Synthesis and characterization of some new fluoroquinolone-barbiturate hybrid systems
-
Kamal Sweidan
 ,
Mustafa M. El-Abadelah
,
Mustafa M. El-Abadelah

Abstract
New hybrid molecules containing fluoroquinolone-barbiturate moieties were synthesized via simple routes, followed by characterization using various spectroscopic approaches. The THF solvent molecule was incorporated into compounds 2 and 3 through its oxygen atom based on its nucleophilicity. Facile displacement of the chloride ion in 4-oxoquinoline-3-carbonyl chloride by the nucleophilic methylene carbon of 1,3-dialkylbarbiturate/thiobarbiturate afforded the respective hybrids 5 and 6.
1 Introduction
Fluoroquinolones, exemplified by ciprofloxacin (1a), play an important role as anti-infective agents and are widely prescribed for various infections, such as, e.g., respiratory and gastrointestinal infections [1]. During the past four decades, several fluoroquinolone derivatives have been synthesized, characterized and examined for their antibacterial and anticancer activities [2–5]; for example, 1-ethyl-6-fluoro-7-{4-[2-(4-chlorophenyl)-2-hydroxyimineothyl]-1-piperazinyl}-4-oxo-1,4-dihydro-3-quinoline carboxylic acid (1b) showed a significant activity against renal cancer cell lines [6].
On the other hand, barbituric acid and its N,N-dialkyl derivatives play an important role in medicinal and pharmaceutical chemistry [7, 8]. For example, 5-benzylidene-barbiturates and thiobarbiturates (1c) show antibacterial activity [9].
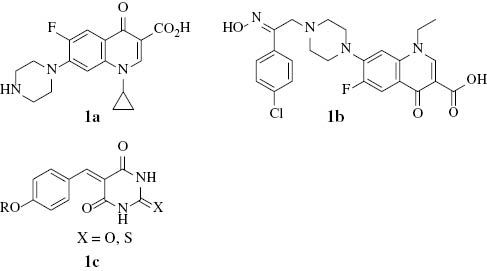
Recently, we described the synthesis and evaluation of the antibacterial activity of model fluoroquinolone-barbiturates [10]. In continuation of our further search for new fluoroquinolone-barbiturate derivatives, we report herein the preparation and characterization of new products of the respective nucleus.
2 Results and discussion
Compound 2 was prepared upon reacting 1 with excess thionyl chloride in dry THF under reflux. Evaporation of excess liquids, followed by the addition of dry triethylamine and 1,3-diethyl-2-thiobarbituric acid onto 2, produced the respective product 3 (Scheme 1).
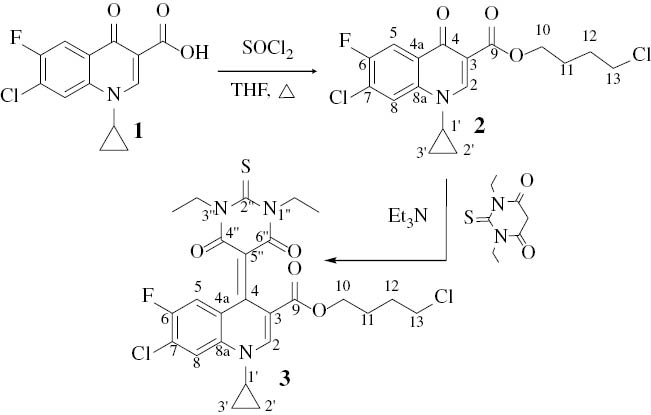
Syntheses of compounds 2 and 3.
The structures of compounds 2 and 3 were characterized by NMR [1H, and distortionless enhancement by polarization transfer (DEPT)-13C], MS and X-ray diffraction analysis. The molecular structure of 2 is shown in Fig. 1. The crystal data and numbers pertinent to data collection and structure refinement of 3 have been reported previously [11], while those for 2 are given herein (Table 1 and Experimental section). There is a short π–π contact between the two inversion-related pyridinic rings with a centroid–centroid distance of 3.562 Å. Related structure determinations have been reported [12, 13].
