Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease
-
Chenhui Ji
 ,
Dongfang Li
,
Dongfang Li

Abstract
Glucose-dependent insulinotropic polypeptide (GIP) is a member of the incretin hormones and growth factors. Neurons express the GIP receptor, and GIP and its agonists can pass through the blood brain barrier and show remarkable neuroprotective effects by protecting synapse function and numbers, promoting neuronal proliferation, reducing amyloid plaques in the cortex and reducing the chronic inflammation response of the nervous system. Long-acting analogues of GIP that are protease resistant had been developed as a treatment for type 2 diabetes. It has been found that such GIP analogues show good protective effects in animal models of Alzheimer’s disease. Novel dual agonist peptides that activate the GIP receptor and another incretin receptor, glucagon-like peptide -1 (GLP-1), are under development that show superior effects in diabetic patients compared to single GLP-1 agonists. The dual agonists also show great promise in treating neurodegenerative disorders, and there are currently several clinical trials ongoing, testing GLP-1 mimetics in people with Alzheimer’s or Parkinson’s disease.
Introduction
Alzheimer’s disease (AD) is a sporadic disease with only few risk gene links associated with it. The inherited forms of AD are very rare, about 1% of all patients (Hutton et al., 1998; Karch and Goate, 2015). Therefore, the key triggers that induce the development of AD are most likely environmental, and it is very difficult to assess what these initial causes may be. Therefore, a promising approach to investigate what potential contributing factors there are is the correlation of AD onset with other factors that increase the risk of developing AD. Several such risk factors have been identified, and type 2 diabetes is one of these. In type 2 diabetes mellitus (T2DM), insulin signaling is impaired, often caused by a de-sensitization of the insulin response (Ott et al., 1999; Peila et al., 2002; Roberts et al., 2014). This observation may shed light on the underlying processes of AD development. Insulin is a growth factor, and a reduction of growth factor signaling will have repercussions on neuronal survival, neuronal repair, synaptic activity and general cell metabolism (Hölscher, 2011). Insulin is crucial for cell growth and survival. Neurons also carry insulin receptors, and activating these induces dendritic sprouting, neuronal stem cell activation, and general cell growth, repair and neuroprotection (Hoyer, 2004; Stockhorst et al., 2004; van Dam and Aleman, 2004; Hölscher, 2005; Li and Hölscher, 2007). In diabetes, research into other signaling pathways that support insulin signaling is ongoing. The use of incretins, a family of peptide hormones that helps to normalize insulin signaling, has proven to be very successful (Holst, 2004; Drucker and Nauck, 2006; Baggio and Drucker, 2007; Campbell and Drucker, 2013).
Insulin in the brain
Insulin signaling has been shown to be impaired in patients with AD, even in the absence of diabetes (Moloney et al., 2010; Talbot, 2014). This de-sensitization may well be one of the pathological factors that underlie cognitive impairments in the early stage of AD. A number of studies have shown that insulin has direct effects on brain activity and cognitive processes. In animal models, a decrease in insulin receptor signaling system produces cognitive impairments and a reduction in hippocampal synaptic plasticity of transmission (LTP) (Trudeau et al., 2004; Biessels et al., 2006). Conversely, insulin injected directly into the brain can improve performance of memory tasks in animals, and when applied as a nasal spray, also the performance of attention tasks in humans (Stockhorst et al., 2004; Freiherr et al., 2013). This effect might also be linked to the fact that LTP of synaptic transmission is impaired if insulin signaling is affected, as shown in animal models of diabetes. Treatments of diabetic animals with insulin or incretins rescued the impairment in neurotransmission and memory impairments (Gispen and Biessels, 2000; Biessels et al., 2004; Porter et al., 2010a,b; Lennox et al., 2013). People with T2DM also have cognitive impairments, and treatment with diabetes medication improves these impairments (Gispen and Biessels, 2000; Hoyer, 2004). Reduced insulin sensitivity and efficacy is also observed in the majority of elderly people (Carro and Torres-Aleman, 2004; Hoyer, 2004). Biochemical analysis of brain tissue has shown that insulin signaling is impaired in patients with AD (Moloney et al., 2010; Bomfim et al., 2012; Talbot et al., 2012). This unexpected connection between T2DM and AD opened up novel research targets to find out what the underlying mechanisms for the development of AD may be. First clinical trials that treated patients with AD with insulin delivered via nasal spray showed encouraging effects. Memory as well as levels of key AD biomarkers in the cerebro-spinal fluid (CSF) was improved (Hölscher, 2014a). Larger clinical trials are currently ongoing.
