
New insights of epigenetics in vascular and cellular senescence
-
Menglin Zhu
 und
Yizhun Zhu
und
Yizhun Zhu

ABSTRACT
Vascular senescence plays a vital role in cardiovascular diseases and it is closely related to cellular senescence. At the molecular level, aging begins with a single cell, and it is characterized by telomere shortening, mitochondrial dysfunction, stem cell exhaustion, epigenetic changes, and so on. Epigenetics is an independent discipline that modifies DNA activity without altering the DNA sequence. The application of epigenetics helps to alleviate the occurrence of human diseases, inhibit senescence, and even inhibit tumor occurrence. Epigenetics mainly includes the modification of DNA, histone, and noncoding RNA. Herein, the application of epigenetics in vascular senescence and aging has been reviewed to provide the prospects and innovative inspirations for future research.
INTRODUCTION
Aging involves a variety of complex physiological mechanisms. It is characterized by a gradual decline of physiological function over time and is a feature of various physiological and pathological processes,[1] and is connected with the risk of cancer, diabetes, cardiovascular and neurodegenerative diseases.[2] So, understanding the mechanisms associated with aging is important. Among the causes of cardiovascular diseases, vascular aging occupies a large proportion. In the process of vascular aging, there will be various changes in vascular function, including vascular homeostasis disorders and vascular remodeling.[3] Previous studies of our group have shown that Smyd3, GATA4, PARP16, and Fra-1 can become new targets for the treatment of vascular aging by regulating vascular inflammation, injury, and function.
There are several senescence hallmarks such as mitochondrial dysfunction, genomic instability, oxidative stress, telomere attrition, stem cell exhaustion, and so on.[4] Senescence is a response to damage; it can play a beneficial role in wound healing, tissue repair, and embryonic development.[1,5,6] The abnormal accumulation of senescent cells with age can have harmful effects. The main factors that cause senescence and induce cell senescence are telomere shortening and mitochondrial dysfunction.[7] Changes in vascular endothelial cells (ECs) and smooth muscle cells (SMCs) play a role in vascular senescence. In turn, senescence can drive the consequential aging hallmarks in response to damage, such as stem cell exhaustion.
In recent years, significant progress has been made in studying vascular aging mechanisms, especially the epigenetic mechanism. Epigenetics is a reversible genetic mechanism responsible for maintaining the stability and availability of genetic information without changing the sequence of DNA. In other words, it is something that inherits genotypes and phenotypes.[8,9] Therefore, understanding the epigenetic changes in the vascular aging process is an important research field that can contribute to the treatment of cardiovascular diseases caused by vascular aging. In this paper, we will introduce vascular senescence from three aspects of the telomere, mitochondrial dysfunction, and stem cell exhaustion, and review the effect of epigenetics in vascular aging and probe into the mechanism of epigenetics in vascular aging.
CELLULAR SENESCENCE AND VASCULAR SENESCENCE
Cellular senescence involves telomere shortening, mitochondrial dysfunction, and stem cell exhaustion, and we briefly illustrate the relationships among them in Figure 1. Cellular senescence was first discovered in fibroblast cells.[10] Cellular senescence is shown to inhibit proliferation and can be used as an idea to inhibit cancer. Previous studies showed that one of the main factors of cell senescence is telomere shortening. When telomere shortening reaches a critical length, DNA damage will be triggered, causing replicative senescence. Other stimuli not related to telomere shortenings, such as oxidative stress, radiation, and cell fusion, are also potent inducers of cellular senescence.[11,12] A striking feature of senescent cells is an increase in reactive oxygen species (ROS), mainly due to mitochondrial dysfunction. ROS can accelerate senescence by exacerbating DNA damage.[13] Autophagy is still a controversial way to regulate senescence, promoting aging through senescence-associated secretory phenotype (SASP) and inhibiting aging through ROS and p53.[14,15] Experimental animal models for aging studies can be used in nematodes and Drosophila with short cycles; more often, animal models such as mice that are genetically similar to humans are used. Mice can be used to build models of gene knockout and premature aging. Cell senescence can be studied using gene silencing and overexpression techniques. Calories, basal metabolic rate, stress response, and changes in nuclear protein balance are all associated with aging, so related genes can be used as research objects in the study of aging.[16] Senescent cells are characterized by the expression of lysosomal β-galactosidase, changes in apoptosis and mitotic indicators, SASP release, and persistent DNA damage.[10] The expression of the cell cycle inhibitors p53, p21, and p16 is upregulated in senescent cells.[17] In vascular cells, ECs and SMCs account for a large proportion and their senescence changes also conform to the senescence as mentioned above.[18] Therefore, we can judge that vascular senescence is connected with cellular senescence. Next, we will summarize cellular senescence from three aspects, telomere shortening, mitochondrial dysfunction, and stem cell exhaustion, and briefly summarize its role in vascular senescence.
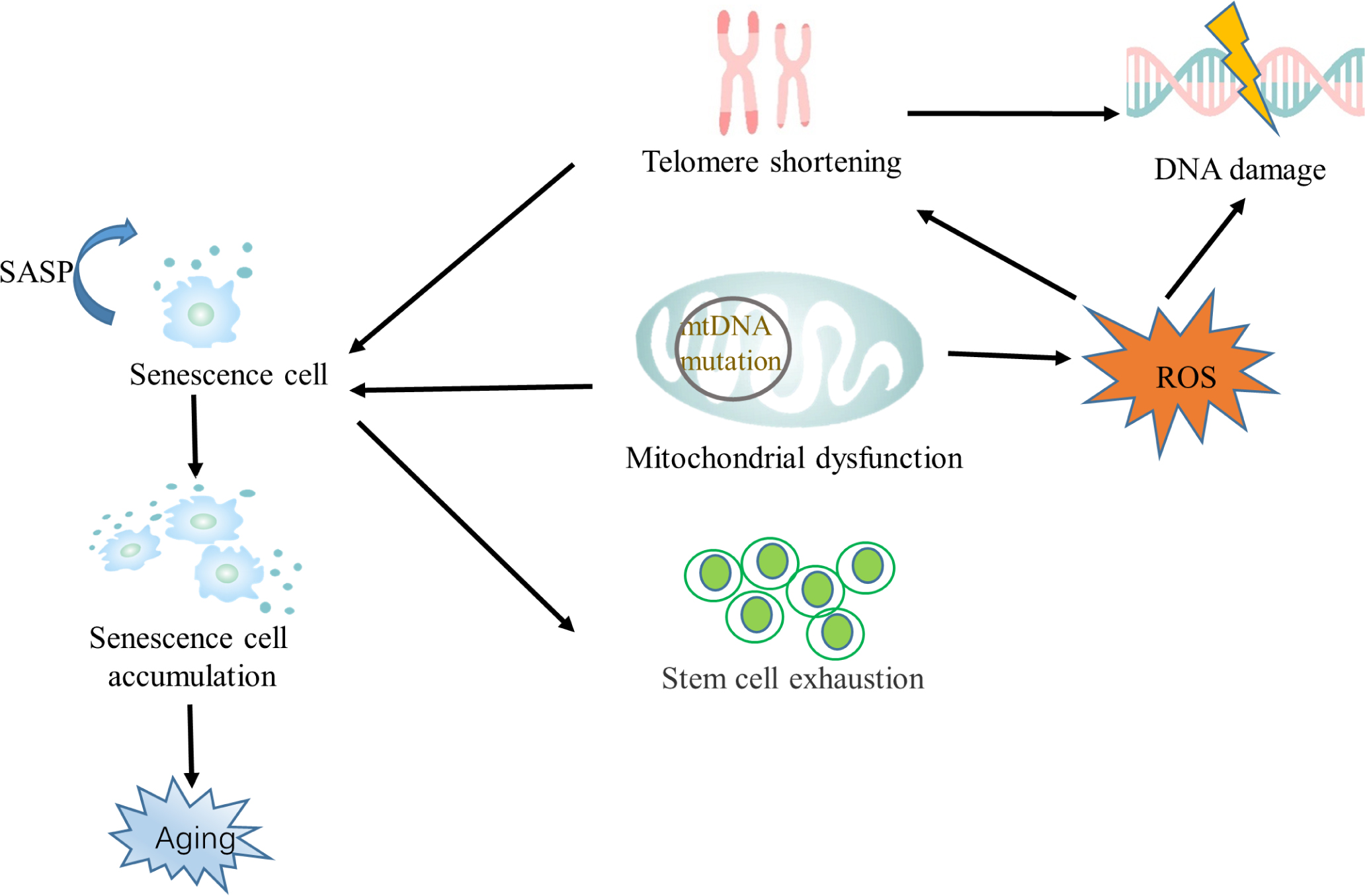
The relationship between senescence and its hallmarks mtDNA, mitochondrial DNA; ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype
Telomere and telomerase