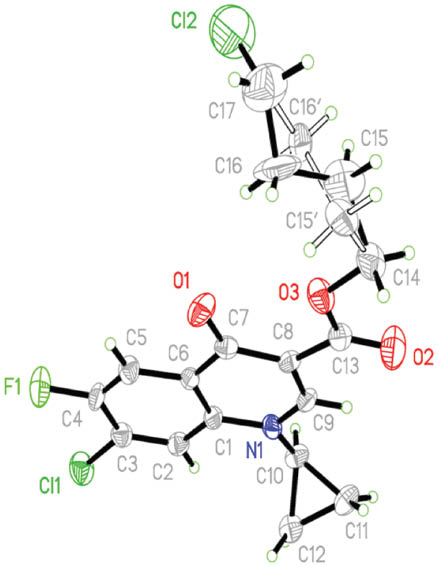
Molecular structure of 2 in the crystal. Both alternatives of the disordered atoms C15 and C16 are shown.
Crystal structure data for 2.
| Empirical formula | C17H16Cl2FNO3 |
| Formula weight Mr | 372.21 |
| Temperature, K | 293 |
| Crystal size, mm3 | 0.29 × 0.03 × 0.03 |
| Crystal system | Monoclinic |
| Space group | P 21/n |
| a, Å | 15.172(4) |
| b, Å | 7.166(2) |
| c, Å | 15.640(6) |
| β, deg | 102.04(3)° |
| V, Å3 | 1663.0(9) |
| Z | 4 |
| Dcalcd., g cm–3 | 1.487 |
| μ(MoKα), mm–1 | 0.416 |
| F(000), e | 768 |
| θ-range data collection, deg | 3.14–25.02 |
| hkl range | –17→18, ±8, –15 → 18 |
| Refl. collected/unique/Rint | 6503/2945/0.11 |
| Refl. with I > 2σ(I) | 905 |
| Refinement method | Full-matrix least squares on F2 |
| Data/restraints/parameters | 2945/0/236 |
| Final R 1/wR 2 (I > 2 σ(I)) | 0.0748/0.0885 |
| Final R 1/wR 2 (all data) | 0.2566/0.1416 |
| Goodness-of-fit on F2 | 0.918 |
| Largest difference peak/hole, e Å–3 | 0.31/–0.25 |
To investigate this result, the reaction of 1 with thionyl chloride in dry THF was conducted using the previously mentioned procedure, and the result was consistent with that obtained for compound 2. It is clear that tetrahydrofuran reacted with the acid chloride 4, once formed in situ, as based on the hard–hard interactions. The oxygen atom of tetrahydrofuran is considered a hard nucleophile and the carbon of the acyl chloride as a hard electrophile; the proposed SN2 mechanism is given (Scheme 2).
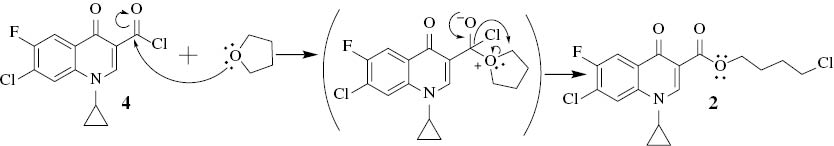
Proposed mechanism for the formation of compound 2.
A Knoevenagel condensation reaction occurred in the formation of 3, whereby a carbon–carbon double bond was formed between the quinoline and thiobarbiturate moieties. This condensation reaction seems to be more favorable than the Michael addition reaction of thiobarbiturate at the β-carbon which permits for extra resonance structures through the lone pair of the quinoline nitrogen atom and subsequently decreasing the electrophilic character of the β-carbon.
The reaction of 1 with excess thionyl chloride in dry toluene was conducted under reflux to afford, as expected, the corresponding acid chloride 4 which was characterized by IR spectroscopy; the progress of the reaction was monitored by TLC. Separately, solutions of barbituric acid and 1,3-dimethylbarbituric acid were added into the solution of 4 in the presence of triethylamine to give the target products 5 and 6, respectively (Scheme 3). The progress of these reactions was monitored by TLC.
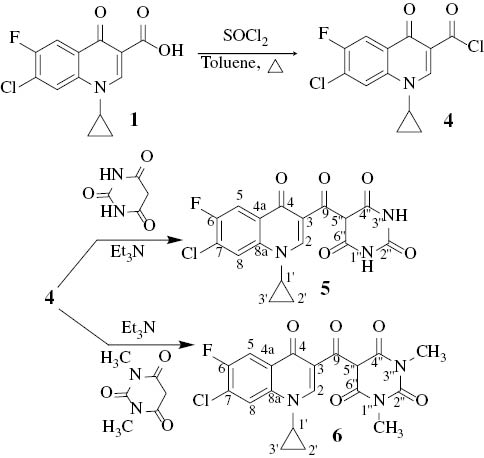
Syntheses of the fluoroquinolene-barbiturate hybrids 5 and 6.