Re-sensitizing insulin signaling in the brain: the incretins
The incretins can re-sensitize insulin, and analogues of these have been developed to treat type 2 diabetes (Campbell and Drucker, 2013). The incretin family contains the peptide glucose-dependent insulinotropic polypeptide (GIP) (Baggio and Drucker, 2007). GIP is a 42-amino-acid incretin hormone which activates pancreatic islets to enhance insulin secretion and to help reduce hyperglycemia, similar to the sister incretin glucagon-like peptide (GLP-1) (Gault et al., 2003; Tan and Bloom, 2013). GIP also has been shown to promote pancreatic β-cell growth, differentiation, proliferation and cell survival, documenting its growth-hormone properties (Gault et al., 2003; Nyberg et al., 2005; Faivre and Hölscher, 2013b). Therefore, research is ongoing to develop novel protease-resistant GIP analogues with enhanced survival times in the blood stream as a therapeutic tool for T2DM treatment (Irwin et al., 2006, 2013; Martin et al., 2013).
The biological characteristics of GIP
GIP is a 42-amino-acid incretin hormone, and its receptor GIP is widely expressed in the body, including in the pancreas, stomach, adrenal cortex, cardiopulmonary cells, endothelial cells, adipose tissue and bone tissue (Yip and Wolfe, 2000), suggesting that it has additional effects other than just to facilitate insulin release. GIP receptors are also expressed in the brain and are found on larger neurons such as the pyramidal cortical neurons (Nyberg et al., 2005), which is very similar to the pattern of expression of GLP-1 receptors (Hamilton and Hölscher, 2009; Lee et al., 2011; Teramoto et al., 2011; Darsalia et al., 2012, 2013). The peptide GIP is also expressed in neurons and serves as a neuronal transmitter (Nyberg et al., 2007). However, GIP has poor stability in blood plasma with a GIP biological half-life of only around 5 min (Hinke et al., 2003). GIP is quickly degraded by the enzyme dipeptidyl peptidase IV (DPP-IV). Complete GIP (1-42) in vivo is cleaved by DPP-IV at the first two amino acid residues at the N-terminal (Tyr1-Ala2) to produce GIP (3-42). GIP (3-42) itself has no biological activity, but it can compete with GIP for the receptor binding site, resulting in GIP receptor (GIPR) inhibition (Hinke et al., 2003). As GIP is quickly degraded by the enzyme DPP-IV, several enzyme-resistant GIP modifications have been designed, such as D-Ala2-GIP and N-AcGIP (Hinke et al., 2003; Irwin et al., 2006), greatly improving the plasma biological half-life (Irwin et al., 2006). In non-obese mice, treatment with GIP analogues did not affect the body weight, blood glucose and plasma insulin levels, and blood glucose levels remained in a normal range (between 4 mmol/l and 8 mmol/l) for any age euglycemic amyloid precursor protein (APP)/presenilin 1 (PS1) transgenic or wild-type mice (Faivre et al., 2012; Faivre and Hölscher, 2013b).
Stable analogues such as D-ala2-GIP or N-glyc-GIP facilitate synaptic plasticity in the hippocampus, while the antagonist Pro3-GIP impairs LTP (Gault and Hölscher, 2008). In a GIPR-KO mouse strain, LTP was also much reduced, and paired-pulse facilitation showed an effect on presynaptic activity, indicating that the release of synaptic vesicles is reduced (Faivre et al., 2011).
Protease-resistant GIP analogues such as D-ala2-GIP cross the blood brain barrier (BBB) and also enhance neuronal progenitor cell proliferation in the brain (Faivre et al., 2011; Hamilton et al., 2011). Furthermore, GIP analogues have clear effects on memory formation, with the GIP receptor agonist D-Ala2GIP facilitating memory, and the GIP receptor antagonist Pro3-GIP impairing memory (Porter et al., 2010a; Faivre et al., 2012; Faivre and Hölscher, 2013b).
