Syntheses, structures and magnetic properties of two mononuclear nickel(II) complexes based on bicarboxylate ligands
-
Jun-Xia Li
 and
Zhong-Xiang Du
and
Zhong-Xiang Du

Abstract
Two new complexes, [Ni(pydc)(H2O)3] (1) and [Ni(cma)(H2O)3] (2) (H2pydc = pyridine-2,6-dicarboxylic acid, H2cma = 2-(carboxymethoxy)benzoic acid), have been synthesized and structurally characterized. In both compounds, NiII is surrounded by one tridentate chelate dianionic ligand (pydc2ˉ for 1 and cma2ˉ for 2) and three coordinated water molecules in a distorted octahedral geometry (NiNO5 for 1 and NiO6 for 2). The magnetic properties for 1 and 2 have been discussed in detail.
1 Introduction
The design, synthesis and investigation of transition metal complexes containing coordinated carboxylate ligands have been a continuously interesting subject in the development of coordination chemistry [1]. Polycarboxylate ligands are used as linkers in solid aggregates. The presence of two or more carboxylate groups in different orientations permits the construction of one-, two- or three-dimensional frameworks, which can be further reinforced by the presence of hydrogen bonds, π···π and other weak interactions [2–4]. In our previous research, we have published several bidentate carboxylate metal compounds: a mononuclear CoII complex built with imidazole-4,5-dicarboxylic acid [5], a CdII polymer constructed from pyrazine-2,3-dicarboxylic acid [5] and several 2,2′-oxydiacetic acid-based compounds [6–9]. In this paper, we describe the syntheses and crystal structures of two new nickel(II) complexes [Ni(pydc)(H2O)3] (1) and [Ni(cma)(H2O)3] (2) constructed with pyridine-2,6-dicarboxylic acid (H2pydc) and 2-(carboxymethoxy)benzoic acid (H2cma), respectively. The magnetism of 1and 2 is also studied.
2 Experimental section
H2cma was prepared by the reaction of chloroacetic acid (Tianjin Damao Chemical Reagent Factory, Tianjin, P. R. China) with 2-hydroxybenzoic acid (Tianjin Bodi Chemical Co., Ltd, Tianjin, P. R. China) [10]. All other reagents were commercially available and used without further purification. Elemental analyses for C, H and N were carried out on an Elementar Vario EL elemental analyzer (Germany Elementar Company, Germany). Temperature-dependent magnetic susceptibilities of solid samples were recorded on an MPMS Multivu magnetometer (American Quantum Design Company, USA) in the temperature range from 2 to 300 K with an applied field of 10 kOe (1 kOe = 7.96 × 104 A m–1). Pascal’s constants were used to estimate the correction for the underlying diamagnetism of the sample. All data except for the magnetic data were collected at room temperature.
2.1 Preparation of tri-aqua-pyridine-2,6-dicarboxylato-nickel(II), [Ni(pydc)(H2O)3] (1)
Ni(CH3COO)2·4H2O (Sinopharm Chemical Reagent Co., Ltd, Shanghai, P. R. China) (0.5 mmol, 0.125 g) and H2pydc (0.5 mmol, 0.084 g) were dissolved in 20 mL of methanol–H2O (Tianjin Fuyu Fine Chemical Co., Ltd, Tianjin, P. R. China) (v/v, 1:3) and then the pH of the mixture was adjusted with KOH (Tianjin De‘en Chemical Reagent Co., Ltd, Tianjin, P. R. China) (0.5 mol L–1) to pH = 7.5. The solution was filtered after being stirred for 4 h at 80 °C. Four days later, green block-shaped crystals had grown from the filtrate by slow evaporation. Yield: 57 mg (41 % based on Ni). – Anal. for C7H9NiNO7 (%): calcd. C 30.23, H 3.24, N 5.04; found: C 30.28, H 3.17, N 5.12.