From DEPT-13C NMR analysis, it is clear that C5 of the barbiturate moiety in 5 and 6 carries one hydrogen atom; both compounds adopted the keto form rather than the enol tautomer [14]. The prepared compounds 3, 5 and 6 will be evaluated as antibacterial agents in due course.
3 Experimental section
All experiments were performed in purified solvents. 7-Chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (1) was purchased from Sigma Aldrich (St. Louis, MO, USA) and 4-chlorobutyl 7-chloro-1-cyclopropyl-4-(1,3-diethyl-4,6-dioxo-2-sulfanylidene-1,3-diazinan-5-ylidene)-6-fluoro-1,4-dihydroquinoline-3-carboxylate (3) was prepared according to the published procedure [11]. The spectral data of 3 are given in the experimental part.
3.1 4-Chlorobutyl 7-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (2)
A mixture of 1 (1.5 g, 5.3 mmol) and SOCl2 (4 mL, 54 mmol) in 25 mL of dry THF was stirred under reflux for 8 h. After cooling to room temperature, THF and excess SOCl2 were evaporated under reduced pressure. The solid residue was purified by column chromatography using MeOH–CHCl3 (10:90) as eluent to afford the title compound as colorless crystals. Yield 1.5 g (95 %); m.p. 167–169 °C. – IR (film): v = 3084, 2988, 2927, 2856, 1727, 1615, 1541, 1455 cm–1. – 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 1.17 (m, 2H) and 1.38 (m, 2H), (2′-H/3′-H), 1.94–2.01 (m, 4H, 11-H + 12-H), 3.49 (m, 1H, 1′-H), 3.65 (t, J = 8 Hz, 2H, 13-H), 4.37 (t, J = 8 Hz, 2H, 10-H), 8.01 (s, 1H, 8-H), 8.15 (d, 3JH–F = 10 Hz, 1H, 5-H), 8.56 (s, 1H, 2-H). – 13C NMR (100 MHz, CDCl3): δ = 8.3 (C-2′/C-3′), 26.1 (C-11), 29.3 (C-12), 34.8 (C-1′), 44.7 (C-13), 64.2 (C-10), 110.6 (C-3), 113.8 (d, 2JC–F = 20 Hz, C-5), 119.0 (C-8), 127.0 (d, 2JC–F = 17.5 Hz, C-7), 128.7 (d, 3JC–F = 5 Hz, C-4a), 137.2 (C-8a), 148.9 (C-2), 156.6 (d, 1JC–F = 260 Hz, C-6), 165.3 (C-9), 172.7 (C-4). – HRMS (ESI): m/z = 372.05879 (calcd. 372.05695 for C17H17Cl2FNO3, [M+H]+). – C17H16Cl2FNO3 (372.2): calcd. C 54.86, H 4.33, N 3.76; found C 54.79, H 4.42, N 3.62.
3.2 4-Chlorobutyl 7-chloro-1-cyclopropyl-4-(1,3-diethyl-4,6-dioxo-2-thioxo-tetrahydropyrimidin-5(6H)-ylidene)-6-fluoro-1,4-dihydroquinoline-3-carboxylate (3) [11]
M.p. 230–232 °C. – IR (film): v = 3081, 2988, 2926, 2859, 1709, 1671, 1625, 1540, 1456, 1281 cm–1. – 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 1.16 (m, 2H) and 1.38 (m, 2H), (2′-H/3′-H), 1.41 (t, J = 8 Hz, 6H, CH3), 1.94–2.03 (m, 4H, 11-H + 12-H), 3.49 (m, 1H, 1′-H), 3.67 (t, J = 8 Hz, 2H, 13-H), 4.38 (t, J = 8 Hz, 2H, 10-H), 4.62 (q, J = 7 Hz, 4H, CH2-Me), 8.00 (s, 1H, 8-H), 8.15 (d, 3JH–F = 10 Hz, 1H, 5-H), 8.59 (s, 1H, 2-H). – 13C NMR (100 MHz, CDCl3): δ = 8.9 (C-2′/C-3′), 12.7 (CH3), 25.7 (C-11), 29.6 (C-12), 38.4 (C-1′), 44.2 (C-13), 45.8 (N-CH2), 65.5 (C-10), 108.2 (C-5″), 110.1 (C-3), 118.0 (d, 2JC–F = 20 Hz, C-5), 119.4 (C-8), 125.9 (d, 2JC–F = 20 Hz, C-7), 129.1 (d, 3JC–F = 5 Hz, C-4a), 136.4 (C-8a), 145.4 (C-2), 151.2 (C-4), 156.5 (d, 1JC–F = 260 Hz, C-6), 160.3 (C-4″/C-6″), 164.7 (C-9), 177.9 (C=S). – HRMS (ESI): m/z = 554.10578 (calcd. 554.10834 for C25H27Cl2FN3O4S, [M+H]+). –C25H26Cl2FN3O4S (554.5): calcd. C 54.15, H 4.73, N 7.58; found C 53.91, H 4.82, N 7.41.