Telomeres are small DNA–protein complexes at the ends of biological linear chromosomes in eukaryotic cells and are used to maintain chromosome integrity and control the cell cycle. Telomeres are protected from DNA damage by a multi-protein complex called shelterin. The ends of chromosomes cannot synthesize DNA without a template, so telomeres shorten with every cell cycle division. Therefore, telomerase should be expressed to circumvent this erosion. Reduced telomerase activity leads to shorter telomeres, which are associated with aging. Telomerase includes the protein telomerase reverse transcriptase (TERT, or hTERT in humans) and a catalytic RNA (TERC).[19] In normal cells, TERT is not expressed, so the activity of telomerase is generally lost. However, the expression of TERT is inducible in SMCs and ECs.[20] In the absence of telomerase, telomere shortening eventually leads to cell growth stagnation.[19] However, studies have revealed that even in the presence of telomerase, some special ribosomal protein complexes can cause irreversible damage to the DNA of telomeres, thus inducing aging.[7,21,22]
In addition to DNA replication problems that shorten telomeres, ROS also cause them to shrink. Telomere shortening is inseparable from chronic inflammation. Studies have shown that chronic inflammation can induce oxidative stress, increase ROS, and exacerbate telomere dysfunction, thereby inducing cell senescence.[23,24]
Lack of telomerase leads to premature aging in mice. There are three types of early senescence: replicative, oncogene-induced, and stress-induced premature senescence (SIPS).[25] However, SIPS is not involved in telomere shortening. Studies have indicated that cells with a short time of DNA damage or oxidative stress can lead to cell senescence with no or only slight shortening of telomeres.[26] So, SIPS occurs independently of telomeres. But there are also different opinions that SIPS is caused by telomere damage (not telomere shortening).[22,26] This means SIPS is also related to telomere. It suggests that there are other mechanisms besides telomere shortening that induce senescence.
Telomere length and activity of telomerase can be used as an indicator of vascular senescence. Studies have shown that TERT can antagonize the expression of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) through the p53 signaling pathway, leading to telomere shortening and mitochondrial dysfunction.[27] Meanwhile, PGC-1α deficiency can aggravate the degree of atherosclerosis. In doxorubicin-induced VSMCs senescence, SRT1720, an activator of SIRT1,[28] can alleviate its senescence phenotype by increasing telomere length and TERT expression.[29]
Mitochondrial dysfunction
It is well known that mitochondria produce ATP and are an essential energy source for cells. Still, they are also important in many other cellular processes, such as the control of cell cycle apoptosis and the regulation of cell metabolism.[1,30] There are multiple mitochondria in a cell, which can be damaged and the number in a cell can be reduced over time.[31] Mitochondrial morphologic and biological disorders are also associated with cellular senescence. Therefore, mitochondrial dysfunction can result in dysfunction of tissues or organs, leading to aging of the body.
Exactly how mitochondria affect aging is still unclear, but recent researches have suggested several possible mechanisms. Harman’s theory of free radicals suggested that the accumulation of ROS (probably by mitochondria) causes aging,[32] but most current studies have demonstrated that ROS do not directly induce aging.[33,34] One of the reasons for mitochondrial DNA (mtDNA) mutation is that DNA damage is unrepaired, and ROS are an inducer of DNA damage. Oxidative DNA damage plays a significant role in senescence. ROS and lipid peroxidation products affect genome and mtDNA, leading to various types of DNA damage.[35] DNA replication before a repair will lead to DNA mutations and genomic instability.[35,36] However, another critical reason for mutations is that errors occur during normal mtDNA replication.[37,38] mtDNA located near ROS-producing sites in mitochondria has a very high mutation rate due to the absence of histone protection, and studies have shown that mitochondrial mutations increase with age.[39,40] To sum up, the root cause of mitochondrial dysfunction is the generation of mitochondrial ROS (mtROS), leading to vascular dysfunction.[39,41] Moreover, there is evidence that mitochondrial damage of ECs and SMCs impacts vascular aging,[42,43] so a variety of drug studies will target mtROS as a direction.[39] PCG-1α is involved in the expression of antioxidant enzymes and has a regulatory effect on mitochondrial function. Studies have shown that AngII can induce mitochondrial dysfunction by inhibiting the activity of PCG-1α and increasing ROS level, to achieve the purpose of vascular senescence.[17,44] SIRT1 can inhibit AngII-induced vascular senescence by activating PCG-1α.[44,45] SIRT3, SIRT4, and SIRT5 are enzymes that regulate cell metabolism, and they also restrict nonmetabolic aspects of the mitochondria to ensure that they maintain homeostasis under stress.[46] Polymerase γ (Polγ) is an essential mitochondrial functional polymerase that repairs mtDNA.[47] Recent studies have shown that the production of mtROS can induce the shortening of telomeres, and thus induce aging.[48]
Meanwhile, mitochondrial damage gradually increases with age, and autophagy plays an important role by cleaning damaged mitochondria.[49] 17β-estradiol (E2) has been demonstrated to mediate the SIRT1/LKB1/AMPK/ULK1 pathway to induce autophagy of HUVECs and VSMCs, thereby alleviating vascular senescence.[50] As an autophagy receptor, SQSTM1 can induce the senescence of VSMCs. Studies have shown that PCG-1α can promote autophagy and alleviate vascular senescence by regulating the expression of SQSTM1.[51]
Changes in mitochondrial homeostasis can maintain cell senescence through various mechanisms, including mtROS production, imbalanced mitochondrial dynamics, electron transport chain defect, bioenergetics imbalance and increased 5′ adenosine monophosphate-activated protein kinase (AMPK) activity, altered mitochondrial metabolite profile (e.g., NAD+), and dysregulated mitochondrial calcium homeostasis.[12,52] There have been a lot of studies on the changes of these characteristics in a variety of diseases. Among cardiovascular diseases, studies on mitochondrial dysfunction mainly focus on atherosclerosis.[53]
The aggravation of mitochondrial dysfunction in ECs and SMCs leads to the aggravation of atherosclerosis.