2.2 Preparation of tri-aqua-2-(carboxylatomethoxy)benzoato-nickel(II), [Ni(cma)(H2O)3] (2)
Ni(CH3COO)2·4H2O (0.5 mmol, 0.125 g) and H2cma (0.5 mmol, 0.098 g) were dissolved in 20 mL of methanol–H2O (v/v, 1:1) and then the pH of the mixture was adjusted with KOH (0.5 mol L–1) to pH = 8.0. The solution was sealed in a 25 mL Teflon reactor and kept under autogeneous pressure at 130 °C for 4 days. After cooling to room temperature at a rate of 6 °C h–1, green block-shaped crystals suitable for X-ray diffraction were grown from the filtrate by slow evaporation. Yield: 74 mg (48 % based on Ni). – Anal. for C9H12NiO8 (%): calcd. C 35.19, H 3.91; found: C 35.25, H 3.84.
2.3 Single-crystal X-ray structure determinations
Two green block-shaped crystals with dimensions of 0.41 × 0.36 × 0.28 mm3 (1) and 0.41 × 0.35 × 0.29 mm3 (2) were selected for measurement. Diffraction data of 1and 2were collected at 296(2) K on a Bruker SMART APEX II CCD diffractometer (Germany) with graphite-monochromatized MoKα radiation (λ = 0.71073 Å). A total of 5805 (1) and 6250 (2) reflections were collected in the range of 3.20 ≤ θ ≤ 28.25° (1) and 2.93 ≤ θ ≤ 25.50° (2) by using ω-2θ scans. Of these reflections, 2418 (1) and 2136 (2) were unique with Rint = 0.0154 (1) and 0.0494 (2), respectively. The structures were solved by direct methods [11, 12] and refined by full-matrix least squares on F2 using the SHELXL-97 software [11, 13]. All non-hydrogen atoms were refined anisotropically. Both crystal structures were tested for higher metrical symmetry with the program Platon [14]. Indications for C-centering were found for 1 which could not be verified by subsequent tests including the routine ADDSYM in PLATON. The hydrogen atoms of the coordinated water molecules in 1 were added from difference Fourier syntheses and needed restraints for their bond lengths in the course of the further refinement. Altogether there were four least-squares restraints. The hydrogen atoms of 2 were generated geometrically and treated by a mixture of independent and constrained refinement. The crystal data and data collection and refinement parameters for 1 and 2 are summarized in Table 1. Selected bond lengths and bond angles are given in Table 2. Hydrogen bond parameters are listed in Table 3.
Crystal structure data for 1 and 2.
| Formula | C7H9NNiO7 | C9H12NiO8 |
| Mr | 277.86 | 306.90 |
| Temperature, K | 296(2) | 296(2) |
| Crystal size, mm3 | 0.41 × 0.36 × 0.28 | 0.41 × 0.35 × 0.29 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P 21/n (no. 14) | P 21/c (no. 14) |
| a, Å | 6.457(2) | 8.681(3) |
| b, Å | 23.144(7) | 6.755(3) |
| c, Å | 6.908(2) | 19.876(7) |
| β, deg | 106.736(3) | 97.614(4) |
| V, Å3 | 988.5(5) | 1155.3(7) |
| Z | 4 | 4 |
| Dcalcd., g cm–3 | 1.87 | 1.76 |
| μ(MoKα), mm–1 | 2.0 | 1.7 |
| F(000), e | 568 | 632 |
| hkl range | –8 ≤ h ≤ 8 | –10 ≤ h ≤ 10 |
| –30 ≤ k ≤ +20 | –7 ≤ k ≤ +8 | |
| –9 ≤ l ≤ +9 | –24 ≤ l ≤ +24 | |
| ((sinθ)/λ)max, Å–1 | 0.666 | 0.606 |
| Refl. measd/unique/Rint | 5805/2418/0.0154 | 6250/2136/0.0494 |
| Param. refined | 157 | 163 |
| R(F)/wR(F2)a, b (all refl.) | 0.0296/0.0660 | 0.0473/0.1134 |
| GoF (F2)c | 1.045 | 1.068 |
| A/B (weighting scheme)b | 0.0398/0.0745 | 0.0674/0.2935 |
| Δρfin (max/min), e Å–3 | 0.23/–0.36 | 0.86/–0.76 |
aR = Σ||Fo| – |Fc||/Σ|Fo|; bwR = [Σw(Fo2 – Fc2)2/Σw(Fo2)2]1/2, w = [σ2(Fo2) + (AP)2 + BP]–1, where P = (Max(Fo2, 0) + 2Fc2)/3; cGoF = [Σw(Fo2 – Fc2)2/(nobs – nparam)]1/2.
