Permanent dynamic transporter-mediated turnover of glutamate across the plasma membrane of presynaptic nerve terminals: arguments in favor and against
-
Tatiana Borisova


Abstract
Mechanisms for maintenance of the extracellular level of glutamate in brain tissue and its regulation still remain almost unclear, and criticism of the current paradigm of glutamate transport and homeostasis has recently appeared. The main premise for this study is the existence of a definite and non-negligible concentration of ambient glutamate between the episodes of exocytotic release in our experiments with rat brain nerve terminals (synaptosomes), despite the existence of a very potent Na+-dependent glutamate uptake. Glutamate transporter reversal is considered as the main mechanisms of glutamate release under special conditions of energy deprivation, hypoxia, hypoglycemia, brain trauma, and stroke, underlying an increase in the ambient glutamate concentration and development of excitotoxicity. In the present study, a new vision on transporter-mediated release of glutamate as one of the main mechanisms involved in the maintenance of definite concentration of ambient glutamate under normal energetical status of nerve terminals is forwarded. It has been suggested that glutamate transporters act effectively in outward direction in a non-pathological manner, and this process is thermodynamically synchronized with uptake and provides effective outward glutamate current, thereby establishing and maintaining permanent and dynamic glutamatein/glutamateout gradient and turnover across the plasma membrane. In this context, non-transporter tonic glutamate release by diffusion, spontaneous exocytosis, cystine-glutamate exchanger, and leakage through anion channels can be considered as a permanently added ‘new’ exogenous substrate using two-substrate kinetic model calculations. Permanent glutamate turnover is of value for tonic activation of post/presynaptic glutamate receptors, long-term potentiation, memory formation, etc. Counterarguments against this mechanism are also considered.
Rationale
Chemical synapses are the main structures responsible for nerve signal transmission in the brain. The basic principles of brain functioning still remain almost uncertain despite great efforts and a lot of experimental data. In this context, it is suggested that some misevaluation of the role of the key processes underlying synaptic transmission takes place. The main premise for this work is the fact that definite ambient concentrations of small amino acid neurotransmitters exist in presynaptic rat brain nerve terminals (synaptosomes) in our experiments albeit potent neurotransmitter’s reuptake. Synaptosomes retain all characteristics of intact nerve terminals, that is, the ability to maintain membrane potential and accomplish uptake and transporter-mediated release of glutamate, exocytosis, etc., and are one of the best systems to explore the relationship between the structure of proteins, its biochemical and cell-biological properties, and physiological role (Südhof, 2004).
The extracellular level of glutamate in nerve terminals
Eighty years ago, it has been accepted that glutamate plays a central metabolic role in the brain (Krebs, 1935; Stern et al., 1949). Then the excitatory action of glutamate in the brain tissue and importance of uptake for its control were discovered (Hayashi, 1954), and after that, glutamate was considered as the major excitatory neurotransmitter in the mammalian nervous system (Danbolt, 2001, for review; Krnjevic, 1970; Schousboe et al., 1976; Schousboe, 1981; Watkins and Evans, 1981; Fonnum et al., 1981). Glutamate is involved in many aspects of normal brain functioning, e.g. cognition, memory, and learning. Disturbance in glutamate homeostasis leads to the development of neurologic consequences and is a common feature of the pathogenesis of major neurological and neurodegenerative disorders and diseases, e.g. Alzheimer’s and Parkinson’s diseases, brain trauma, schizophrenia, epilepsy, etc. (Danbolt, 2001, for review).
Chemical synapses release neurotransmitters by exocytosis of synaptic vesicles. Nerve signal transmission is initiated by depolarization of the plasma membrane of presynaptic nerve terminals followed by Ca2+ entrance, fusion of synaptic vesicles with the plasma membrane, and exocytotic release of the neurotransmitters to the synaptic cleft, which then interact with the postsynaptic receptors and activate the signaling pathways. After exocytosis, termination of neurotransmission occurs by neurotransmitter reuptake to the neurons and glial cells. It is so because there are no enzymes in the synaptic cleft responsible for degradation of glutamate [γ-aminobutyric acid (GABA), glycine, aspartate, etc.]. Glutamate transporters are integral membrane proteins with eight putative transmembrane domains, which locate in the plasma membrane of neurons and glial cells, and use Na+/K+ electrochemical gradient as energy source for transport of glutamate from the synaptic cleft into the cytosol against its gradient (Danbolt, 2001, for review; Kasatkina and Borisova, 2010). Km values of glutamate uptake reported for brain synaptosome preparations vary between 1–5 μm, 13.9 μm, 25 μm, and 36 μm (Robinson, 1998; Debler and Lajtha, 1987; Bennett et al., 1973; Logan and Snyder, 1972; Danbolt, 2001, for review). In our experiments with synaptosomes, Km was equal to 10.7±2.5 μm, and Vmax, -12.5±3.2 nmol/min/mg of protein (Borisova et al., 2004; Borisova and Himmelreich, 2005). The glutamate binding sites in the transporters are optimized for binding of extracellular but not intracellular glutamate, and the affinity of the transporters for glutamate, when the substrate binding site faces the extracellular space, is about 50 times higher as compared to the intracellular glutamate binding, thereby promoting dissociation of glutamate into the cytoplasm against a high intracellular glutamate concentration (Watzke and Grewer, 2001). The roles of glutamate transporters in synaptic transmission are far from being completely understood.
The ambient concentration of glutamate between the episodes of its exocytotic release is a crucial parameter of synaptic neurotransmission. The mechanisms of its establishment and maintenance were assessed in a restricted number of publications. It is commonly accepted that the extracellular glutamate level is a balance between neurotransmitter uptake and release. There is a ‘dual’ contradicted significance of the extracellular level of glutamate in the synaptic cleft. From one side, a low extracellular glutamate concentration maintained between episodes of exocytotic release prevents continual activation of postsynaptic glutamate receptors and protects neurons from excitotoxic injury and death and also provides a high signal-to-noise ratio for glutamatergic transmission (Jabaudon et al., 1999; Cavelier et al., 2005). From the other side, the certain level of ambient glutamate is very important for tonic activation of presynaptic and postsynaptic glutamate receptors, e.g. NMDA receptors in hippocampal slices, AMPA, kainite, and mGlu receptors, which is a way of regulation of glutamatergic transmission (Sah et al., 1989; Dalby and Mody, 2003; Cavelier and Attwell, 2005), and knowledge of the precise ambient extracellular glutamate concentration in the brain is required for understanding its potential impacts on tonic and phasic receptor signaling (Suna et al., 2014). Definite extracellular neurotransmitter concentration in vivo is significant for several neurological processes such as migration and differentiation of brain cells during development and synaptic plasticity (LoTurco et al., 1995; Nguyen et al., 2001; Manent and Represa, 2007; Featherstone and Shippy, 2008).