Stem cell exhaustion
Stem cell exhaustion will become more and more severe with increasing age, and its function and renewal ability will decline and finally lead to tissue degradation.[54] However, the reasons for stem cell exhaustion are also manifold, such as increase in the number of senescent cells, diminished immunity, and decline in the self-renewal ability of cells. Generally speaking, stem cells are divided into two types. One is infrequently renewed stem cells that need to maintain cell function by removing metabolic waste or repairing DNA damage in the absence of cell division.[55] For instance, after the loss of autophagy, skeletal muscle stem cells (SMSCs) will enter the senescence state. In addition, studies have shown that DNA hygromethylation at the SPRY1 gene locus can downregulate sprouty1, resulting in the loss of the self-renewal ability of SMSC.[55] The other type is cells that renew more frequently, such as the hematopoietic stem cells (HSCs). The function of HSCs declines with age due to the decline in the dividing ability of the senescence stem cells, resulting from the accumulation of DNA damage.[56] It is unclear why stem cells show exhaustion during senescence and whether the decline in stem cell function is a cause or a result of aging. Studies have used repeated consumption of stem cells SOX2+ to cause stem cell exhaustion and the results have shown that SOX2+ exhaustion can cause cell senescence and premature senility in mice.[57] Due to the deacetylation of mitochondrial proteins, overexpression of SIRT3 can reverse the regenerative capacity of senescent HSCs.[7,58] miRNAs associated with the aging process can also influence longevity by regulating stem cells.[59]
The role of endothelial progenitor cells (EPCs) in angiogenesis has been studied extensively. The number and function of EPCs reflect the ability of endogenous vascular cells to repair, and the number of EPCs decreases with age.[60] EPCs derived from bone marrow are involved in vascular repair and are associated with atherosclerosis,[61] and their number is inversely correlated with the degree of atherosclerosis.[60] The degeneration of EPCs causes stagnation of vascular repair and renewal. Therefore, studies have shown that bone marrow–derived EPCs obtained from young ApoE-/- mice can treat atherosclerosis.[61]
EPIGENETIC CHANGES IN VASCULAR SENESCENCE
Epigenetic changes in aging can be inherited. Longevity is largely determined by epigenetics rather than innate genes. Epigenetics is influenced by a variety of factors, including diet and environment.[62] Epigenetics includes histone modification, DNA methylation, and noncoding RNA modification. Epigenetics has been manifested in abnormal gene expression, reactivation of transcription factors, and genomic instability during aging. Changes in transcription factors and chromatin can influence transcription and translation mechanisms. These epigenomic changes can be observed during vascular senescence and play an important role. They can mediate changes in cell function, and thus affect cell senescence.[7,63] A variety of aging-related diseases can be caused by cell senescence, for example, Alzheimer’s disease, stroke, atherosclerosis, and so on. Epigenetic modifications can regulate diseases caused by vascular senescence. Next, we will introduce the epigenetic changes in vascular senescence.
Histone modification
Histone is a cylindrical octamer composed of H2A, H2B, H3, and H4. It is a protein component of the nucleosome, and posttranslational modification (PTM) often occurs in this region. PTMs control the life span of living organisms by regulating transcriptional activity, modifying chromosomes, and repairing DNA damage. Functionally, histone modification has two main aspects: one is to destroy chromosomes and the other is to recruit new proteins for chromosomes to provide new binding surfaces.[2] PTMs mainly include acetylation, methylation, phosphorylation, ADP-ribose, and ubiquitination. Among general histone modifications, methylation and acetylation are the most common and important. A large number of experiments have shown that trimethylation of histone H4 at lysine 20 (H4K20me3) and trimethylation of histone H3 at lysine 9 (H3K9me3) and 27 (H3K27me3) can be regarded as markers of the aging process.[64] Studies have shown that the expression of H3K27me3 is downregulated in the aging HSCs. In contrast, the expression of H3K9me3 is downregulated in the mesenchymal stem cells (MSCs) from aged individuals.[65]
SIRT3 is a mitochondrial deacetylase that can inhibit tumors by regulating pyruvate dehydrogenase (PDH), succinate dehydrogenase (SDH), and 3-hydroxy-3-methylglutaryl COA synthase (HMGCS2).[66] The oxidized form (NAD+) of nicotinamide adenine dinucleotide (NAD) is a cofactor of sirtuins and PARP, and NAD is consumed in the process deacetylation of sirtuins and ADP-ribosylation of PARP.[67] Therefore, sirtuins and PARP regulate the cell function from gene expression and DNA damage through fatty acid metabolism.[67] A study by our group in the direction of vascular dysfunction showed that Smyd3, a histone methyltransferase, could increase the expression of H3K4me3 by binding with p21, and we finally obtained the results of alleviation of the senescence phenotype of vascular ECs and aging mice induced by AngII.[68] In addition, we found that Smyd3 can directly bind to the promoter region of H3K4me3 to increase the expression of H3M4me3, and these changes can also affect the vascular remodeling and the proliferation and migration of VSMCs induced by PDGF-BB.[69] While studying the effect of Smyd3 on vascular senescence, we found that PARP16, a member of the PARP family, can be upregulated by Smyd3 to exert an effect on vascular aging through UPR.[70] JMJD3 is a histone demethylase, and we found that inhibition of its expression could reduce the occurrence of autophagy, to achieve the purpose of reducing angiogenesis.[71] Interestingly, JMJD3 can also regulate the occurrence of rheumatoid arthritis and cardiac fibrosis.[72] We also found that Fra-1 can aggravate the vascular senescence phenotype by directly binding to the p16 and p21 promoter regions.[73] The discovery of these targets provides new ideas for the treatment of vascular diseases.
The AMPK/Snf1 signaling pathway can regulate the synthesis of acetyl-CoA, oxidation of NAD, and the activity of histone deacetylases (HDACs).[64] Our group found HDAC4 can affect the autophagy of vascular ECs by regulating the transcription factor FoxO3a, thereby inducing the occurrence of vascular inflammation.[74] One of the causes of cell senescence is cell growth arrest caused by DNA damage, which is due to the upregulation of CDKN1A/p21CIP1/WAF1 by p53 binding to the p21 promoter. SIRT1 has been shown to inhibit apoptosis or senescence by inhibiting the transcriptional activity of p53.[75] However, our study on Smyd3 has demonstrated that the Smyd3–p21 axis accelerates cell senescence without passing through the p53 pathway.[76] This provides a new way to study the mechanism of cell senescence.