Selected bond lengths (Å) and bond angles (deg) for 1 and 2.
| Complex 1 | Complex 2 | ||
|---|---|---|---|
| Ni(1)–O(3) | 1.903 (1) | Ni(1)–O(6) | 1.986(2) |
| Ni(1)–N(1) | 1.909(2) | Ni(1)–O(5) | 2.019(2) |
| Ni(1)–O(5) | 2.040 (1) | Ni(1)–O(3) | 2.023(2) |
| Ni(1)–O(4) | 2.040 (1) | Ni(1)–O(1) | 2.049(2) |
| Ni(1)–O(1) | 2.385 (1) | Ni(1)–O(2) | 2.058(2) |
| Ni(1)–O(2) | 2.509(1) | Ni(1)–O(7) | 2.097(2) |
| N(1)–Ni(1)–O(4) | 80.44(4) | O(6)–Ni(1)–O(3) | 96.85(9) |
| N(1)–Ni(1)–O(5) | 80.48(4) | O(6)–Ni(1)–O(2) | 89.67(9) |
| O(3)–Ni(1)–O(5) | 99.47(4) | O(1)–Ni(1)–O(2) | 179.27(9) |
| O(3)–Ni(1)–O(4) | 99.58(4) | O(6)–Ni(1)–O(5) | 167.77(9) |
| O(1)–Ni(1)–O(2) | 172.39(5) | O(3)–Ni(1)–O(7) | 175.17(9) |
| O(3)–Ni(1)–N(1) | 177.44(5) | O(5)–Ni(1)–O(3) | 95.34(9) |
| O(5)–Ni(1)–O(4) | 160.92(5) | O(5)–Ni(1)–O(2) | 89.30(9) |
Hydrogen bond geometries (Å, deg) for 1 and 2.
| Donor–H···Acceptor | d(D–H) | d(H···A) | d(D···A) | ∠(D–H···A) |
|---|---|---|---|---|
| Complex 1 | ||||
| O(3)–H(3A)···O(4)a | 0.85 | 1.82 | 2.671(1) | 175.0 |
| O(3)–H(3B)···O(5)b | 0.85 | 1.82 | 2.671(1) | 176.6 |
| O(1)–H(1A)···O(7)c | 0.86 | 2.03 | 2.893(2) | 171.0 |
| O(1)–H(1B)···O(2)d | 0.88 | 1.98 | 2.855(2) | 177.0 |
| O(2)–H(2A)···(6)e | 0.87 | 2.04 | 2.914(2) | 171.0 |
| O(2)–H(2B)···O(1)f | 0.85 | 2.19 | 3.032(2) | 170.3 |
| Complex 2 | ||||
| O(3)–H(6W)···O(4)g | 0.85 | 1.92 | 2.768(3) | 173.6 |
| O(3)–H(5W)···O(8)h | 0.85 | 1.88 | 2.698(3) | 160.0 |
| O(2)–H(4W)···O(5)g | 0.85 | 1.91 | 2.755(3) | 169.6 |
| O(2)–H(3W)···O(4)i | 0.85 | 2.02 | 2.835(3) | 160.2 |
| O(1)–H(2W)···O(6)h | 0.85 | 1.99 | 2.810(3) | 163.3 |
| O(1)–H(1W)···O(8)j | 0.85 | 1.90 | 2.723(3) | 162.9 |
Symmetry operations: ax – 1/2, −y + 3/2, z − 1/2; bx + 1/2, −y + 3/2, z + 1/2; cx−1, y, z; dx, y, z + 1; ex + 1, y, z; fx + 1/2, −y + 3/2, z − 1/2; g−x, y + 1/2, −z + 1/2; h−x + 1, y − 1/2, −z + 1/2; ix, y + 1, z; jx, y − 1, z.