The GIP receptor signaling pathway
We know that GIP receptors on β-cells in the pancreas modulate insulin release via a mechanism that involves closure of K-channels, depolarization of the cell membrane that activates voltage-dependent calcium channels (VDCCs) and the increase of cyclic adenosine monophosphate (cAMP) levels. The subsequent influx of Ca2+ then activates Ca2+-sensitive enzymes such as phospholipase A2 (PLA2), phospholipase C, adenylate cyclase that forms cAMP and protein kinase A (PKA), and activates the mechanisms of vesicle exocytosis to release insulin into the extracellular space (Leech and Habener, 1997; Suzuki et al., 1997; Green et al., 2004; Kim et al., 2005a,b). The same biochemical mechanisms that control the release of neurotransmitters into the synaptic cleft via vesicles are found in neurons (Winder and Conn, 1993; Okamoto et al., 1994; Wheeler et al., 1994). Indeed, it has been shown that in neuronal cell cultures, the incretin GLP-1 modulates glutamate-induced Ca2+ influx. This effect was due to altered VDCC activity. Ca2+ influx induced by K+ conductance was also altered. GLP-1 furthermore induced cAMP formation, activated PKA, mitogen-activated protein kinases (MAPKs) and more (Gilman et al., 2003). It is therefore likely that the mechanism by which GIP increases insulin release in the pancreas is similar to the effects on LTP and synaptic transmission observed in the present study in the brain.
β-Amyloid fragments also have been shown to affect synaptic transmission. β-Amyloid has detrimental effects on LTP (Freir et al., 2001). The underlying mechanism of this impairment includes the change of K+-channel activity (Jalonen et al., 1997; Pannaccione et al., 2004), reduction of VDCC activity and Ca2+ influx (Abe and Kimura, 1996; Freir and Herron, 2003), which in turn affects Ca2+-sensitive enzyme activity (Wang et al., 2004) and reduces vesicle release (Arias et al., 1995; Harris et al., 1996).
Neuroprotective effects of GIP
The role of GIP in progenitor cell proliferation and neurogenesis
GIP is a neurotrophic factor that can inhibit apoptosis of cerebellar granule cells (Maino et al., 2014). Activation of GIPR leads to proliferation of neuronal progenitor cells and therefore may contribute to neurogenesis. Moreover, GIP has neuroprotective and regenerative properties (Figure 1).
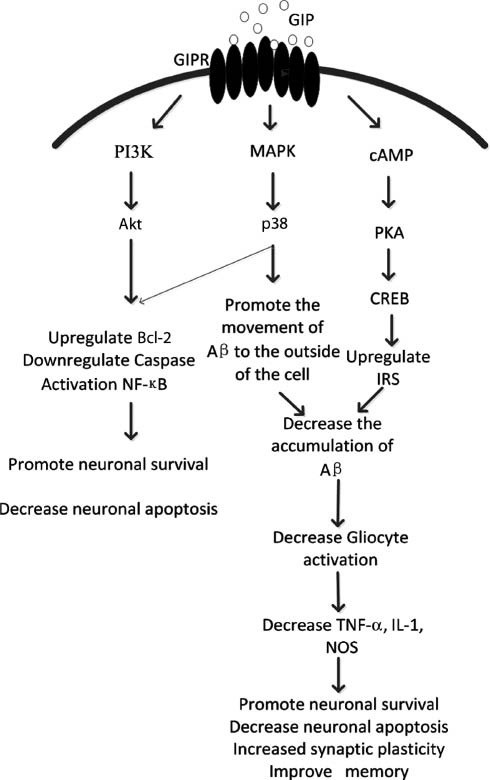
After GIP activation of the GIPR, multi-directional pathways are activated by cAMP/PKA/CREB (Kim et al., 2008), cAMP/Akt/PKB (Kim et al., 2005b) and MAPK (Ehses et al., 2003), and other signaling pathways. P38 can be activated to facilitate extracellular Aβ transport, and in addition, to activate adenylate cyclase (AC), which increases intracellular cAMP levels and further activation of cAMP-dependent PKA. PKA activation increases cAMP response element binding protein (CREB) transcriptional activity, increases insulin receptor substrate 1 (Irs1) (Trümper et al., 2002) expression, together with the activation of p38 reduces the deposition of amyloid, decreases inflammatory cytokine TNF-α, IL-1, NOS release, and thereby can improve cognition, memory, and synaptic plasticity increase while decreasing neuronal necrosis and apoptosis (Ehses et al., 2003; McIntosh et al., 2009).