DNA methylation
DNA methylation is one of the most studied topics in epigenetics. It plays a very vital role in the development process. It can silence some genes that do not need to be expressed; but over time, some DNA methylation is directional, so this suggests that DNA methylation is not random, or some of it is not random, and it may be related to the mechanisms of aging. In addition, there are random bidirectional changes in epigenetic variability called epigenetic drift.[15] These changes in DNA methylation are associated with age and have important significance for transcriptional regulation and protein expression. DNA methylation generally involves de novo methylation and maintenance methylation. DNA methyltransferase (DNMT) can add methyl groups to DNA, in which DNMT3a and DNMT3b are de novo DNMTs and DNMT1 is a maintenance DNMT.[77] However, the ten– eleven translocation (TET) family enzymes are the opposite of DNMTs.[78] The oxidation of TET1/2/3 dioxygenase demethylates 5-methylcytosine (5MC). DNMT3A and TET2, as regulators of DNA methylation, can be mutated in a variety of blood diseases. Their loss may lead to an increase in the number of stem cells and progenitor cells and hinder the ability of stem cells and progenitor cells to differentiate in mice.[65]
The most common form of DNA methylation is adding the methyl group to a 5′ cytosine of C–G dinucleotides, known as CpGs. In mammals, CpG exists in two main forms: one is dispersed in DNA sequences and the other is in a highly aggregated state, known as CpG island. CpG is mainly located in the promoter and exon regions of genes. DNA methylation in the promoter region is generally related to transcription factor binding and gene transcription.[79] CpG islands are less methylated than non-CpG islands and are usually associated with promoters. Some CpG islands and gene-rich regions become highly methylated with age.[62,80] Therefore, detecting DNA methylation can predict age, as evidenced by a large amount of data on age-related CpG from a variety of tissues.[81]
DNA methylation is also associated with DNA damage repair. Double-strand breaks can induce DNA hypermethylation during tumor development.[82] DNMT occupies an important position in vascular inflammation and aging. It affects the function of SMCs and ECs, thereby inducing the occurrence of vascular diseases. 8-Hydroxyguanine (8-OHdG), an indicator of ROS, can change the methylation status of DNA through oxidative damage; so, studies now suggest that oxidative stress inhibits DNA methylation.[83] Similarly, we can think of DNA methylation as affecting oxidative stress. Moreover, it has been proved that the DNMT inhibitor 5-azacytidine (5-AZA) can induce superoxide dismutase 2 (SOD2) expression and reduce SMC proliferation.[84] DNA methylation can also affect senescence through SASP. Studies have shown that the DNA desmethylation agent 5-aza-2-deoxycytidine (5-Aza-dC) can upregulate the SASP expression of epithelioma papulosum cyprini (EPC), inhibit the transcription of TERT, and reduce the activity of telomerase, thus causing the occurrence of cell senescence.[85]
Noncoding RNA
Noncoding RNA (ncRNA) is the type of RNA that does not code for proteins. In recent years, the research of ncRNA in epigenetics has been increasing gradually. ncRNAs, including long RNAs and microRNAs (miRNAs), are involved in regulating cellular processes and are important regulators of transcription networks and chromatin status.[86] Recent studies have shown that ncRNAs can affect the aging process, and senescence can also affect ncRNA levels. Different ncRNAs are key regulators of gene expression and chromatin remodeling and function by binding to their targets. ncRNAs regulate various physiological processes, including development, stress response, tumorigenesis, and immune response.[87,88] There are more researches on miRNA in ncRNA. miRNAs inhibit the production of proteins by affecting the translation and stability of messenger RNA. It has been demonstrated that miRNAs can be used to study replication senescence of various types of cells, such as replicating CD8(+) T cells, skin fibroblasts, renal proximal tubular epithelial cells, and arterial and umbilical vein-derived ECs.[89]
Tumor suppressors play an important role in cellular senescence and can affect a variety of cell functions. The common suppressors, such as p21, p53, and p16, are regulated by miRNA.[90] Previous evidence has shown that miR-24 can regulate p16 and miR-885-5p can regulate p53.[91] miRNA expression changes in disease; it is usually upregulated in aging. miR-217 was detected in atherosclerosis and was found to induce EC senescence by inhibiting the expression of SIRT1.[92] miR-145 and miR-143 were found to be downregulated in atherosclerotic vessels.[93] Kallistatin, an endogenous protein, alleviates vascular senescence by inhibiting the synthesis of miR-34a.[94] A variety of miRNAs can regulate the aging of human aortic ECs (HAECs).
Studies on lncRNAs have also provided ideas for finding new therapeutic targets for cardiovascular diseases in recent years. lncRNAs can act on vascular diseases by regulating the different functions of ECs and VSMCs.[95] The loss of lncRNA H19 can increase the expression of p21 and p16, thus controlling the senescence of ECs, and this conclusion has also been proved by in vitro experiments.[96] MEG3 has been widely studied in cardiovascular diseases, and its expression is elevated in the atrium and HUVECs of the elderly. Moreover, many studies have found that MEG3 can affect ECs and SMCs by regulating a variety of miRNAs, thus exerting an impact on senescence.[97] GAS5 is an lncRNA that has been widely studied in the brain. Moreover, transcriptome studies have found that the expression of GAS5 is increased in atherosclerotic plaques in rats and patients. However, its specific mechanism is not clear and further studies are needed.[98]
CONCLUSION AND PROSPECTS
In this review, we provide an overview of markers of cellular senescence, telomere shortening, mitochondrial dysfunction, and stem cell exhaustion and explain their roles in vascular senescence. Among them, telomere shortening and mitochondrial dysfunction induce cellular senescence, while stem cell exhaustion is one of the aging characteristics caused by cellular senescence. The inability of chromosome causes telomere shortening ends to synthesize DNA without a template, and mitochondrial dysfunction is also associated with ROS and DNA damage. DNA damage is a negative hallmark that can lead to cellular dysfunction. However, ROS is an antagonistic hallmark, which has opposite effects on cells at different levels of expression. The low-level expression can mediate cell signal transduction and survival, while high-level expression can lead to cell damage.
p53, p21, and p16 are very important in cell senescence and many studies on vascular senescence have taken them as the main research object. Cells control senescence mainly through p53–p21 and p16–retinoblastoma protein (pRB) pathways.[99] The transcription factor p53 induces cell growth cycle stagnation by controlling cell apoptosis, participating in DNA damage repair, and cell stress response. Senescence is an irreversible growth cycle stagnation, so p53 can activate cell senescence and aging.[100] It is well known that p21 is a target gene of p53 and causes cell senescence by inducing cell cycle stagnation in the G1/S phase. At the same time, p21 can induce cell senescence without p53.[101] When induced, p16 binds to cyclin-dependent kinase 4/6 (CDK4/6) and inhibits its activity, thus causing retinoblastoma (RB)-dependent cell cycle to stagnate.[102] This mechanism is common in inhibiting tumor growth. However, most studies on senescence cannot avoid these two typical pathways. Therefore it is of great significance to find the targets of these two pathways. In Figure 2, the role of p53/p21 and p16 pathways in vascular senescence is briefly summarized.