CCDC 917854 (1) and 960696 (2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
3 Results and discussion
3.1 Structure description for [Ni(pydc)(H2O)3] (1)
The molecular structure determination revealed that compound 1 is a mononuclear species and the asymmetric unit is comprised of one NiII ion, one fully deprotonated pyridine-2,6-dicarboxylate ligand (pydc2ˉ) and three coordinated water molecules.
The coordination sphere of the NiII center is a slightly distorted octahedron comprised of two carboxylate O (O4, O5) atoms and one pyridyl N (N1) atom of one pydc2ˉ dianion and three H2O ligands. As it is induced by the pydc2ˉ ligand, both sets of coordinating atoms are arranged in a meridional fashion (Table 2 and Fig. 1). The pydc2ˉ ligand binds to the NiII center in a tridentate chelate mode forming two five-membered chelate rings. If the atoms N1, O4, O3, O5 define an equatorial plane, the atoms O1, O2 occupy the axial positions. Ni1 deviates from the equatorial least-squares plane N1/O4/O3/O5 by 0.0297 Å toward O1, with an O1–Ni1–O2 angle of 172.39(5)°.

The molecular structure of 1 with the crystallographic atom numbering scheme adopted. Displacement ellipsoids are shown at a 30 % probability level.
The Ni–N and Ni–O(COO) bond lengths of 1 are in the range of 1.903–2.040 Å, in agreement with those of the previously published triclinic complex [Ni(pydc)(H2O)2] (1.903–2.006 Å) [15] which contains penta-coordinated NiII in an approximately square-pyramidal geometry. The Ni–O(H2O) bond lengths of 1(2.385 Å, 2.509 Å) are much longer than those in [Ni(pydc)(H2O)2] (2.150 Å) which reflects the higher coordination number in 1 as compared to [Ni(pydc)(H2O)2].
The coordinated H2O ligands are involved as donors in intermolecular hydrogen bonding interactions with coordinated and uncoordinated carboxylate O atoms of pydc2ˉ and another coordinated H2O as acceptors (Table 3). The adjacent mononuclear units of 1 are stabilized and constructed into a three-dimensional architecture by these O–H···O hydrogen bonds (Fig. 2).

3D supramolecular architecture formed by hydrogen bonding interactions between molecular units of 1.
Compound 1 is another hydrate closely related to the complex [Ni(pydc)(H2O)2] (see above) [15], but with three H2O ligands instead of two. The formation of 1 and [Ni(pydc)(H2O)2] can be ascribed to the differences in the synthesis conditions: the starting reagents (Ni(CH3COO)2 for 1, NiCl2 for [Ni(pydc)(H2O)2]), the solvent (presence or absence of methanol) and the preparative methods (conventional solution reaction for 1, hydrothermal reaction for [Ni(pydc)(H2O)2]). It proves again that the synthesis conditions may have a decisive influence on slight differences in the composition of complexes.