A lack of consensus regarding the value of the extracellular glutamate level is observed among groups (Pendyam et al., 2012). The existence of numerous nonlinear mechanisms makes the characterization of glutamate homeostasis in the synapses very difficult (Pendyam et al., 2012), and the concentration of glutamate in the central nervous system varies in dependence on the biological compartment being measured (Moussawi et al., 2011). Glutamate levels in cerebrospinal fluid are around 10 μm; intracellular glutamate concentrations in the brain are approximately 10 mm (Danbolt, 2001, for review; Featherstone and Shippy, 2008). Glutamate concentration in the synaptic cleft following action potential-mediated release exceeds 1 mm for <10 ms and quickly returns to <20 nm between release events due to glutamate uptake by neurons and glia (Dzubay and Jahr, 1999). The extracellular glutamate concentration that resulted from electrophysiological estimates of tissue slices in vitro was 0.02–0.1 μm, whereas in vivo measurements using microdialysis or voltammetry varied between 1 and 30 μm (Moussawi et al., 2011). Thus, estimates of the basal concentration of glutamate within the extracellular space vary over three orders of magnitude, ranging from 0.02 to 30 μm, and depend on the methods of measurement (Herman and Jahr, 2007; Chefer et al., 2009; Moussawi et al., 2011). Estimates of ambient glutamate based on microdialysis measurements are approximately 100-fold higher than those based on electrophysiological measurements of tonic NMDA receptor activity (Suna et al., 2014). It is so because a thin layer of damaged tissue with disrupted glutamate transport could underlie the significant quantitative discrepancy between the ambient glutamate estimates provided by electrophysiological studies in slices and those from microdialysis studies (Suna et al., 2014). For monoamine-, acetylcholine-, and purine-type neurotransmitters, the extracellular concentrations are also maintained at low levels (<100 nm); however, it is determined presumably by neurotransmitter release (van der Zeyden et al., 2008).
The mechanism maintaining correct extracellular level and homeostasis of glutamate in the central nervous system and its regulation is crucial for understanding normal and pathological synaptic neurotransmission, and the absence of consensus in estimations of ambient glutamate concentration reveals the multiplicity of these mechanisms. Several publications with critics of the current paradigm of glutamate transport and homeostasis have recently appeared (Funicello et al., 2004; Pendyam et al., 2012; Makarov et al., 2013). In our experiments, an increase in the ambient level of glutamate under conditions of cholesterol deficiency was associated with weak uptake in nerve terminals, and also tonic (Borisova et al., 2010a,b) and transporter-mediated (Krisanova et al., 2012) release of l-[14C]glutamate depended on the ratio of glutamatein/glutamateout in nerve terminals. However, changes in transporter-mediated uptake of glutamate were not always accompanied by alterations in the ambient level of glutamate that was shown under specific conditions of centrifuge-induced hypoxia (Borisova et al., 2004; Borisova and Himmelreich, 2005; Borisova and Krisanova, 2008; Krisanova et al., 2009). Average basal extracellular concentration of l-[14C]glutamate in l-[14C]glutamate-preloaded synaptosomes in our experiments was equal to 0.193±0.013 nmol/mg protein (Borisova et al., 2010a). This ambient level of l-[14C]glutamate estimated as percentage of total label incorporated in nerve terminals consisted of 18%±1% and was similar in Ca2+-free and Ca2+-containing media and depended on synaptosomal protein concentration and so on the number of glutamate transporters in the preparation (Borisova et al., 2010a). The dependence of ambient glutamate on membrane densities of expressed glutamate transporters was shown in Xenopus oocytes (Suna et al., 2014).
According to the existing theory, the establishment and maintenance of the extracellular level of glutamate are determined by a balance between non-transporter tonic release (occurred through diffusion, spontaneous exocytosis, anion channels, cystine-glutamate exchanger (Bouvier et al., 1992; Danbolt, 2001, for review; Cavelier and Attwell 2005) and uptake of glutamate that can be represented as follows:
To maintain ambient l-[14C]glutamate at the permanent level of 18%±1% of total label incorporated in nerve terminals, tonic non-transporter release should significantly exceed the uptake of l-[14C]glutamate. However, in our experiments, tonic non-transporter release of l-[14C]glutamate in the presence of glutamate transporter inhibitor dl-threo-β-benzyloxyaspartate (dl-TBOA) (200 μm) consisted of 7.8%±0.6% of total label for 6 min and was accompanied with a consequent increase in extracellular l-[14C]glutamate (Borisova and Krisanova, 2008; Borisova et al., 2010a). So, tonic release during blockage of glutamate transporters is at least two times lower than the extracellular level of l-[14C]glutamate in synaptosomes, which can be represented as follows:
Therefore, there is no balance between the extracellular level of l-[14C]glutamate and its tonic release from synaptosomes under conditions of blocked glutamate transporter functioning. So, the contribution of transporter-mediated release to the establishment of the extracellular level of glutamate can be predicted (see the next sections).
The absolute value of the ambient level of glutamate in the parallel synaptosomal preparations was determined in our experiments by different methods, that is, amino acid analyzer, spectrofluorimetric glutamate dehydrogenase assay, and amperometric glutamate-sensitive biosensor based on the analysis of the activity of glutamate oxidase, and it was equal to 2.8 μm (Borisova et al., 2010a; Borisova, 2013), 2.3 μm (Krisanova et al., 2012), and 7.9 μm (Soldatkin et al., 2015), respectively. A comparative study of glutamate uptake using l-[14C]glutamate and glutamate biosensor (Soldatkin et al., 2015) revealed that almost all exogenously added glutamate (10 μm) was utilized by glutamate transporters for several minutes. Similarly with abovementioned experiments with radiolabeled l-[14C]glutamate, it is difficult to explain the fact that the extracellular level of glutamate in our biosensor experiments was almost similar (~8 μm) before the addition of exogenous glutamate (10 μm) for initiation of uptake process and after completion of uptake. So, a constant amount of ambient glutamate remains in the medium before and after uptake measurements (stimulated by the addition of exogenous neurotransmitter at a concentration of 10 μm).
Permanent glutamate turnover across the plasma membrane of nerve terminals
As it can be concluded from the abovementioned section, it is absolutely unclear why definite and non-negligible concentration of extracellular glutamate exists in our experiments and why ambient glutamate is not deleted from the extracellular space, despite the availability of strong and potent mechanism of transporter-mediated uptake. Also, there is no balance between non-transporter tonic release and the extracellular level of l-[14C]glutamate in our experiments (Equation 2). In this context, use of isolated nerve terminals allows to overcome contribution of glial uptake, thereby uncovering and bringing new insight to the mechanisms of the establishment of ambient glutamate level.