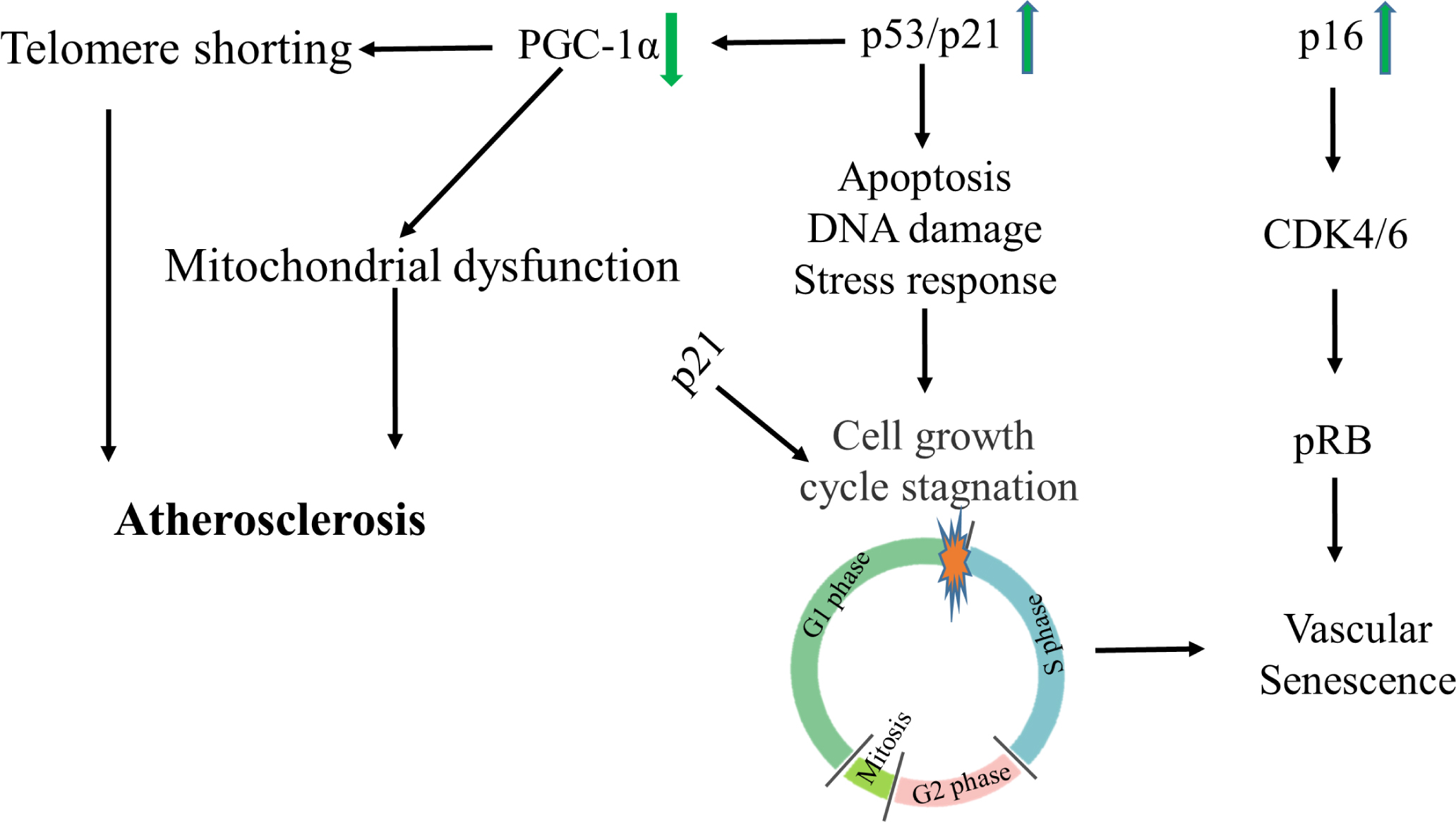
p53/p21 and p16 pathways in vascular senescence CDK, cyclin-dependent kinase; PGC-1α, peroxisome proliferator-activated receptor γ coactivator-1α; pRB, p16–retinoblastoma protein
We also introduce the effects of epigenetics on vascular senescence from three aspects, histone modification, DNA methylation, and ncRNA modification, and relevant targets are summarized in Table 1. Existing studies have found the mechanism of coping with vascular senescence from different targets, providing a direction for the follow-up treatment of vascular senescence-related diseases.
Related targets of epigenetics in vascular senescnece
| Targets | Description | Effects |
|---|---|---|
| SIRT1 | Sirtuin 1 | Alleviating apoptosis or senescence by inhibiting the transcriptional activity of p53 |
| SIRT3 | Sirtuin 3 | Reducing vascular dysfunction |
| SMYD3 | SET And MYND Domain Containing 3 | Increasing the expression of H3K4me3 by binding with p21 and inducing vascular senescence and vascular remodeling |
| JMJD3 | Jumonji Domain Containing 3 | Inhibition of its expression can reduce the occurrence of autophagy and achieve the purpose of reducing angiogenesis |
| HDAC4 | Histone Deacetylase 4 | Affecting the autophagy of vascular endothelial cells by regulating the transcription factor FoxO3a Inducing the occurrence of vascular inflammation |
| GATA4 | GATA Binding Protein 4 | Induceing angiogenesis and inflammation of the blood vessels |
| Fra-1 | Fos-related antigen 1 | Inhibition of its expression alleviates vascular inflammation and senescence phenotype in mice |
| PARP16 | Poly (ADP-Ribose) Polymerase Family Member 16 | Regulating the unfolded protein response of endoplasmic reticulum and promoting vascular senescence |
| miR-24 | MicroRNA 24 | Regulation of senescence through the p16 pathway |
| miR-885-5p | MicroRNA 885 | Regulation of senescence through the p53 pathway |
| miR-217 | MicroRNA 217 | Induction of endothelial cell senescence through inhibition of SIRT1 expression |
| miR145/miR143 | MicroRNA 145/MicroRNA 143 | Inhibition of Mir-34A alleviates vascular Aging in atherosclerosis |
| H19 | H19 Imprinted Maternally Expressed Transcript | Regulating endothelial cells and smooth muscle cells |
| MEG3 | Maternally expressed gene 3 | Regulation of endothelial cells proliferation and capillary angiogenesis |
| GAS5 | Growth arrest specific 5 | Expression of GAS5 is increased in atherosclerotic plaques |
The research on vascular senescence can provide new ideas for the treatment of cardiovascular diseases. However, the epigenetic mechanism of vascular senescence is still incomplete, and vast unknown areas are waiting to be explored. Currently, senescent cells are mostly studied, but how senescent cells promote senescence in vascular system remains a hot scientific area to be disclosed. We can target senescent cells to alleviate vascular senescence and reduce the occurrence of vascular diseases.
Epigenetics can provide more targets for the study of vascular senescence, and the full application of modern technology can also facilitate our research. For example, the new sequencing technology has been widely used, making it more convenient and accurate to evaluate the epigenetic changes in single cells of aging. Therefore, using epigenetics to find new targets for the treatment of cardiovascular diseases has a good prospect.
Author Contributions
-
All authors contributed to the article and approved the submitted version.
Conflict of Interest
-
The authors have declared no conflict of interests.
Source of Funding
-
This work was supported by the National Natural Science Foundation of China (81173054), Macau FDCT grants (067/2018A2, 033/2017/AMJ, 0007/2019/AKP, 0052/2020/A), the key laboratory program of the Education Commission of Shanghai Municipality (No. ZDSYS14005), and the Technology Base and Talents Special Program of Guangxi Province (Guike AD20238024).