Compound 1should also be compared to the complex cation of the bimetallic compound [Ni(H2pydc)(H2O)3][Ce(pydc)3]·3H2O [16]. In [Ni(H2pydc)(H2O)3][Ce(pydc)3]·3H2O, the H2pydc ligand is fully protonated at Ni, while it is fully deprotonated at Ce. The Ni–N and Ni–O(COO) bond lengths in 1 (1.903–2.040 Å) are shorter than those in [Ni(H2pydc)(H2O)3][Ce(pydc)3]·3H2O (1.994–2.178 Å), while the Ni–O(H2O) bond lengths in 1(2.385, 2.509 Å) are much longer than those in [Ni(H2pydc)(H2O)3][Ce(pydc)3]·3H2O (2.030, 2.049 Å). The corresponding bond angles in 1 also show larger differences as compared with those in [Ni(H2pydc)(H2O)3][Ce(pydc)3]·3H2O.
3.2 Structure description for [Ni(cma)(H2O)3] (2)
Compound 2 is also mononuclear and the asymmetric unit contains one NiII ion, one deprotonated 2-(carboxymethoxy)benzoate dianion (cma2ˉ) and three water ligands. It is isostructural with the analogous CoII complex [Co(cma)(H2O)3] [17].
The NiII ion is coordinated by two carboxylate O (O5, O6) atoms and one ether O (O7) atom of one cma2ˉ ligand and three water molecules, exhibiting a distorted NiO6 octahedral environment (Table 2 and Fig. 3). The cma2ˉ anion coordinates to the NiII center in a tridentate chelate pattern forming one five-membered and one six-membered chelate ring. The O3, O5, O6, O7 atoms are in the equatorial plane and the O1, O2A atoms occupy the axial sites. The two sets of ligating atoms (O atoms of the cma2ˉ ligand and the three water oxygens) are arranged meridionally each. Ni1 deviates from the equatorial least-squares plane O3/O5/O6/O7 by 0.0116 Å toward O1, with an O1–Ni1–O2 angle of 179.27(9)°. The bond lengths are in the range from 1.986(2) to 2.097(2) Å, and agree well with that of the isostructural cobalt complex [Co(cma)(H2O)3] [17].
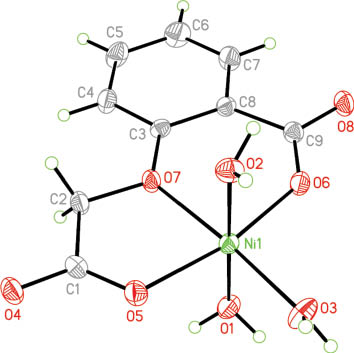
The molecular structure of 2 with the atom numbering scheme adopted. Displacement ellipsoids are shown at a 30 % probability level.
The coordinated water molecules serve as donors in intermolecular hydrogen bonds with coordinated and uncoordinated carboxylate O atoms of cma2ˉ as acceptors (Table 3). Neighboring mononuclear units of 1 are stabilized and joined into a three-dimensional network structure via these O–H···O hydrogen bonds (Fig. 4).

3D network structure formed by hydrogen bonding interactions in 2.
3.3 Magnetic properties
The magnetic susceptibility of complexes 1 and 2is measured at 1 T in the temperature range of 2–300 K. The plots of χMT versus T and 1/χM versus T are shown in Figs. 5 and 6 with χM being the molar magnetic susceptibility and T the temperature.
For 1, the molar magnetic susceptibility, χM, increases gradually as the temperature is lowered. It reaches the maximum of 0.056 cm3 mol−1 at 5 K, and then decreases sharply. The χMT value increases slowly from 300 K to 75 K. As the temperature is lowered continuously, it sharply declines to 0.099 cm3 K mol−1 at 2 K. Weak ferromagnetic exchange interactions are evident at lower temperatures. The thermal variation of the molar susceptibility obeys the Curie–Weiss law (1/χM = (T–θ)/Cm) in the temperature range of 25–250 K (inset in Fig. 5). The values of the Curie and Curie–Weiss constants are θ = 1.54 K, Cm = 0.69 cm3 K mol−1. At 300 K, χMT = 0.66 cm3 K mol−1, the magnetic moment (μeff) per NiII, which is determined by the equation μeff = 2.828(χMT)1/2, is 2.30 μB. This value is smaller than that expected for an isolated divalent high-spin NiII system with μeff = 2.83 μB.