The abovementioned facts and the existence of definite level of ambient l-[14C]glutamate (Borisova et al., 2010a) and also of definite extracellular level of endogenous glutamate in nerve terminals determined by amino acid analyzer, spectrofluorimetric glutamate dehydrogenase assay, and amperometric glutamate-sensitive biosensor in our experiments (Borisova et al., 2010a; Krisanova et al., 2012; Soldatkin et al., 2015) forced the author to forward new hypothesis. The existence of the efficient permanent extracellular/intracellular glutamate exchange and so the permanent dynamic glutamate gradient across the plasma membrane of nerve terminals and the central role of glutamate transporters in this glutamate turnover is suggested. As glutamate transporter functioning is driven by Na+/K+ electrochemical gradient across the plasma membrane created by Na/K ATPase, it is clear that the maintenance of such permanent glutamate gradient requires a significant energy supplementation.
The maintenance of a permanent glutamate gradient and turnover across the plasma membrane requires specific mechanisms providing both the inward and outward glutamate current in nerve terminals. Below, we discuss potency, strength, and weakness of all prospective candidates ensuring the direct and reverse glutamate current in presynapse.
Suggested mechanisms for permanent inward glutamate transport in nerve terminals
This is a formal subsection concerning inward glutamate transport mechanisms, because between three possible candidates, that is, high-affinity Na+-dependent glutamate transporters, low-affinity glutamate transporters, and glutamate-glutamine neuronal-glial cycle, only high-affinity Na+-dependent glutamate transporters in nerve terminals are able to provide effective, highly regulated permanent inward movement of glutamate molecules across the plasma membrane. It is commonly accepted that, namely, Na+-dependent glutamate transporters provide the maintenance of low extracellular glutamate concentration in the synaptic cleft. Low-affinity uptake exhibits Km values above 500 μm (Johnston, 1981). This uptake system has been suggested to supply brain cells with amino acids for metabolic purposes (for review, see studies by Erecinska and Silver [1990] and Danbolt [2001]). In vivo, high-affinity Na+-dependent glutamate transporters of glial cells and glutamate-glutamine cycle can also contribute to inward glutamate transport. The role of glia in the maintenance of proper extracellular glutamate level is to delete an excess of glutamate from the extracellular space and to prevent uncontrolled leakage of glutamate from the synaptic cleft to the intersynaptic space (see Danbolt, 2001, for review).
Suggested mechanisms for permanent outward glutamate transport in nerve terminals
The most complicated issue is to specify the potential mechanisms providing permanent outward glutamate current in nerve terminals. One of the main requirements is that this process should be potent enough as compared to uptake system, be highly regulated, and reacts immediately on changes in the functional state of nerve terminals. It is clear that any misregulation in outward glutamate current can lead to serious consequences impairing neuronal communication. The suggested mechanisms maintaining permanent outward glutamate transport from nerve terminals are the following: (1) non-pathological transporter-mediated release of glutamate synchronized with uptake, (2) spontaneous exocytosis, (3) diffusion, (4) cystine-glutamate exchange, (5) release through anion channels, and also glutamate release as consequence of activation of presynaptic glutamate receptors. Below, we examined the strong and weak points of all these candidates for outward glutamate current.
Non-pathological transporter-mediated glutamate release synchronized with uptake
It is accepted that glutamate transporter reversal is the main mechanism of glutamate release resulting in an increase in ambient glutamate and development of excitotoxicity under special conditions of energy deprivation, hypoxia, hypoglycemia, ischemia, brain trauma, stroke, etc. (Grewer et al., 2008).
Our data showed that ‘cold’ glutamate-, dl-threo-β-hydroxyaspartate (dl-THA)- (Krisanova et al., 2012), and d-aspartate (unpublished data) substituted l-[14C]glutamate in synaptosomes and induced a comparable increase in the ambient level of l-[14C]glutamate (and also d-[3H]aspartate) in nerve terminals. So, a quick redistribution of l-[14C]glutamate between intracellular and extracellular spaces, the new intracellular/extracellular ratio of glutamate/l-[14C]glutamate, and efficient synaptosomal extracellular/intercellular glutamate turnover take place in nerve terminals. The common feature of all studied substances, i.e. glutamate, dl-THA, and d-aspartate, is that they are substrates of glutamate transporters. The resemblance of the data obtained with above agents, some of which are the receptor agonists or the substrates for different enzymes and exchangers, shows that registered dynamic changes in extracellular/intracellular distribution of radioactive l-[14C]glutamate are due to the functioning of glutamate transporters only. It is hypothesized that glutamate transporters provide dynamics extracellular/intracellular l-[14C]glutamate turnover after the application of different transporter substrates. The similarity in the effectiveness of different transporter substrates in the establishment of new extracellular level of the neurotransmitter indicates the central role of the transporters in this efficient glutamate exchange between the extracellular and intercellular compartments of nerve terminals.
Cavelier and Attwell (2005) considered a possibility that exogenously applied glutamate might act on cell in the slice to release further glutamate. Recently, Makarov et al. (2013) have proposed a complex two-substrate kinetic model that takes into account transporter reversal. It has been suggested that the Michaelis-Menten kinetic scheme, which is popular in transporter studies, does not include transporter reversal and completely neglects the possibility of equilibrium between the substrate concentrations on both sides of the membrane. According to Makarov et al. (2013), one transporter substrate can release another one already accumulated inside the cell. This model allows the calculation of a ‘heteroexchange’ and ‘transacceleration’ using standard Michaelis coefficients for respective substrates. Transport depends on the energy balance of coupled transport of the substrate and simultaneously transported ions. The main conclusion of Makarov et al. (2013) is that when the substrates in both sides of the membrane are in equilibrium, the addition of extra substrate leads to re-equilibrium and release of the substrate (the same one already accumulated in the cytosol) from the cell.
In the present study, a new vision on glutamate transporter functioning in non-pathological reverse mode is forwarded. The author suggests that glutamate transporters act effectively in reverse direction in non-pathological manner; this process is thermodynamically synchronized with uptake, providing effective outward glutamate flow from nerve terminals, thereby maintaining permanent and dynamic glutamatein/glutamateout gradient across the plasma membrane and supporting the establishment and maintenance of a definite extracellular level of the neurotransmitter. Equation (1) can be corrected as follows:
Spontaneous exocytosis of synaptic vesicles
Despite the selection of abovementioned candidate responsible for effective outward glutamate flow, that is, non-pathological transporter-mediated release of glutamate, there are other candidates for this position, which cannot be excluded from consideration. These mechanisms should afford efficient and regulated release of glutamate, and several more or less eligible candidates, e.g. diffusion, spontaneous exocytosis, cystine-glutamate exchanger, and release through anion channels, are analyzed below.