REFERENCES
1. Chapman J, Fielder E, Passos JF. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett 2019;593:1566-79.10.1002/1873-3468.13498Suche in Google Scholar PubMed
2. Pal S, Tyler JK. Epigenetics and aging. Sci Adv 2016;2:e1600584.10.1126/sciadv.1600584Suche in Google Scholar PubMed PubMed Central
3. Ding Q, Shao C, Rose P, Zhu YZ. Epigenetics and Vascular Senescence-Potential New Therapeutic Targets?. Front Pharmacol 2020;11:535395.10.3389/fphar.2020.535395Suche in Google Scholar PubMed PubMed Central
4. Picca A, Guerra F, Calvani R, et al. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int J Mol Sci 2019;20:805.10.3390/ijms20040805Suche in Google Scholar PubMed PubMed Central
5. Muñoz-Espín D, Cañamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell 2013;155:1104-18.10.1016/j.cell.2013.10.019Suche in Google Scholar PubMed
6. Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014;31:722-33.10.1016/j.devcel.2014.11.012Suche in Google Scholar PubMed PubMed Central
7. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194-217.10.1016/j.cell.2013.05.039Suche in Google Scholar PubMed PubMed Central
8. O’Sullivan RJ, Karlseder J. The great unravelling: chromatin as a modulator of the aging process. Trends Biochem Sci 2012;37:466-76.10.1016/j.tibs.2012.08.001Suche in Google Scholar PubMed PubMed Central
9. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell 2007;128:635-8.10.1016/j.cell.2007.02.006Suche in Google Scholar PubMed
10. Yang N, Sen P. The senescent cell epigenome. Aging (Albany NY) 2018;10:3590-609.10.18632/aging.101617Suche in Google Scholar PubMed PubMed Central
11. Gorgoulis VG, Pefani DE, Pateras IS, Trougakos IP. Integrating the DNA damage and protein stress responses during cancer development and treatment. J Pathol 2018;246:12-40.10.1002/path.5097Suche in Google Scholar PubMed PubMed Central
12. Vasileiou PVS, Evangelou K, Vlasis K, et al. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019;8:686.10.3390/cells8070686Suche in Google Scholar PubMed PubMed Central
13. Victorelli S, Passos JF. Reactive Oxygen Species Detection in Senescent Cells. Methods Mol Biol 2019;1896:21-9.10.1007/978-1-4939-8931-7_3Suche in Google Scholar PubMed
14. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 2019;15:565-81.10.1038/s41582-019-0244-7Suche in Google Scholar PubMed
15. Jones MJ, Goodman SJ, Kobor MS. DNA methylation and healthy human aging. Aging Cell 2015;14:924-32.10.1111/acel.12349Suche in Google Scholar PubMed PubMed Central
16. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic Mechanisms of Longevity and Aging. Cell 2016;166:822-39.10.1016/j.cell.2016.07.050Suche in Google Scholar PubMed PubMed Central
17. Salazar G. NADPH Oxidases and Mitochondria in Vascular Senescence. Int J Mol Sci 2018;19:1327.10.3390/ijms19051327Suche in Google Scholar PubMed PubMed Central
18. Minamino T, Komuro I. Vascular aging: insights from studies on cellular senescence, stem cell aging, and progeroid syndromes. Nat Clin Pract Cardiovasc Med 2008;5:637-48.10.1038/ncpcardio1324Suche in Google Scholar PubMed
19. Hornsby PJ. Telomerase and the aging process. Exp Gerontol 2007;42:575-81.10.1016/j.exger.2007.03.007Suche in Google Scholar PubMed PubMed Central
20. Mendelsohn AR, Larrick JW. Telomerase Reverse Transcriptase and Peroxisome Proliferator-Activated Receptor γ Co-Activator-1α Cooperate to Protect Cells from DNA Damage and Mitochondrial Dysfunction in Vascular Senescence. Rejuvenation Res 2015;18:479-83.10.1089/rej.2015.1780Suche in Google Scholar PubMed
21. Fumagalli M, Rossiello F, Clerici M, et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 2012;14:355-65.10.1038/ncb2466Suche in Google Scholar PubMed PubMed Central
22. Hewitt G, Jurk D, Marques FD, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 2012;3:708.10.1038/ncomms1708Suche in Google Scholar PubMed PubMed Central
23. Saretzki G. Telomeres, Telomerase and Ageing. Subcell Biochem 2018;90:221-308.10.1007/978-981-13-2835-0_9Suche in Google Scholar PubMed
24. Jung HM, Phillips BL, Chan EK. miR-375 activates p21 and suppresses telomerase activity by coordinately regulating HPV E6/E7, E6AP, CIP2A, and 14-3-3ζ. Mol Cancer 2014;13:80.10.1186/1476-4598-13-80Suche in Google Scholar PubMed PubMed Central
25. Gea J, Ausín P, Martínez-Llorens JM, Barreiro E. Respiratory muscle senescence in ageing and chronic lung diseases. Eur Respir Rev 2020;29:200087.10.1183/16000617.0087-2020Suche in Google Scholar PubMed
26. de Magalhães JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev 2018;170:2-9.10.1016/j.mad.2017.07.001Suche in Google Scholar PubMed
27. Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011;470:359-65.10.1038/nature09787Suche in Google Scholar PubMed PubMed Central
28. Mitchell SJ, Martin-Montalvo A, Mercken EM, et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep 2014;6:836-43.10.1016/j.celrep.2014.01.031Suche in Google Scholar PubMed PubMed Central
29. Sung JY, Kim SG, Cho DH, Kim JR, Choi HC. SRT1720-induced activation of SIRT1 alleviates vascular smooth muscle cell senescence through PKA-dependent phosphorylation of AMPKα at Ser485. FEBS Open Bio 2020;10:1316-25.10.1002/2211-5463.12895Suche in Google Scholar PubMed PubMed Central
30. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell 2012;148:1145-59.10.1016/j.cell.2012.02.035Suche in Google Scholar PubMed PubMed Central
31. Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 2016;17:308-21.10.1038/nrm.2016.14Suche in Google Scholar PubMed PubMed Central
32. Harman D. The biologic clock: the mitochondria?. J Am Geriatr Soc 1972;20:145-7.10.1111/j.1532-5415.1972.tb00787.xSuche in Google Scholar PubMed
33. Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med 2011;51:327-36.10.1016/j.freeradbiomed.2011.05.010Suche in Google Scholar PubMed
34. Jang JY, Blum A, Liu J, Finkel T. The role of mitochondria in aging. J Clin Invest 2018;128:3662-70.10.1172/JCI120842Suche in Google Scholar PubMed PubMed Central
35. Kudryavtseva AV, Krasnov GS, Dmitriev AA, et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016;7:44879-905.10.18632/oncotarget.9821Suche in Google Scholar PubMed PubMed Central
36. Dehennaut V, Loison I, Dubuissez M, Nassour J, Abbadie C, Leprince D. DNA double-strand breaks lead to activation of hypermethylated in cancer 1 (HIC1) by SUMOylation to regulate DNA repair. J Biol Chem 2013;288:10254-64.10.1074/jbc.M112.421610Suche in Google Scholar PubMed PubMed Central
37. Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest 2013;123:951-7.10.1172/JCI64125Suche in Google Scholar PubMed PubMed Central
38. Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 2010;79:683-706.10.1146/annurev-biochem-060408-093701Suche in Google Scholar PubMed
39. Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of Vascular Aging. Circ Res 2018;123:849-67.10.1161/CIRCRESAHA.118.311378Suche in Google Scholar PubMed PubMed Central
40. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 2005;39:359-407.10.1146/annurev.genet.39.110304.095751Suche in Google Scholar PubMed PubMed Central
41. Csiszar A, Gautam T, Sosnowska D, et al. Caloric restriction confers persistent anti-oxidative, pro-angiogenic, and anti-inflammatory effects and promotes anti-aging miRNA expression profile in cerebromicrovascular endothelial cells of aged rats. Am J Physiol Heart Circ Physiol 2014;307:H292-306.10.1152/ajpheart.00307.2014Suche in Google Scholar PubMed PubMed Central