Temperature dependence of χM (triangle block) and χMT (square block) for 1.

Temperature dependence of χM (triangle block) and χMT (square block) for 2.
For 2, χM remains nearly constant from 300 to 25 K. As the temperature descends continuously, it sharply increases to 0.57 cm3 mol−1 at 2 K. The χMT value increases gradually when the temperature drops. It reaches the peak value of 1.32 cm3 K mol−1 at 6 K, and then decreases sharply down to 2 K. The thermal variation of the molar susceptibility obeys the Curie–Weiss law over the whole range (2–300 K) (inset in Fig. 6). The values of the Curie and Curie–Weiss constants are θ = 1.92 K, Cm = 1.19 cm3 K mol−1. The positive θ value supports the presence of intermolecular ferromagnetic interactions in 2. At 300 K, χMT = 1.20 cm3 K mol−1, the μeff of per NiII is 3.10 μB. This value is higher than that expected for a spin-only isolated NiII ion in the ground state with μeff = 2.83 μB.
4 Conclusions
In summary, we have presented two new mononuclear NiII compounds 1and 2. Complex 1was obtained by the solution reaction of Ni(CH3COO)2, H2pydc and KOH in methanol–H2O, while 2 was synthesized by the solvothermal reaction of Ni(CH3COO)2, H2cma and KOH in methanol–H2O. Both complexes show intermolecular ferromagnetic interactions at lower temperatures. Although there are already many characterized structural types originating from H2pydc or H2cma as ligands, it is anticipated that new structures still can be assembled when appropriate reaction conditions are adopted.
References
[1] R. C. Mehrotra, R. Bohra, Metal Carboxylates, Academic Press, London, 1983, p. 63.Search in Google Scholar
[2] S. Konar, S. C. Manna, E. Zangrando, N. R. Chaudhuri, Inorg. Chim. Acta2004, 357, 1593.10.1016/j.ica.2003.12.003Search in Google Scholar
[3] O. Z. Yeşilel, A. Mutlu, O. Büyükgüngör, Polyhedron2009, 28, 437.10.1016/j.poly.2008.11.044Search in Google Scholar
[4] R. Noguchi, A. Sugie, A. Hara, K. Nomiya, Inorg. Chem. Commun. 2006, 9, 107.10.1016/j.inoche.2005.10.003Search in Google Scholar
[5] J.-X. Li, Z.-X. Du, L.-Z. Wang, W.-P. Huang, Inorg. Chim. Acta2011, 376, 479.10.1016/j.ica.2011.07.013Search in Google Scholar
[6] J.-X. Li, Z.-X. Du, W.-P. Huang, Z. Naturforsch.2011, 66b, 1029.10.5560/ZNB.2011.66b1029Search in Google Scholar
[7] J.-X. Li, Z.-X. Du, B.-L. Zhu, H.-Q. An, J.-X. Dong, X.-J. Hu, W.-P. Huang, Inorg. Chem. Commun.2011, 14, 522.Search in Google Scholar
[8] J.-X. Li, W.-B. Guo, Z.-X. Du, W.-P. Huang, Inorg.Chim. Acta2011, 375, 290.10.1016/j.ica.2011.05.018Search in Google Scholar
[9] J.-X. Li, Z.-X. Du, Z. Kristallogr. NCS2010, 225, 795.Search in Google Scholar
[10] Y. Liang, X.-P. Shi, Appl. Chem. Indus.2001, 30, 31.Search in Google Scholar
[11] G. M. Sheldrick, Shelxs/l-97, Programs for Crystal Structure Determination, University of Göttingen, Göttingen (Germany) 1997.Search in Google Scholar
[12] G. M. Sheldrick, Acta Crystallogr. 1990, A46, 467.10.1107/S0108767390000277Search in Google Scholar
[13] G. M. Sheldrick, Acta Crystallogr. 2008, A64, 112.10.1107/S0108767307043930Search in Google Scholar
[14] A. L. Spek, Acta Crystallogr.2009, D65, 148.10.1107/S090744490804362XSearch in Google Scholar