Neuronal communication is provided by precisely timed stimulated compound exocytosis of synaptic vesicles containing neurotransmitters, which is triggered by an intracellular Ca2+ spike that follows action potentials (Südhof, 2004; Rizzoli, 2014). Synaptic vesicles are subsequently retrieved from the plasma membrane of the presynaptic terminals, that is, endocytosis, and are turned into new fusion-competent synaptic vesicles (synaptic vesicle recycling). Rizzoli (2014) has underlined that synaptic vesicle recycling is one of the best controlled processes in cell biology; however, it is still unclear how this process is controlled. A special case of fusion of synaptic vesicles is the ‘kiss and run’ mechanism, in which the vesicles fuse only transiently with the plasma membrane, release their content to the extracellular space via fusion pore, after which the fusion pore is closed (Alabi and Tsien, 2013). The evidence in favor of the existence of ‘kiss and run’ mechanism presented over the last decade has been subsequently argued against on technical grounds (Rizzoli, 2014).
In parallel to stimulated exocytotic release, all synapses also exhibit spontaneous release of neurotransmitters (Truckenbrodt and Rizzoli, 2014; Kavalali, 2015). The major difference between these two mechanisms of the release is that stimulated exocytosis is directly coupled with action potentials, while spontaneous release is suggested to take place without any triggers. Spontaneous release of neurotransmitters occurs in the absence of stimulation in virtually all synaptic boutons (Truckenbrodt and Rizzoli, 2014). Spontaneous release can be increased by Ca2+-dependent manner, e.g. caffeine-triggered Ca2+ release from internal stores (Zefirov et al., 2006), and by Ca2+-independent way, e.g. by the application of lanthanum ions (Heuser and Miledi, 1971), α-latrotoxin (Ceccarelli et al., 1973; Betz and Henkel, 1994), or hyperosmotic sucrose (Rosenmund and Stevens, 1996; Truckenbrodt and Rizzoli, 2014). It has long been thought that this exocytosis is driven by fluctuations in local Ca2+ levels. The vesicles responding to these fluctuations are suggested to be the same ones that release upon stimulation, albeit potentially triggered by different Ca2+ sensors. This view has been challenged by several recent works, in which it has been suggested that spontaneous release is driven by a separate pool of synaptic vesicles (Truckenbrodt and Rizzoli, 2014). Many proteins, which constitute the core of fusion machinery, are required for exocytotic process, that is, SNAREs syntaxin 1, SNAP-25, and synaptobrevin. Holt et al. (2008) found that synaptic vesicles are constitutively active fusion machines, which function independently of Ca2+ needing only synaptobrevin for activity. The final step in fusion does not involve the regulatory activities of other vesicle constituents, although these may be involved in regulating earlier processes (Holt et al., 2008). Taking into account the abovementioned facts, spontaneous exocytosis cannot be excluded from consideration as one of the candidates ensuring permanent outward glutamate flow. However, Jabaudon et al. (1999) have demonstrated that tonic glutamate release does not reflect potential-evoked and spontaneous exocytosis in cultural hippocampal slices because it is not blocked by TTX, botulinum, and tetanus neurotoxins.
Compartmentalization of glutamate in synaptic vesicles gives an excellent opportunity to ‘mask’ the major part of cytosolic neurotransmitter, thereby withdrawing vesicular glutamate at least partially from thermodynamic reliance of glutamate transporters. Glutamate transporters work only with existent extracellular and cytosolic pool of glutamate performing effective glutamate turnover, followed by the establishment of definite glutamate gradient. However, it should be underlined that the process of glutamate uptake by synaptic vesicles should be also taken into consideration in calculations.
Other mechanisms of non-transporter tonic release
A cystine-glutamate antiporter/exchanger carries cystine into the cell in exchange of internal glutamate (Bannai, 1986) and couples the uptake of one molecule of cystine to the release of one molecule of glutamate (Bannai, 1986; Baker et al., 2003; Bridges et al., 2012a,b). The physiological role of this exchanger is to act as a cystine transporter that uses the transmembrane gradient of glutamate as driving force. Extracellular glutamate inhibits cystine uptake, and when the extracellular concentration of glutamate became higher, the transporter may actually catalyze a release of cystine (see study of Danbolt [2001] for review). However, according to Cavelier and Attwell (2005), cystine-glutamate exchange does not generate tonic glutamate release. Theoretical calculations suggest that a significant fraction of tonic glutamate release in hippocampal slices could occur via diffusion of glutamate across the lipid membranes (Cavelier and Attwell, 2005); however, it is clear that this process is uncontrolled. 4,4′-Diisothiocyano-2,2′-stillbene-disulfonic acid, DIDS, has been found to block numerous anion transporters and channels, which also represent a potential way of outward glutamate current. According to Cavelier and Attwell (2005), tonic glutamate release is reduced by DIDS, and so it is mediated by DIDS-sensitive mechanism. In our experiments, the application of 1 mm DIDS for inhibition of anion exchange in nerve terminals did not significantly affect (approximately by 10%) the extracellular level of glutamate in synaptosomes (unpublished data).
Activation of presynaptic glutamate receptors by ambient glutamate can stimulate glutamate outflow from nerve terminals, resulting from receptor-dependent Na+, and Ca2+ entrance to the cytosol, activation of signaling pathways, ROS formation, etc. However, the application of NMDA (250 μm), AMPA (250 μm), and kainite (250 μm) did not increase significantly, e.g. by <15%, the extracellular level of l-[14C]glutamate in nerve terminals in our experiments as compared to 100 μm glutamate-induced enlargement in this level, i.e. by 100% (unpublished data). The latest is a result of not only activation of presynaptic glutamate receptors but also transporter-mediated redistribution of the neurotransmitter between the intracellular and extracellular spaces.