42. Springo Z, Tarantini S, Toth P, et al. Aging Exacerbates Pressure-Induced Mitochondrial Oxidative Stress in Mouse Cerebral Arteries. J Gerontol A Biol Sci Med Sci 2015;70:1355-9.10.1093/gerona/glu244Suche in Google Scholar PubMed PubMed Central
43. Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, et al. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell 2018;17:e12731.10.1111/acel.12731Suche in Google Scholar PubMed PubMed Central
44. Xiong S, Salazar G, Patrushev N, et al. Peroxisome proliferator-activated receptor γ coactivator-1α is a central negative regulator of vascular senescence. Arterioscler Thromb Vasc Biol 2013;33:988-98.10.1161/ATVBAHA.112.301019Suche in Google Scholar PubMed PubMed Central
45. Gerhart-Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J 2007;26:1913-23.10.1038/sj.emboj.7601633Suche in Google Scholar PubMed PubMed Central
46. van de Ven RAH, Santos D, Haigis MC. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol Med 2017;23:320-31.10.1016/j.molmed.2017.02.005Suche in Google Scholar PubMed PubMed Central
47. Krasich R, Copeland WC. DNA polymerases in the mitochondria: A critical review of the evidence. Front Biosci (Landmark Ed) 2017;22:692-709.10.2741/4510Suche in Google Scholar PubMed PubMed Central
48. Passos JF, Saretzki G, Ahmed S, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 2007;5:e110.10.1371/journal.pbio.0050110Suche in Google Scholar PubMed PubMed Central
49. Berridge MJ. Vitamin D deficiency accelerates ageing and age-related diseases: a novel hypothesis. J Physiol 2017;595:6825-36.10.1113/JP274887Suche in Google Scholar PubMed PubMed Central
50. Sasaki Y, Ikeda Y, Uchikado Y, Akasaki Y, Sadoshima J, Ohishi M. Estrogen Plays a Crucial Role in Rab9-Dependent Mitochondrial Autophagy, Delaying Arterial Senescence. J Am Heart Assoc 2021;10:e019310.10.1161/JAHA.120.019310Suche in Google Scholar PubMed PubMed Central
51. Salazar G, Cullen A, Huang J, et al. SQSTM1/p62 and PPARGC1A/PGC-1alpha at the interface of autophagy and vascular senescence. Autophagy 2020;16:1092-110.10.1080/15548627.2019.1659612Suche in Google Scholar PubMed PubMed Central
52. Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 2015;14:1-7.10.1111/acel.12287Suche in Google Scholar PubMed PubMed Central
53. Yu E, Mercer J, Bennett M. Mitochondria in vascular disease. Cardiovasc Res 2012;95:173-82.10.1093/cvr/cvs111Suche in Google Scholar PubMed
54. McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol 2018;217:65-77.10.1083/jcb.201708092Suche in Google Scholar PubMed PubMed Central
55. Mendelsohn AR, Larrick JW. Rejuvenating Muscle Stem Cell Function: Restoring Quiescence and Overcoming Senescence. Rejuvenation Res 2016;19:182-6.10.1089/rej.2016.1829Suche in Google Scholar PubMed
56. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007;447:725-9.10.1038/nature05862Suche in Google Scholar PubMed
57. Vilas JM, Carneiro C, Da Silva-Álvarez S, et al. Adult Sox2+ stem cell exhaustion in mice results in cellular senescence and premature aging. Aging Cell 2018;17:e12834.10.1111/acel.12834Suche in Google Scholar PubMed PubMed Central
58. Brown K, Xie S, Qiu X, et al. SIRT3 reverses aging-associated degeneration. Cell Rep 2013;3:319-27.10.1016/j.celrep.2013.01.005Suche in Google Scholar PubMed PubMed Central
59. Boulias K, Horvitz HR. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab 2012;15:439-50.10.1016/j.cmet.2012.02.014Suche in Google Scholar PubMed PubMed Central
60. Xiao Q, Kiechl S, Patel S, et al. Endothelial progenitor cells, cardiovascular risk factors, cytokine levels and atherosclerosis--results from a large population-based study. PLoS One 2007;2:e975.10.1371/journal.pone.0000975Suche in Google Scholar PubMed PubMed Central
61. Rauscher FM, Goldschmidt-Clermont PJ, Davis BH, et al. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation 2003;108:457-63.10.1161/01.CIR.0000082924.75945.48Suche in Google Scholar PubMed
62. Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol 2015;16:593-610.10.1038/nrm4048Suche in Google Scholar PubMed PubMed Central
63. Booth LN, Brunet A. The Aging Epigenome. Mol Cell 2016;62:728-44.10.1016/j.molcel.2016.05.013Suche in Google Scholar PubMed PubMed Central
64. Wang Y, Yuan Q, Xie L. Histone Modifications in Aging: The Underlying Mechanisms and Implications. Curr Stem Cell Res Ther 2018;13:125-35.10.2174/1574888X12666170817141921Suche in Google Scholar PubMed
65. Goodell MA, Rando TA. Stem cells and healthy aging. Science 2015;350:1199-204.10.1126/science.aab3388Suche in Google Scholar PubMed
66. Ristic B, Bhutia YD, Ganapathy V. Cell-surface G-protein-coupled receptors for tumor-associated metabolites: A direct link to mitochondrial dysfunction in cancer. Biochim Biophys Acta Rev Cancer 2017;1868:246-57.10.1016/j.bbcan.2017.05.003Suche in Google Scholar PubMed PubMed Central
67. Newman JC, Verdin E. β-Hydroxybutyrate: A Signaling Metabolite. Annu Rev Nutr 2017;37:51-76.10.1146/annurev-nutr-071816-064916Suche in Google Scholar PubMed PubMed Central
68. Long F, Yang D, Wang J, Wang Q, Ni T, Wei G, et al. SMYD3-PARP16 axis accelerates unfolded protein response and mediates neointima formation. Acta pharmaceutica Sinica B 2021;11:1261-73.10.1016/j.apsb.2020.12.010Suche in Google Scholar PubMed PubMed Central
69. Yang D, Su Z, Wei G, et al. H3K4 Methyltransferase Smyd3 Mediates Vascular Smooth Muscle Cell Proliferation, Migration, and Neointima Formation. Arterioscler Thromb Vasc Biol 2021;41:1901-14.10.1161/ATVBAHA.121.314689Suche in Google Scholar PubMed
70. Yang D, Wang Q, Wei G, et al. Smyd3-PARP16 axis accelerates unfolded protein response and vascular aging. Aging (Albany NY) 2020;12:21423-45.10.18632/aging.103895Suche in Google Scholar PubMed PubMed Central
71. Luo X, Yang D, Wu W, et al. Critical role of histone demethylase Jumonji domain-containing protein 3 in the regulation of neointima formation following vascular injury. Cardiovasc Res 2018;114:1894-906.10.1093/cvr/cvy176Suche in Google Scholar PubMed
72. Long F, Wang Q, Yang D, et al. Targeting JMJD3 histone demethylase mediates cardiac fibrosis and cardiac function following myocardial infarction. Biochem Biophys Res Commun 2020;528:671-7.10.1016/j.bbrc.2020.05.115Suche in Google Scholar PubMed
73. Yang D, Xiao C, Long F, et al. Fra-1 plays a critical role in angiotensin II-induced vascular senescence. FASEB J 2019;33:7603-14.10.1096/fj.201801671RRRRSuche in Google Scholar PubMed
74. Yang D, Xiao C, Long F, et al. HDAC4 regulates vascular inflammation via activation of autophagy. Cardiovasc Res 2018;114:1016-28.10.1093/cvr/cvy051Suche in Google Scholar PubMed
75. Ong ALC, Ramasamy TS. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res Rev 2018;43:64-80.10.1016/j.arr.2018.02.004Suche in Google Scholar PubMed
76. Yang D, Wei G, Long F, et al. Histone methyltransferase Smyd3 is a new regulator for vascular senescence. Aging Cell 2020;19:e13212.10.1111/acel.13212Suche in Google Scholar PubMed PubMed Central
77. Tabaei S, Tabaee SS. DNA methylation abnormalities in atherosclerosis. Artif Cells Nanomed Biotechnol 2019;47:2031-41.10.1080/21691401.2019.1617724Suche in Google Scholar PubMed
78. Rahman MM, Brane AC, Tollefsbol TO. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells 2019;8:1214.10.3390/cells8101214Suche in Google Scholar PubMed PubMed Central
79. Kochmanski J, Marchlewicz EH, Cavalcante RG, Sartor MA, Dolinoy DC. Age-related epigenome-wide DNA methylation and hydroxymethylation in longitudinal mouse blood. Epigenetics 2018;13:779-92.10.1080/15592294.2018.1507198Suche in Google Scholar PubMed PubMed Central