[15] Y. Liu, J.-M. Dou, D. Wang, X.-X. Zhang, L. Zhou, Acta Crystallogr. 2006, E62, m2208.10.1107/S1600536806032119Search in Google Scholar
[16] T. K. Prasad, M. V. Rajasekharan, Polyhedron2006, 26, 1364.Search in Google Scholar
[17] F.-Y. Meng, L.-H. Zhu, M.-H. Zeng, S. W. Ng, Acta Crystallogr. 2005, E61, m1126.10.1107/S1600536805012961Search in Google Scholar
©2015 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- EPR studies on carboxylic esters, 23 [1]. Preparation of new dialkyl azulenedicarboxylates and EPR-spectroscopic study of their radical anions
- Synthesis and crystal structure of a 3D copper(II)–silver(I) coordination polymer assembled through hydrogen bonding, π–π stacking and metal–π interactions, {[Cu(phen)2(CN)][Ag(CN)2] · 3H2O}n (phen = 1,10-phenanthroline)
- A polyoxometalate-based inorganic–organic hybrid material: synthesis, characterization structure and photocatalytic study
- Hydrogen-bonded assemblies of two organically templated borates: syntheses and crystal structures of [(1,10-phen)(H3BO3)2] and [2-EtpyH][(B5O6(OH)4]
- Catalytic activity of the nanoporous MCM-41 surface for the Paal–Knorr pyrrole cyclocondensation
- A new route for the synthesis of 4-arylacetamido-2-aminothiazoles and their biological evaluation
- Structural and spectroscopic characterization of isotypic sodium, rubidium and cesium acesulfamates
- Nd39Ir10.98In36.02 – A complex intergrowth structure with CsCl- and AlB2-related slabs
- Syntheses, structures and magnetic properties of two mononuclear nickel(II) complexes based on bicarboxylate ligands
- Synthesis and characterization of some new fluoroquinolone-barbiturate hybrid systems
- Synthesis of pyrazoles containing benzofuran and trifluoromethyl moieties as possible anti-inflammatory and analgesic agents
Articles in the same Issue
- Frontmatter
- In this Issue
- EPR studies on carboxylic esters, 23 [1]. Preparation of new dialkyl azulenedicarboxylates and EPR-spectroscopic study of their radical anions
- Synthesis and crystal structure of a 3D copper(II)–silver(I) coordination polymer assembled through hydrogen bonding, π–π stacking and metal–π interactions, {[Cu(phen)2(CN)][Ag(CN)2] · 3H2O}n (phen = 1,10-phenanthroline)
- A polyoxometalate-based inorganic–organic hybrid material: synthesis, characterization structure and photocatalytic study
- Hydrogen-bonded assemblies of two organically templated borates: syntheses and crystal structures of [(1,10-phen)(H3BO3)2] and [2-EtpyH][(B5O6(OH)4]
- Catalytic activity of the nanoporous MCM-41 surface for the Paal–Knorr pyrrole cyclocondensation
- A new route for the synthesis of 4-arylacetamido-2-aminothiazoles and their biological evaluation
- Structural and spectroscopic characterization of isotypic sodium, rubidium and cesium acesulfamates
- Nd39Ir10.98In36.02 – A complex intergrowth structure with CsCl- and AlB2-related slabs
- Syntheses, structures and magnetic properties of two mononuclear nickel(II) complexes based on bicarboxylate ligands
- Synthesis and characterization of some new fluoroquinolone-barbiturate hybrid systems
- Synthesis of pyrazoles containing benzofuran and trifluoromethyl moieties as possible anti-inflammatory and analgesic agents