3.3 7-Chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carbonyl chloride (4)
A mixture of 1 (1.5 g, 5.3 mmol) and thionyl chloride (SOCl2) (4 mL, 54 mmol) in 20 mL of dry toluene was stirred under reflux for 8 h. After cooling to room temperature, toluene and excess SOCl2 were evaporated under reduced pressure. The solid residue was suspended in dry hexane and the suspension was evaporated to dryness to afford the solid residue which was used without further purification. Yield 1.5 g (95 %). – IR (KBr): v = 3071, 2982, 1726, 1705, 1460, 1361 cm–1. – 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 1.17 (m, 2H) and 1.37 (m, 2H), (2′-H/3′-H), 3.51 (m, 1H, 1′-H), 8.00 (s, 1H, 8-H), 8.17 (d, 3JH–F = 10 Hz, 1H, H-5), 8.53 (s, 1H, 2-H).
3.4 5-(7-Chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carbonyl)pyrimidine-2,4,6(1H,3H,5H)-trione (5)
A mixture of 4 (0.6 g, 2 mmol), barbituric acid (0.28 g, 2 mmol) and 0.9 mL of Et3N in 25 mL of dry CH2Cl2 was stirred at room temperature for 8 h and then warmed (40–45 °C) for 6 h. After cooling to room temperature, the precipitated solid product was collected by suction filtration, washed with CH2Cl2 and dried in vacuo to afford the title product as red precipitate. Yield 0.61 g (78 %); m.p. 212–213 °C. – IR (film): v = 3425, 3114, 2929, 2998, 1707, 1624, 1505, 1455 cm–1. – 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 1.16 (m, 2H) and 1.38 (m, 2H), (2′-H/3′-H), 3.49 (m, 1H, 1′-H), 4.45 (s, 1H, 5″-H), 8.02 (s, 1H, 8-H), 8.17 (d, 3JH–F = 10 Hz, 1H, H-5), 8.56 (s, 1H, 2-H), 9.45 (s, 2H, NH). – 13C NMR (100 MHz, [D6]DMSO): δ = 8.2 (C-2′/C-3′), 35.7 (C-1′), 59.7 (C-5″), 109.7 (C-3), 113.7 (d, 2JC–F = 20 Hz, C-5), 118.8 (C-8), 127.9 (d, 2JC–F = 20 Hz, C-7), 128.1 (d, 3JC–F = 5 Hz, C-4a), 137.2 (C-8a), 148.9 (C-2), 152.1 (C-2″), 155.6 (d, 1JC–F = 260 Hz, C-6), 162.5 (C-4″/C-6″), 172.4 (C-4), 194.3 (C-9). – HRMS (ESI): m/z = 390.02995 (calcd. 390.02930 for C17H10ClFN3O5, [M–H]–). – C17H11ClFN3O5 (391.7): calcd. C 52.12, H 2.83, N 10.73; found C 51.89, H 3.19, N 10.61.