80. Zampieri M, Ciccarone F, Calabrese R, Franceschi C, Bürkle A, Caiafa P. Reconfiguration of DNA methylation in aging. Mech Ageing Dev 2015;151:60-70.10.1016/j.mad.2015.02.002Suche in Google Scholar PubMed
81. Jung SE, Lim SM, Hong SR, Lee EH, Shin KJ, Lee HY. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci Int Genet 2019;38:1-8.10.1016/j.fsigen.2018.09.010Suche in Google Scholar PubMed
82. Hayashi K, Hishikawa A, Itoh H. DNA Damage Repair and DNA Methylation in the Kidney. Am J Nephrol 2019;50:81-91.10.1159/000501356Suche in Google Scholar PubMed
83. Weitzman SA, Turk PW, Milkowski DH, Kozlowski K. Free radical adducts induce alterations in DNA cytosine methylation. Proc Natl Acad Sci U S A 1994;91:1261-4.10.1073/pnas.91.4.1261Suche in Google Scholar PubMed PubMed Central
84. O’Hagan HM, Wang W, Sen S, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011;20:606-19.10.1158/1538-7445.AM2011-LB-185Suche in Google Scholar
85. Futami K, Maita M, Katagiri T. DNA demethylation with 5-aza-2’-deoxycytidine induces the senescence-associated secretory phenotype in the immortal fish cell line, EPC. Gene 2019;697:194-200.10.1016/j.gene.2019.02.048Suche in Google Scholar PubMed
86. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem 2012;81:145-66.10.1146/annurev-biochem-051410-092902Suche in Google Scholar PubMed PubMed Central
87. Fernandes JCR, Acuña SM, Aoki JI, Floeter-Winter LM, Muxel SM. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Noncoding RNA 2019;5:17.10.3390/ncrna5010017Suche in Google Scholar PubMed PubMed Central
88. Shin H, Kim Y, Kim M, Lee Y. BC200 RNA: An Emerging Therapeutic Target and Diagnostic Marker for Human Cancer. Mol Cells 2018;41:993-9.Suche in Google Scholar
89. Lopez MF, Niu P, Wang L, et al. Opposing activities of oncogenic MIR17HG and tumor suppressive MIR100HG clusters and their gene targets regulate replicative senescence in human adult stem cells. NPJ Aging Mech Dis 2017;3:7.10.1038/s41514-017-0006-ySuche in Google Scholar PubMed PubMed Central
90. Reddy PH, Williams J, Smith F, et al. MicroRNAs, Aging, Cellular Senescence, and Alzheimer’s Disease. Prog Mol Biol Transl Sci 2017;146:127-71.10.1016/bs.pmbts.2016.12.009Suche in Google Scholar PubMed
91. Munk R, Panda AC, Grammatikakis I, Gorospe M, Abdelmohsen K. Senescence-Associated MicroRNAs. Int Rev Cell Mol Biol 2017;334:177-205.10.1016/bs.ircmb.2017.03.008Suche in Google Scholar PubMed PubMed Central
92. Menghini R, Casagrande V, Cardellini M, et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009;120:1524-32.10.1161/CIRCULATIONAHA.109.864629Suche in Google Scholar
93. Cordes KR, Sheehy NT, White MP, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009;460:705-10.10.1038/nature08195Suche in Google Scholar
94. Guo Y, Li P, Gao L, et al. Kallistatin reduces vascular senescence and aging by regulating microRNA-34a-SIRT1 pathway. Aging Cell 2017;16:837-46.10.1111/acel.12615Suche in Google Scholar
95. Simion V, Haemmig S, Feinberg MW. LncRNAs in vascular biology and disease. Vascul Pharmacol 2019;114:145-56.10.1016/j.vph.2018.01.003Suche in Google Scholar
96. Hofmann P, Sommer J, Theodorou K, et al. Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc Res 2019;115:230-42.10.1093/cvr/cvy206Suche in Google Scholar
97. Boon RA, Hofmann P, Michalik KM, et al. Long Noncoding RNA Meg3 Controls Endothelial Cell Aging and Function: Implications for Regenerative Angiogenesis. J Am Coll Cardiol 2016;68:2589-91.10.1016/j.jacc.2016.09.949Suche in Google Scholar
98. Chen L, Yao H, Hui JY, et al. Global transcriptomic study of atherosclerosis development in rats. Gene 2016;592:43-8.10.1016/j.gene.2016.07.023Suche in Google Scholar
99. Chi C, Li DJ, Jiang YJ, et al. Vascular smooth muscle cell senescence and age-related diseases: State of the art. Biochim Biophys Acta Mol Basis Dis 2019;1865:1810-21.10.1016/j.bbadis.2018.08.015Suche in Google Scholar
100. Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene 2013;32:5129-43.10.1038/onc.2012.640Suche in Google Scholar
101. Wyllie F, Haughton M, Bartek J, Rowson J, Wynford-Thomas D. Mutant p53 can delay growth arrest and loss of CDK2 activity in senescing human fibroblasts without reducing p21(WAF1) expression. Exp Cell Res 2003;285:236-42.10.1016/S0014-4827(03)00050-8Suche in Google Scholar
102. Burd CE, Sorrentino JA, Clark KS, et al. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013;152:340-51.10.1016/j.cell.2012.12.010Suche in Google Scholar PubMed PubMed Central
How to cite this article: Zhu M, Ding Q, Lin Z, Chen X, Chen S, Zhu Y. New insights of epigenetics in vascular and cellular senescence. J Transl Intern Med 2021; 9: 239-48.
© 2021 Menglin Zhu et al., published by Sciendo
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 3.0 License.
Artikel in diesem Heft
- Editorial
- Translational research in anti-pancreatic fibrosis drug discovery and development
- Dawning precision treatment for gastric cancer: The latest biomarkers
- Commentary
- Can gamma entrainment of the brain rhythms prevent or alleviate Alzheimer’s disease?
- Review
- The challenges and optimization of cell-based therapy for cardiovascular disease
- New insights of epigenetics in vascular and cellular senescence
- Original Article
- Lung adenocarcinoma-specific three-integrin signature contributes to poor outcomes by metastasis and immune escape pathways
- Relationship between homocysteine levels and post-stroke cognitive impairment in female and male population: from a prospective multicenter study
- Assessment of 17 clinically available renal biomarkers to predict acute kidney injury in critically ill patients
- Elevated resting heart rates are a risk factor for mortality among patients with coronavirus disease 2019 in Wuhan, China
- Lipid-related protein NECTIN2 is an important marker in the progression of carotid atherosclerosis: An intersection of clinical and basic studies
- Effect of local anti-vascular endothelial growth factor therapy to prevent the formation of stenosis in outflow vein in arteriovenous fistula
- Case Report
- Hyperoxaluria: An early diagnosis will allow a timely combined renal and liver transplantations to avoid irreversible damages to all other organs
Artikel in diesem Heft
- Editorial
- Translational research in anti-pancreatic fibrosis drug discovery and development
- Dawning precision treatment for gastric cancer: The latest biomarkers
- Commentary
- Can gamma entrainment of the brain rhythms prevent or alleviate Alzheimer’s disease?
- Review
- The challenges and optimization of cell-based therapy for cardiovascular disease
- New insights of epigenetics in vascular and cellular senescence
- Original Article
- Lung adenocarcinoma-specific three-integrin signature contributes to poor outcomes by metastasis and immune escape pathways
- Relationship between homocysteine levels and post-stroke cognitive impairment in female and male population: from a prospective multicenter study
- Assessment of 17 clinically available renal biomarkers to predict acute kidney injury in critically ill patients
- Elevated resting heart rates are a risk factor for mortality among patients with coronavirus disease 2019 in Wuhan, China
- Lipid-related protein NECTIN2 is an important marker in the progression of carotid atherosclerosis: An intersection of clinical and basic studies
- Effect of local anti-vascular endothelial growth factor therapy to prevent the formation of stenosis in outflow vein in arteriovenous fistula
- Case Report
- Hyperoxaluria: An early diagnosis will allow a timely combined renal and liver transplantations to avoid irreversible damages to all other organs