Role of transporter-mediated glutamate release and non-transporter tonic release in outward glutamate current
The author hypothesizes that the abovementioned mechanisms of non-transporter tonic release of glutamate can be considered in two-substrate kinetic model of Makarov et al. (2013), as the addition of new exogenous substrate that changes the existing equilibrium of glutamate, which is maintained by glutamate transporters in both sides of the plasma membrane, and induces uptake of glutamate from the extracellular space and the consequent release of cytosolic glutamate by transporters. Kinetics of this possibility can be taken from following theoretical calculations, i.e. when the substrates in both sides of the membrane are in equilibrium, and then the addition of new portion of the substrate leads to re-equilibrium and release of the intrinsic ‘second’ substrate (Makarov et al., 2013). The abovementioned non-transporter mechanisms (together with spontaneous exocytosis) providing for glutamate leakage from nerve terminals and an increase in ambient glutamate can be also considered in kinetic calculations as ‘the new exogenous substrate’ and so cause simultaneous transporter-mediated uptake and release and thus re-equilibrium of glutamate between the extracellular space and the cytosol. It should be noted that in our experiments, the extracellular level of l-[14C]glutamate in nerve terminals under conditions of inhibition of glutamate transporters by 200 μmdl-TBOA increased by 10.8%±0.6% of total label accumulated by synaptosomes for 12 min (p≤0.05, Student’s t-test, n=4) (Borisova et al., 2010a). This value corresponds to net tonic glutamate release not only by spontaneous exocytosis of synaptic vesicles filled with glutamate but also by all possible mechanisms (cystine-glutamate exchanger, diffusion, anion channels), and so the amount of ‘the new exogenous substrate’ is non-negligible in kinetic calculations. Regulated exocytosis leads to the release of bulk glutamate to the synaptic cleft that also can be considered as the ‘the new exogenous substrate’ and so can cause a re-equilibrium of glutamate between the extracellular space and the cytosol resulting from simultaneous uptake and release of cytosolic glutamate by transporters. Also, it should be kept in mind that the local concentrations of glutamate at the transporter site could differ from those in the whole compartments.
Continuous non-transporter tonic release of glutamate in outer space of the plasma membrane and also glutamate uptake by synaptic vesicles inside of nerve terminals permanently disbalance transporter-dependent glutamate gradient across the plasma membrane, making this system very dynamic and dependent not only on Na+/K+ electrochemical gradient, and thus on the potential of the plasma membrane, but also on the functional state of synaptic vesicles and their ability for consequent uptake of cytosolic glutamate, efficiency of spontaneous exocytosis, lipid composition of the membranes, and thus plasma membrane permeability for glutamate, and effectiveness of diffusion, etc.
A kinetic algorithm at steady-state velocities can be presented as follows:
where Tr is the glutamate transporters and glu is the l-glutamate.
A kinetic algorithm with consideration of permanent tonic glutamate release as new added substrate can be written as follows:
where Tr is the glutamate transporters and glu is the l-glutamate.
Accordingly, a kinetic algorithm with consideration of vesicular glutamate uptake can be written as follows:
Physiological significance of permanent glutamate turnover
The physiological importance of permanent intensive glutamate turnover across the plasma membrane and maintenance of definite glutamate concentration in the synaptic cleft (but at low levels) is underscored by several factors. First, the significance of proper extracellular level of glutamate for tonic activation of postsynaptic receptors has already been underlined in several publications (Sah et al., 1989; Dalby and Mody, 2003; Cavelier and Attwell, 2005), which may be of value for long-term potentiation and memory formation, making synaptic contacts permanently active. The regulatory role of activation of presynaptic glutamate auto- and hetero-receptors in the transport of glutamate and other neurotransmitters can be also considered. Second, permanent glutamate turnover makes nerve terminals very flexible and able to react immediately on the changes in their functional state, excess of exogenous glutamate, hypoxia, hypoglycemia, etc. Third, ambient glutamate is strongly dependent on the number of glutamate transporters expressed in the plasma membrane of nerve terminals (Borisova et al., 2010a; Suna et al., 2014), and so the concentration of glutamate in different synaptic clefts and extracellular spaces of the brain regions can be different and can correlate with glutamate transporter density and so may be one of the main regulatory mechanisms of brain functioning. The molecular mechanisms regulating transporter intracellular trafficking have been identified (González et al., 2002), and surface diffusion of astrocytic glutamate transporters and the fact that GLT-1 transporters are highly mobile on rat astrocytes have been recently demonstrated (Murphy-Royal et al., 2015).
The suggestion on the existence of a permanent dynamic gradient of neurotransmitters can be actual for other secondary-active neurotransmitter transporters, e.g. GABA, glycine, and possibly dopamine, whereas the physiological significance of these gradients is different from that of glutamate. It is so because in our experiments, the extracellular level of [3H]GABA in synaptosomes is also maintained at a definite and non-negligible level (Pozdnyakova et al., 2014; Borisova et al., 2015). The extracellular level of [14C]glycine is unstable, presumably because of its very intensive diffusion through the plasma membrane (unpublished data). The main uncertainty of the abovementioned suggestion is the fact that the author based it on her own experiments performed in isolated rat brain nerve terminals. However, the use of nerve terminals allowed the author to avoid consideration of glial processes and to concentrate attention on presynaptic nerve terminals only. Also, the author compared her own results obtained on synaptosomes with the data of other methodological approaches.
In conclusion, a new vision on non-pathological functioning of glutamate transporters in reverse direction between the episodes of exocytotic release was forwarded, in addition to the previous one, where glutamate transporter reversal was considered as the main mechanism of glutamate release resulting in an increase in ambient glutamate and development of excitotoxicity under special conditions of energy deprivation, hypoxia, hypoglycemia, brain trauma, stroke, etc. It has been suggested that glutamate transporters act effectively in outward direction, and this process is thermodynamically synchronized with uptake and provides glutamate outflow, thereby establishing and maintaining permanent and dynamic glutamatein/glutamateex gradient and turnover across the plasma membrane. In this context, glutamate enriching the extracellular space through tonic release by means of diffusion, spontaneous exocytosis, cystine-glutamate exchanger, and leakage through anion channels can be considered as permanently added exogenous substrate in two-substrate kinetic model calculation (Figure 1). Permanent glutamate turnover may be of value for tonic activation of postsynaptic/presynaptic glutamate receptors, long-term potentiation, memory formation, etc. This mechanism can be actual for secondary neurotransmitter transporters, e.g. GABA.
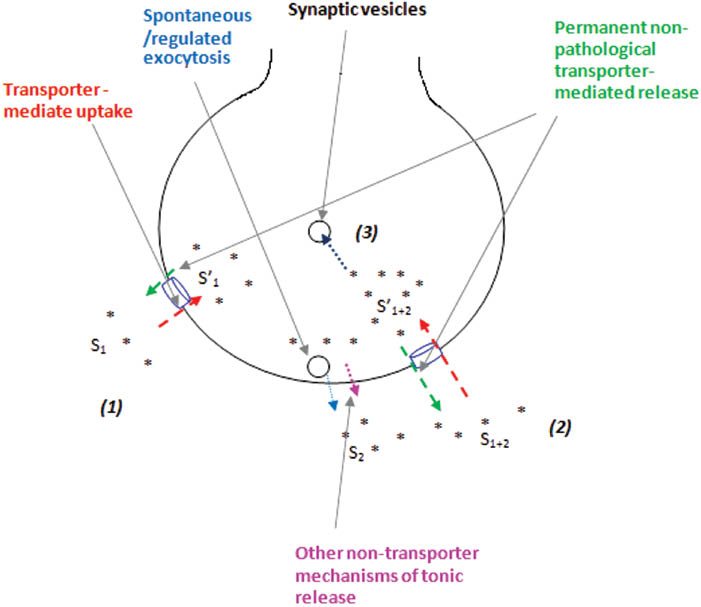
The mechanisms establishing permanent glutamate turnover in nerve terminals.
