. Corma, Chem. Rev.1997, 97, 2373.10.1021/cr960406nSearch in Google Scholar
[32] P. Selvam, S. K. Bhatia, C. G. Sonwane, Ind. Eng. Chem. Res.2001, 40, 3237.10.1021/ie0010666Search in Google Scholar
[33] A. Taguchi, F. Schüth, Microporous Mesoporous Mater.2005, 77, 1.10.1016/j.micromeso.2004.06.030Search in Google Scholar
[34] S. Bhattacharyya, G. Lelong, M. L. Saboungi, J. Exp. Nanosci.2006, 1, 375.10.1080/17458080600812757Search in Google Scholar
[35] L. Nagarapu, M. Dharani Kumari, N. Vijaya Kumari, S. Kantevari, Catal. Commun.2007, 8, 1871.10.1016/j.catcom.2007.03.004Search in Google Scholar
[36] R. Hekmat Shoar, M. Heidary, M. Farzaneh, R. Malakouti, Synth. Commun.2009, 39, 1742.10.1080/00397910802585910Search in Google Scholar
[37] R. Hekmat Shoar, G. Rahimzadeh, F. Derikvand, M. Farzaneh, Synth. Commun.2010, 40, 1270.10.1080/00397910903068204Search in Google Scholar
[38] H. R. Darabi, M. R. Poorheravi, K. Aghapoor, A. Mirzaee, F. Mohsenzadeh, N. Asadollahnejad, H. Taherzadeh, Y. Balavar, Environ. Chem. Lett.2012, 10, 5.10.1016/j.jgo.2012.09.115Search in Google Scholar
[39] K. Aghapoor, L. Ebadi-Nia, F. Mohsenzadeh, M. Mohebi Morad, Y. Balavar, H. R. Darabi, J. Organomet. Chem.2012, 708–709, 25.10.1016/j.jorganchem.2012.02.008Search in Google Scholar
[40] H. R. Darabi, K. Aghapoor, A. Darestani Farahani, F. Mohsenzadeh, Environ. Chem. Lett.2012, 10, 369.10.1016/j.jgo.2012.09.115Search in Google Scholar
[41] C. D. Nunes, A. A. Valente, M. Pillinger, A. C. Fernandes, C. C. Romao, J. Rocha, I. S. Gonçalves, J. Mater. Chem.2002, 12, 1735.10.1039/b109678hSearch in Google Scholar
[42] S. Wang, Y. Shi, X. Ma, J. Gong, ACS Appl. Mater. Interfaces2011, 3, 2154.10.1021/am200380aSearch in Google Scholar PubMed
[43] G. Busca, Heterogeneous Catalytic Materials: Solid State Chemistry, Surface Chemistry and Catalytic Behaviour, Elsevier, Amsterdam, 2014, chapter 6.4, pp. 151.Search in Google Scholar
[44] H. Lee, B. H. Kim, Tetrahedron.2013, 69, 6698.10.1016/j.tet.2013.05.113Search in Google Scholar
©2015 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- EPR studies on carboxylic esters, 23 [1]. Preparation of new dialkyl azulenedicarboxylates and EPR-spectroscopic study of their radical anions
- Synthesis and crystal structure of a 3D copper(II)–silver(I) coordination polymer assembled through hydrogen bonding, π–π stacking and metal–π interactions, {[Cu(phen)2(CN)][Ag(CN)2] · 3H2O}n (phen = 1,10-phenanthroline)
- A polyoxometalate-based inorganic–organic hybrid material: synthesis, characterization structure and photocatalytic study
- Hydrogen-bonded assemblies of two organically templated borates: syntheses and crystal structures of [(1,10-phen)(H3BO3)2] and [2-EtpyH][(B5O6(OH)4]
- Catalytic activity of the nanoporous MCM-41 surface for the Paal–Knorr pyrrole cyclocondensation
- A new route for the synthesis of 4-arylacetamido-2-aminothiazoles and their biological evaluation
- Structural and spectroscopic characterization of isotypic sodium, rubidium and cesium acesulfamates
- Nd39Ir10.98In36.02 – A complex intergrowth structure with CsCl- and AlB2-related slabs
- Syntheses, structures and magnetic properties of two mononuclear nickel(II) complexes based on bicarboxylate ligands
- Synthesis and characterization of some new fluoroquinolone-barbiturate hybrid systems
- Synthesis of pyrazoles containing benzofuran and trifluoromethyl moieties as possible anti-inflammatory and analgesic agents
Articles in the same Issue
- Frontmatter
- In this Issue
- EPR studies on carboxylic esters, 23 [1]. Preparation of new dialkyl azulenedicarboxylates and EPR-spectroscopic study of their radical anions
- Synthesis and crystal structure of a 3D copper(II)–silver(I) coordination polymer assembled through hydrogen bonding, π–π stacking and metal–π interactions, {[Cu(phen)2(CN)][Ag(CN)2] · 3H2O}n (phen = 1,10-phenanthroline)
- A polyoxometalate-based inorganic–organic hybrid material: synthesis, characterization structure and photocatalytic study
- Hydrogen-bonded assemblies of two organically templated borates: syntheses and crystal structures of [(1,10-phen)(H3BO3)2] and [2-EtpyH][(B5O6(OH)4]
- Catalytic activity of the nanoporous MCM-41 surface for the Paal–Knorr pyrrole cyclocondensation
- A new route for the synthesis of 4-arylacetamido-2-aminothiazoles and their biological evaluation
- Structural and spectroscopic characterization of isotypic sodium, rubidium and cesium acesulfamates
- Nd39Ir10.98In36.02 – A complex intergrowth structure with CsCl- and AlB2-related slabs
- Syntheses, structures and magnetic properties of two mononuclear nickel(II) complexes based on bicarboxylate ligands
- Synthesis and characterization of some new fluoroquinolone-barbiturate hybrid systems
- Synthesis of pyrazoles containing benzofuran and trifluoromethyl moieties as possible anti-inflammatory and analgesic agents