3.5 5-(7-Chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carbonyl)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (6)
A mixture of 4 (0.6 g, 2 mmol), 1,3-dimethylbarbituric acid (0.31 g, 2 mmol) and 0.9 mL of Et3N in 25 mL of dry CH2Cl2 was stirred at room temperature for 8 h and then warmed (40–45 °C) for 6 h. After cooling to room temperature, the precipitated solid product was collected by suction filtration, washed with CH2Cl2 and dried in vacuo to afford the title product as red precipitate. Yield 0.58 g (69 %); m.p. 188–190 °C. – IR (film): v = 3125, 3010, 2978, 1719, 1642, 1505, 1451 cm–1. – 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 1.17 (m, 2H) and 1.31 (m, 2H), (2′-H/3′-H), 3.12 (s, 6H, 2CH3), 3.44 (m, 1H, 1′-H), 4.51 (s, 1H, 5″-H), 8.08 (s, 1H, 8-H), 8.15 (d, 3JH–F = 10 Hz, 1H, 5-H), 8.49 (s, 1H, 2-H), 9.38 (s, 2H, NH). – 13C NMR (100 MHz, [D6]DMSO): δ = 8.3 (C-2′/C-3′), 29.5 (CH3), 35.7 (C-1′), 60.1 (C-5″), 108.6 (C-3), 113.3 (d, 2JC–F = 20 Hz, C-5), 118.8 (C-8), 128.0 (d, 2JC–F = 20 Hz, C-7), 128.4 (d, 3JC–F = 5 Hz, C-4a), 137.0 (C-8a), 149.2 (C-2), 151.7 (C-2″), 155.6 (d, 1JC–F = 260 Hz, C-6), 161.3 (C-4″/C-6″), 172.3 (C-4), 192.8 (C-9). – HRMS (ESI): m/z = 420.07241 (calcd. 420.07625 for C19H16ClFN3O5, [M+H]+). – C19H15ClFN3O5 (419.8): calcd. C 54.36, H 3.60, N 10.01; found C 54.02, H 3.89, N 9.81.
3.6 X-Ray structure determination of 2
A suitable colorless tiny crystal of 2 with approximate dimensions 0.29 × 0.03 × 0.03 mm3 was epoxy-mounted on a glass fiber and the diffraction data were collected at room temperature employing enhanced MoKα radiation, λ = 0.71073 Å, and a Calibur/Oxford diffractometer equipped with an Eos CCD detector [15]. Three ω-scan runs, 228 frames, were collected after optimization with an exposure time of 19.92 s, 1° frame width, and a detector distance of 45 mm. An absorption correction obtained by applying multi-scan was done with minimum and maximum transmission factors of 0.8892 and 0.9878, respectively. The tiny crystal size resulted in a data set with a large number of weak reflections. Data sets were measured of several crystals. The best results are the basis for the published structure determination. The CH2CH2CH2Cl ether-linked branch in 2 exhibited large displacement parameters, and C15 and C16 therein are best refined as a split-atom model over two positions with occupancy factors of 0.607 and 0.393, respectively. The cell parameters were refined using all observed reflections, the data were reduced using Olex2 [16], and the structure was solved and refined with the Shelxtl program package [17]. Table 1 summarizes the crystal data and data collection and structure refinement parameters.
CCDC 1042243 (2) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
Acknowledgment
The authors gratefully acknowledge the financial support from the Deanship of Scientific Research at the University of Jordan.
References
[1] L. A. Mitscher, Chem. Rev. 2005, 105, 559.10.1021/cr030101qSuche in Google Scholar PubMed
[2] C. C. Tzeng, Y. L. Chen, Chin. Pharm. J.2002, 54, 229.Suche in Google Scholar
[3] J. Y. Sheu, Y. L. Chen, K. C. Fang, T. C. Wang, C. F. Peng, C. C. Tzeng, J. Heterocycl. Chem.1998, 35, 955.Suche in Google Scholar
[4] Y. L. Chen, K. C. Fang, J. Y. Sheu, S. L. Hsu, C. C. Tzeng, J. Med. Chem. 2001, 44, 2374.10.1021/jm0100335Suche in Google Scholar PubMed
[5] J. Y. Sheu, Y. L. Chen, C. C. Tzeng, S. L. Hsu, K. C. Fang, T. C. Wang, Helv. Chim. Acta.2003, 86, 2481.10.1002/hlca.200390201Suche in Google Scholar
[6] K. C. Fang, Y. L. Chen, J. Y. Sheu, T. C. Wang, C. C. Tzeng, J. Med. Chem.2000, 43, 3809.10.1021/jm000153xSuche in Google Scholar PubMed
[7] S. von Angerer, Sci. Synth. 2004, 16, 379.Suche in Google Scholar
[8] M. Holtkamp, H. Meierkord, Cell Mol. Life Sci. 2007, 64, 2023.10.1007/s00018-007-7021-2Suche in Google Scholar PubMed
[9] Q. Yan, R. Cao, W. Yi, Z. Chen, H. Wen, L. Ma, H. Song, Eur. J. Med. Chem.2009, 44, 4235.10.1016/j.ejmech.2009.05.023Suche in Google Scholar PubMed