(1), (2), and (3) correspond to Equations (4), (5), and (6).
Acknowledgments
This work was supported by the Science and Technology Center in Ukraine project #6055; Projects of the National Academy of Sciences of Ukraine – in the frame of the following programs: ‘Scientific Space Research’; ‘Molecular and Cellular Biotechnologies for Medicine, Industry and Agriculture’; ‘Sensors for Medicine, Ecology, Industry, and Technology’; and ‘Fundamental Problems of Synthesis of New Chemical Substances’ – and Cedars Sinai Medical Center’s International Research and Innovation Management Program, the Association for Regional Cooperation in the Fields of Health, Science and Technology (RECOOP HST Association), and the participating Cedars–Sinai Medical Center–RECOOP Research Centers.
Author disclosure statement: The author declares no competing financial and personal interests exist.
References
Alabi, A.A. and Tsien, R.W. (2013). Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu. Rev. Physiol. 7, 393–422.10.1146/annurev-physiol-020911-153305Suche in Google Scholar
Baker, D.A., McFarland, K., Lake, R.W., Shen, H., Tang, X.C., Toda, S., and Kalivas, P.W.. (2003). Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neurosci. 6, 743–749.10.1038/nn1069Suche in Google Scholar
Bannai, S. (1986). Exchange of cystine and glutamate across plasma membrane of humanfibroblasts. J. Biol. Chem. 261, 2256–2263.10.1016/S0021-9258(17)35926-4Suche in Google Scholar
Bennett, J.P., Jr, Logan, W.J., and Snyder, S.H. (1973). Amino acids as central nervous transmitters: the influence of ions, amino acid analogues and ontogeny on transport systems for L-glutamic and L-aspartic acids and glycine into central nervous synaptosomes of the rat. J. Neurochem. 21, 1533–1550.10.1111/j.1471-4159.1973.tb06037.xSuche in Google Scholar PubMed
Betz, W.J. and Henkel, A.W. (1994). Okadaic acid disrupts clusters of synaptic vesicles in frog motor nerve terminals. J. Cell Biol. 124, 843–854.10.1083/jcb.124.5.843Suche in Google Scholar PubMed PubMed Central
Borisova T. (2013). Cholesterol and presynaptic glutamate transport in the brain (New York: Springer).10.1007/978-1-4614-7759-4Suche in Google Scholar
Borisova, T. and Himmelreich, N. (2005). Centrifuge-induced hypergravity: [3H]GABA and l-[14C]glutamate uptake, exocytosis and efflux mediated by high-affinity, sodium-dependent transporters. Adv. Space Res. 36, 1340–1345.10.1016/j.asr.2005.10.007Suche in Google Scholar
Borisova, T. and Krisanova, N. (2008). Presynaptic transporter-mediated release of glutamate evoked by the protonophore FCCP increases under altered gravity conditions. Adv. Space Res. 42, 1971–1979.10.1016/j.asr.2008.04.012Suche in Google Scholar
Borisova, T., Krisanova, N., and Himmelreich, N. (2004). Exposure of animals to artificial gravity conditions leads to the alteration of the glutamate release from rat cerebral hemispheres nerve terminals. Adv. Space Res. 33, 1362–1367.10.1016/j.asr.2003.09.039Suche in Google Scholar PubMed
Borisova, T., Krisanova, N., Sivko, R., and Borysov, A. (2010a). Cholesterol depletion attenuates tonic release but increases the ambient level of glutamate in rat brain synaptosomes. Neurochem. Int. 6, 466–478.10.1016/j.neuint.2009.12.006Suche in Google Scholar PubMed
Borisova, T., Sivko, R., Borysov, A., and Krisanova N. (2010b). Diverse presynaptic mechanisms underlying methyl-β-cyclodextrin-mediated changes in glutamate transport. Cell. Mol. Neurobiol. 30, 1013–1023.10.1007/s10571-010-9532-xSuche in Google Scholar PubMed
Borisova, T., Nazarova, A., Dekaliuk, M., Krisanova, N., Pozdnyakova, N., Borysov, A., Sivko, R., and Demchenko, A.P. (2015). Neuromodulatory properties of fluorescent carbon dots: effect on exocytotic release, uptake and ambient level of glutamate and GABA in brain nerve terminals. Int. J. Biochem. Cell Biol. 59, 203–215.10.1016/j.biocel.2014.11.016Suche in Google Scholar PubMed
Bouvier, M., Szatkowski, M., Amato, A., and Attwell, D. (1992). The glial cell glutamate uptake carrier countertransports pH-changing anions. Nature 360, 471–474.10.1038/360471a0Suche in Google Scholar PubMed
Bridges, R.J., Natale, N.R., and Patel, S.A. (2012a). System xc– cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 165, 20–34.10.1111/j.1476-5381.2011.01480.xSuche in Google Scholar PubMed PubMed Central
Bridges, R., Lutgen, V., Lobner, D., and Baker, D.A. (2012b). thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System xc–) to normal and pathological glutamatergic signaling. Pharmacol. Rev. 64, 780–802.10.1124/pr.110.003889Suche in Google Scholar PubMed PubMed Central
Cavelier, P. and Attwell, D. (2005). Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J. Physiol. 564, 397–410.10.1113/jphysiol.2004.082131Suche in Google Scholar PubMed PubMed Central
Cavelier, P., Hamann, M., Rossi, D., Mobbs, P., and Attwell, D. (2005). Tonic excitation and inhibition of neurons: ambient transmitter sources and computational consequences. Prog. Biophys. Mol. Biol. 87, 3–16.10.1016/j.pbiomolbio.2004.06.001Suche in Google Scholar PubMed PubMed Central
Ceccarelli, B., Hurlbut, W.P., and Mauro, A. (1973). Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J. Cell Biol. 57, 499–524.10.1083/jcb.57.2.499Suche in Google Scholar PubMed PubMed Central
Chefer, V.I., Denoroy, L., Zapata, A., and Shippenberg, T.S. (2009). Mu opioid receptor modulation of somatodendritic dopamine overflow: GABAergic and glutamatergic mechanisms. Eur. J. Neurosci. 30, 272–278.10.1111/j.1460-9568.2009.06827.xSuche in Google Scholar PubMed PubMed Central
Dalby, N.O. and Mody, I. (2003). Activation of NMDA receptors in rat dentate gyrus granule cells by spontaneous and evoked transmitter release. J. Neurophysiol. 90, 786–797.10.1152/jn.00118.2003Suche in Google Scholar PubMed