Polarized localization of GIPR in the abaxonal Schwann cell membranes, plasma membrane-associated GIPR expression of satellite cells and ependymal GIPR expression strongly suggests complex cell type-specific functions of GIP and GIPR in the adult nervous system that are presumably mediated by autocrine and paracrine interactions, respectively. Notably, in vivo analyses with GIPR-deficient mice suggest a critical role of GIP/GIPR signal transduction in promoting spontaneous recovery after nerve crush, insofar as traumatic injury of GIPR-deficient mouse sciatic nerve revealed impaired axonal regeneration compared with wild-type mice (Buhren et al., 2009).
Hippocampal expression of GIP varied strongly in parallel with cell-proliferation rates in the adult rat hippocampal dentate gyrus (DG). The GIP receptor is expressed by cultured adult hippocampal progenitors and throughout the granule cell layer of the DG, including progenitor cells. Thus, these cells have the ability to respond to GIP. Indeed, exogenously delivered GIP induced proliferation of adult-derived hippocampal progenitors in vivo as well as in vitro, and adult GIP receptor knock-out mice exhibit a significantly lower number of newborn cells in the hippocampal DG compared with wild-type mice (Nyberg et al., 2005; Faivre et al., 2011).
Studies show that the GIP receptor is expressed in the hippocampus, and in vitro experiments show that GIP can promote the proliferation of neural progenitor cells. Therefore, GIPR activation can help to promote neurogenesis (Nyberg et al., 2005; Faivre et al., 2011, 2012; Faivre and Hölscher, 2013b).
GIP reduces neuronal inflammation
Progressive neurodegenerative diseases as well as stroke induce a chronic inflammatory response in the brain (Akiyama et al., 2000; Perry et al., 2007). This secondary downstream process causes further neurodegenerative effects via the activation of immune cells such as microglia in the brain (Tansey and Goldberg, 2010; Vaz et al., 2011; Zhao et al., 2011). These cells release pro-inflammatory cytokines and free radicals such as nitric oxide (NO), which is neurotoxic (Hölscher, 1997; Lee et al., 2010; Vaz et al., 2011). Once the chronic inflammation is established, the process becomes neurotoxic (Perry, 2007; Perry et al., 2007; Holmes et al., 2009; Tansey and Goldberg, 2010).
Studies confirm that GIP analogues not only have neuroprotective but also have anti-inflammatory properties. In APP/PS1 transgenic mice that develop a robust chronic inflammation response, D-ALA2-GIP reduced the activation of microglial and astrocytes in the brain. As a consequence, the oxidative stress in the brain was also reduced (Duffy and Hölscher, 2013a,b). We have shown that D-Ala2-GIP protects memory formation and synaptic plasticity, reduces amyloid plaque load, normalizes the proliferation of stem cells and prevents the loss of synapses in the cortex of the APPswe/PS1deltaE9 mouse model of AD (Duffy and Hölscher, 2013a,b; Faivre and Hölscher, 2013a,b) (Figure 2). Thus, part of the neuroprotective effect of D-ALA2-GIP in APP/PS1 mice may result from reducing the chronic neuroinflammation.
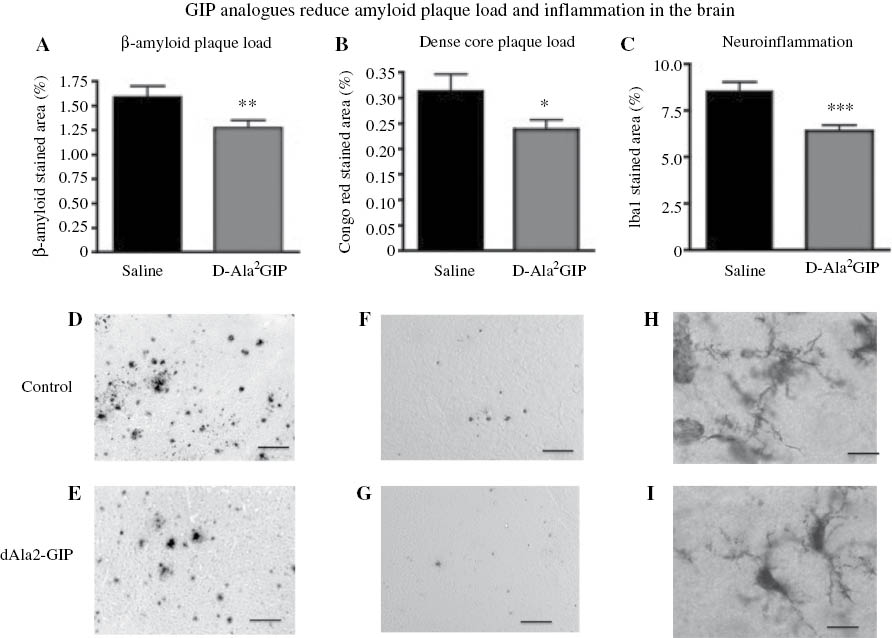
The protease-resistant GIP analogue dAla2-GIP protected against key AD biomarkers. (A) The number of amyloid plaques in the cortex of 12-month-old APP/PS1 mice was much reduced after 8 weeks of once-daily i.p. treatment. (B) The dense-core amyloid plaques which induce a chronic inflammation response were also reduced in numbers. (C) Chronic inflammation by activation of microglia is detrimental to neuronal survival. Drug treatment greatly reduced the amount of activated microglia in the cortex. Adapted from Faivre and Hölscher (2013b).