[10] K. Sweidan, W. Abu Rayyan, M. Abu Zarga, M. M. El-Abadelah, H. A. Y. Mohammad, Lett. Org. Chem.2014, 11, 422.10.2174/1570178611666140401220850Suche in Google Scholar
[11] K. Sweidan, S. F. Haddad, M. A. AlDamen, A. Al-Sheikh, Acta Crystallogr. 2013, E69, o1191.Suche in Google Scholar
[12] M. Nieger, B. Traufetter, H. Wamhoff, private communication, refcode: IQONUX, Cambridge Structural Database, Cambridge (U.K.) 2004.Suche in Google Scholar
[13] R. A. Al-Qawasmeh, Acta Crystallogr. 2012, E68, o2533.Suche in Google Scholar
[14] K. Sweidan, Q. Abu-Salem, A. Al-Sheikh, G. Abu Sheikha, Lett. Org. Chem.2009, 6, 669.10.2174/157017809790442934Suche in Google Scholar
[15] CrysalisPro Software System (version 1.171.35.11; release 16-05-2011), Intelligent Data Collection and Processing Software for Small Molecule and Protein Crystallography, Agilent Technologies Ltd., Yarnton, Oxfordshire (U.K.) 2011.Suche in Google Scholar
[16] O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, H. Puschmann, J. Appl. Cryst.2009, 42, 339.10.1107/S0021889808042726Suche in Google Scholar
[17] G. M. Sheldrick, Shelxtl (version 6.10), Bruker Analytical X-ray Instruments Inc., Madison, WI (USA) 2000.Suche in Google Scholar
©2015 by De Gruyter
Artikel in diesem Heft
- Frontmatter
- In this Issue
- EPR studies on carboxylic esters, 23 [1]. Preparation of new dialkyl azulenedicarboxylates and EPR-spectroscopic study of their radical anions
- Synthesis and crystal structure of a 3D copper(II)–silver(I) coordination polymer assembled through hydrogen bonding, π–π stacking and metal–π interactions, {[Cu(phen)2(CN)][Ag(CN)2] · 3H2O}n (phen = 1,10-phenanthroline)
- A polyoxometalate-based inorganic–organic hybrid material: synthesis, characterization structure and photocatalytic study
- Hydrogen-bonded assemblies of two organically templated borates: syntheses and crystal structures of [(1,10-phen)(H3BO3)2] and [2-EtpyH][(B5O6(OH)4]
- Catalytic activity of the nanoporous MCM-41 surface for the Paal–Knorr pyrrole cyclocondensation
- A new route for the synthesis of 4-arylacetamido-2-aminothiazoles and their biological evaluation
- Structural and spectroscopic characterization of isotypic sodium, rubidium and cesium acesulfamates
- Nd39Ir10.98In36.02 – A complex intergrowth structure with CsCl- and AlB2-related slabs
- Syntheses, structures and magnetic properties of two mononuclear nickel(II) complexes based on bicarboxylate ligands
- Synthesis and characterization of some new fluoroquinolone-barbiturate hybrid systems
- Synthesis of pyrazoles containing benzofuran and trifluoromethyl moieties as possible anti-inflammatory and analgesic agents
Artikel in diesem Heft
- Frontmatter
- In this Issue
- EPR studies on carboxylic esters, 23 [1]. Preparation of new dialkyl azulenedicarboxylates and EPR-spectroscopic study of their radical anions
- Synthesis and crystal structure of a 3D copper(II)–silver(I) coordination polymer assembled through hydrogen bonding, π–π stacking and metal–π interactions, {[Cu(phen)2(CN)][Ag(CN)2] · 3H2O}n (phen = 1,10-phenanthroline)
- A polyoxometalate-based inorganic–organic hybrid material: synthesis, characterization structure and photocatalytic study
- Hydrogen-bonded assemblies of two organically templated borates: syntheses and crystal structures of [(1,10-phen)(H3BO3)2] and [2-EtpyH][(B5O6(OH)4]
- Catalytic activity of the nanoporous MCM-41 surface for the Paal–Knorr pyrrole cyclocondensation
- A new route for the synthesis of 4-arylacetamido-2-aminothiazoles and their biological evaluation
- Structural and spectroscopic characterization of isotypic sodium, rubidium and cesium acesulfamates
- Nd39Ir10.98In36.02 – A complex intergrowth structure with CsCl- and AlB2-related slabs
- Syntheses, structures and magnetic properties of two mononuclear nickel(II) complexes based on bicarboxylate ligands
- Synthesis and characterization of some new fluoroquinolone-barbiturate hybrid systems
- Synthesis of pyrazoles containing benzofuran and trifluoromethyl moieties as possible anti-inflammatory and analgesic agents