Danbolt, N.C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105.10.1016/S0301-0082(00)00067-8Suche in Google Scholar
Debler, E.A. and Lajtha, A. (1987). High-affinity transport of gammaaminobutyric acid, glycine, taurine, L-aspartic acid, and L-glutamic acid in synaptosomal (P2) tissue: a kinetic and substrate specificity analysis. J. Neurochem. 48, 1851–1856.10.1111/j.1471-4159.1987.tb05747.xSuche in Google Scholar
Dzubay, J.A. and Jahr, C.E. (1999). The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J. Neurosci. 19, 5265–5274.10.1523/JNEUROSCI.19-13-05265.1999Suche in Google Scholar
Erecinska, M. and Silver, I.A. (1990). Metabolism and role of glutamate in mammalian brain. Prog. Neurobiol. 35, 245–296.10.1016/0301-0082(90)90013-7Suche in Google Scholar
Featherstone, D.E. and Shippy, S.A. (2008). Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist 14, 171–181.10.1177/1073858407308518Suche in Google Scholar
Fonnum, F., Storm-Mathisen, J., and Divac, I. (1981). Biochemical evidence for glutamate as neurotransmitter in corticostriatal and corticothalamic fibres in rat brain. Neuroscience 6, 863–873.10.1016/0306-4522(81)90168-8Suche in Google Scholar
Funicello, M., Conti, P., Amici, M.D., Micheli C.D., Mennini, T., and Gobbi, M. (2004). Dissociation of [3H]L-glutamate uptake from L-glutamate-induced [3H]D-aspartate release by 3-hydroxy-4,5,6,6a-tetrahydro-3aH-pyrrolo[3,4-d]isoxazole-4-carboxylic acid and 3-hydroxy-4,5,6,6a-tetrahydro-3aH-pyrrolo[3,4-d]isoxazole-6-carboxylic acid, two conformationally constrained aspartate and glutamate analogs. Mol. Pharmacol. 66, 522–529.Suche in Google Scholar
González, M.I., Kazanietz, M.G., and Robinson, M.B. (2002). Regulation of the neuronal glutamate transporter excitatory amino acid carrier-1 (EAAC1) by different protein kinase C subtypes. Mol. Pharmacol. 62, 901–910.10.1124/mol.62.4.901Suche in Google Scholar PubMed
Grewer, C., Gameiro, A., Zhang, Z., Tao, Z., Braams, S., and Rauen, T. (2008). Glutamate forward and reverse transport: From molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 60, 609–619.10.1002/iub.98Suche in Google Scholar PubMed PubMed Central
Hayashi, T. (1954). Effects of sodium glutamate on the nervous system. Keio J. Med. 3, 183–192.10.2302/kjm.3.183Suche in Google Scholar
Herman, M.A. and Jahr, C.E. (2007). Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 27, 9736–9741.10.1523/JNEUROSCI.3009-07.2007Suche in Google Scholar PubMed PubMed Central
Heuser, J. and Miledi, R. (1971). Effect of lanthanum ions on function and structure of frog neuromuscular junctions. Proc. R. Soc. Lond. B Biol. Sci. 179, 247–260.10.1098/rspb.1971.0096Suche in Google Scholar
Holt, M., Riedel, D., Stein, A., Schuette, C., and Jahn, R. (2008). Synaptic vesicles are constitutively active fusion machines, which function independently of Ca2+. Curr. Biol. 18, 715–722.10.1016/j.cub.2008.04.069Suche in Google Scholar
Jabaudon, D., Shimamoto, K., and Yasuda-Kamatani, Y. (1999). Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc. Natl. Acad. Sci. USA 96, 8733–8738.10.1073/pnas.96.15.8733Suche in Google Scholar
Johnston, G.A.R. (1981). Glutamate uptake and its possible role in neurotransmitter inactivation. In: Glutamate: Transmitter in the Central Nervous System, P.J. Roberts, J. Storm-Mathisen, and G.A.R. Johnston, eds. (UK: Wiley, Chichester), pp. 77–87.Suche in Google Scholar
Kasatkina, L. and Borisova, T. (2010). Impaired Na+- dependent glutamate uptake in platelets during depolarization of their plasma membrane. Neurochem. Int. 56, 711–719.10.1016/j.neuint.2010.02.008Suche in Google Scholar
Kavalali, E.T. (2015). The mechanisms and functions of spontaneous neurotransmitter release. Nat. Rev. Neurosci. 16, 5–16.10.1038/nrn3875Suche in Google Scholar
Krebs, H.A. (1935). Metabolism of amino acids. IV. Synthesis of glutamine from glutamic acid and ammonia, and the enzymatic hydrolysis of glutamine in animal tissue. Biochem. J. 29, 1951–1969.10.1042/bj0291951Suche in Google Scholar
Krisanova, N., Trikash, I., and Borisova, T. (2009). Synaptopathy under conditions of altered gravity: changes in synaptic vesicle fusion and glutamate release. Neurochem. Int. 55, 724–731.10.1016/j.neuint.2009.07.003Suche in Google Scholar
Krisanova, N., Sivko, R., Kasatkina, L., and Borisova, T. (2012). Neuroprotection by lowering cholesterol: a decrease in membrane cholesterol content reduces transporter-mediated glutamate release from brain nerve terminals. Biochim. Biophys. Acta Mol. Basis Dis. 1822, 1553–1561.10.1016/j.bbadis.2012.06.005Suche in Google Scholar
Krnjevic, K. (1970). Glutamate and γ-aminobutyric acid in brain. Nature 228, 119–124.10.1038/228119a0Suche in Google Scholar
Logan, W.J. and Snyder, S.H. (1972). High affinity uptake systems for glycine, glutamic and aspartic acids in synaptosomes of rat central nervous tissues. Brain Res. 42, 413–431.10.1016/0006-8993(72)90540-9Suche in Google Scholar
LoTurco, J.J., Owens, D.F., Heath, M.J., Davis, M.B., and Kriegstein, A.R. (1995). GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298.10.1016/0896-6273(95)90008-XSuche in Google Scholar
Manent, J.B. and Represa, A. (2007). Neurotransmitters and brain maturation: early paracrine actions of GABA and glutamate modulate neuronal migration. Neuroscientist 13, 268–279.10.1177/1073858406298918Suche in Google Scholar
Makarov, V., Kucheryavykh, L., Kucheryavykh, Y., Rivera, A., Eaton, M.J., Skatchkov, S.N., and Inyushin, M. (2013). Transport reversal during heteroexchange: a kinetic study. J. Biophys., Article ID 683256.10.1155/2013/683256Suche in Google Scholar
Moussawi, K., Riegel, A., Nair, S., and Kalivas, P.W. (2011). Extracellular glutamate: Functional compartments operate in different concentration ranges. Front. Syst. Neurosci. 5, 94.10.3389/fnsys.2011.00094Suche in Google Scholar
Murphy-Royal, C., Dupuis, J.P., Varela, J.A., Panatier, A., Pinson, B., Baufreton, J., Groc, L., and Oliet S.H. R. (2015). Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 18, 219–228.10.1038/nn.3901Suche in Google Scholar
Nguyen, L., Rigo, J.M. and Rocher, V., Belachew, S., Malgrange, B., Rogister, B., Leprince, P., and Moonen, G. (2001). Neurotransmitters as early signals for central nervoussystemdevelopment. Cell Tissue Res. 305, 187–202.10.1007/s004410000343Suche in Google Scholar
Pendyam, S., Mohan, A., Kalivas, P.W., and Nair, S.S. (2012). Role of perisynaptic parameters in neurotransmitter homeostasis – computational study of a general synapse. Synapse 66, 608–621.10.1002/syn.21547Suche in Google Scholar
Pozdnyakova, N., Dudarenko, M., Yatsenko, L., Himmelreich, N., Krupko O., and Borisova T. (2014). Perinatal hypoxia: Different effects of the inhibitors of GABA transporters GAT-1 and GAT-3 on the initial velocity of [3H]GABA uptake by cortical, hippocampal and thalamic nerve terminals. Croat. Med. J. 55, 250–258.10.3325/cmj.2014.55.250Suche in Google Scholar
Rizzoli, S.O. (2014). Synaptic vesicle recycling: steps and principles. EMBO J. 33, 788–822.10.1002/embj.201386357Suche in Google Scholar
Robinson, M.B. (1998). Examination of glutamate transporter heterogeneity using synaptosomal preparations. Methods Enzymol. 296, 189–202.10.1016/S0076-6879(98)96015-3Suche in Google Scholar
Rosenmund, C. and Stevens, C. F. (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16, 1197–1207.10.1016/S0896-6273(00)80146-4Suche in Google Scholar
Sah, P., Hestrin, S., and Nicoll, R.A. (1989). Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246, 815–818.10.1126/science.2573153Suche in Google Scholar
Schousboe, A. (1981). Transport and metabolism of glutamate and GABA in neurons and glial cells. Int. Rev. Neurobiol. 22, 1–45.10.1016/S0074-7742(08)60289-5Suche in Google Scholar
Schousboe, A., Lisy, V., and Hertz, L. (1976). Postnatal alterations in effects of potassium on uptake and release of glutamate and GABA in rat brain cortex slices. J. Neurochem. 26, 1023–1027.10.1111/j.1471-4159.1976.tb06487.xSuche in Google Scholar PubMed
Soldatkin, O., Nazarova, A., Krisanova, N., Borysov, A., Kucherenko, D., Kucherenko, I., Pozdnyakova, N., Soldatkin, A., and Borisova, T. (2015). Monitoring of the velocity of high-affinity glutamate uptake by isolated brain nerve terminals using amperometric glutamate biosensor. Talanta 135, 67–74.10.1016/j.talanta.2014.12.031Suche in Google Scholar PubMed
Stern, J.R., Eggleston, L.V., Hems, R., and Krebs, H.A. (1949). Accumulation of glutamic acid in isolated brain tissue. Biochem. J. 44, 410–418.10.1042/bj0440410Suche in Google Scholar
Südhof, T.C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547.10.1146/annurev.neuro.26.041002.131412Suche in Google Scholar PubMed
Suna, W., Shchepakin, D., Kalachev, L.V., and Kavanaugh, M.P. (2014). Glutamate transporter control of ambient glutamate levels. Neurochem. Int. 73, 146–151.10.1016/j.neuint.2014.04.007Suche in Google Scholar PubMed
Truckenbrodt, S. and Rizzoli, S.O. (2014). Spontaneous vesicle recycling in the synaptic bouton. Front. Cell Neurosci. 8, 409.10.3389/fncel.2014.00409Suche in Google Scholar PubMed PubMed Central
Watkins, J.C. and Evans, R.H. (1981). Excitatory amino acid transmitters. Annu. Rev. Pharmacol. Toxicol. 21, 165–204.10.1146/annurev.pa.21.040181.001121Suche in Google Scholar PubMed
Watzke, N. and Grewer, C. (2001). The anion conductance of the glutamate transporter EAAC1 depends on the direction of glutamate transport. FEBS Lett. 503, 121–125.10.1016/S0014-5793(01)02715-6Suche in Google Scholar
Zefirov, A.L., Abdrakhmanov, M.M., Mukhamedyarov, M.A., and Grigoryev, P.N. (2006). The role of extracellular calcium in exo- and endocytosis of synaptic vesicles at the frog motor nerve terminals. Neuroscience 143, 905–910.10.1016/j.neuroscience.2006.08.025Suche in Google Scholar PubMed
van der Zeyden, M., Oldenziel, W.H., Rea, K., Cremers, T.I., and Westerink, B.H. (2008). Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol. Biochem. Behav. 90, 135–147.10.1016/j.pbb.2007.09.004Suche in Google Scholar PubMed
©2016 by De Gruyter
Artikel in diesem Heft
- Frontmatter
- Role of iso-receptors in receptor-receptor interactions with a focus on dopamine iso-receptor complexes
- Different patterns of 5-HT receptor and transporter dysfunction in neuropsychiatric disorders – a comparative analysis of in vivo imaging findings
- Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease
- Permanent dynamic transporter-mediated turnover of glutamate across the plasma membrane of presynaptic nerve terminals: arguments in favor and against
- CXCL12/CXCR4 axis: an emerging neuromodulator in pathological pain
- Traumatic brain injury: a risk factor for neurodegenerative diseases
- Excitatory and inhibitory conversive experiences: neurobiological features involving positive and negative conversion symptoms
Artikel in diesem Heft
- Frontmatter
- Role of iso-receptors in receptor-receptor interactions with a focus on dopamine iso-receptor complexes
- Different patterns of 5-HT receptor and transporter dysfunction in neuropsychiatric disorders – a comparative analysis of in vivo imaging findings
- Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease
- Permanent dynamic transporter-mediated turnover of glutamate across the plasma membrane of presynaptic nerve terminals: arguments in favor and against
- CXCL12/CXCR4 axis: an emerging neuromodulator in pathological pain
- Traumatic brain injury: a risk factor for neurodegenerative diseases
- Excitatory and inhibitory conversive experiences: neurobiological features involving positive and negative conversion symptoms