GIP analogues are neuroprotective in AD
Cognitive impairments in AD are most likely the result of extensive synaptic loss in the brain (Selkoe, 2002). β-Amyloid has been shown to impair synaptic communication in the brain (Hartley et al., 1999). GIP analogues that are resistant to DPP-IV protease degradation show good effects in preventing the amyloid-induced impairment in synaptic plasticity (LTP) in the hippocampus (Gault and Hölscher, 2008). The novel long-lived GIP analogue dAla2-N-AcGIP(Lys37MYR) showed clear protection against the impairment of synaptic plasticity that is induced by soluble β-amyloid oligomers (see Figure 3). The drug effect was only visible when GIP analogues were infused 30 min before treatment with amyloid. As amyloid peptides have a very different amino acid sequence than GIP, it is unlikely that GIP and amyloid bind to the same binding sites. Instead, it is more likely that the upregulation of cAMP levels by GIP receptor activation enhanced synaptic vesicle release probability and prevented the amyloid-induced impairment of LTP (Gault and Hölscher, 2008).
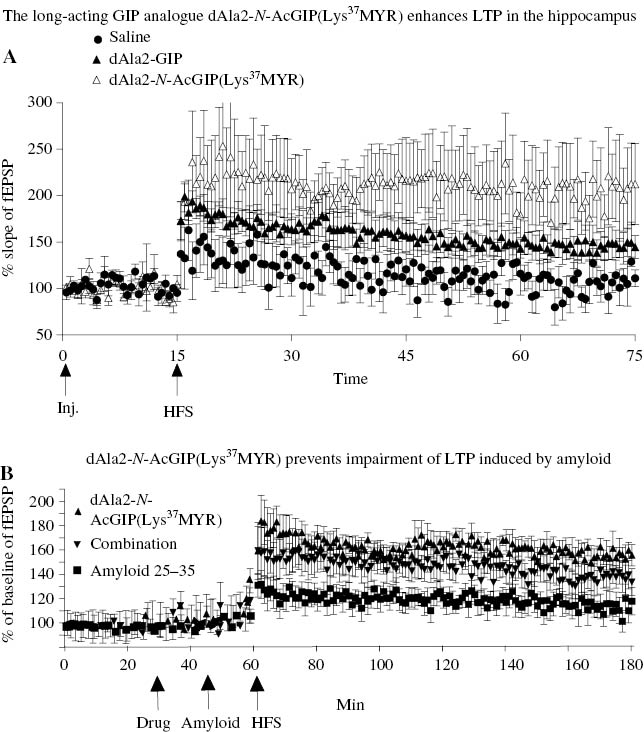
GIP protects synaptic transmission and plasticity from the detrimental effects of β-amyloid. (A) dAla2-N-AcGIP(Lys37MYR) or dAla2-GIP injected i.c.v. (15 nmol in 5 μl) increased LTP more than dAla2-GIP. LTP was induced by a weak-stimulation protocol. (B) Injection of dAla2-N-AcGIP(Lys37MYR) 30 min before β-amyloid injection reversed the impairment of LTP induced by the amyloid (Duffy and Hölscher, 2013a,b).
Furthermore, the protease-resistant GIP analogue D-Ala2GIP showed good effects on key AD hallmarks in the APP/PS1 transgenic mouse model of AD. In one study, D-Ala2GIP injected i.p. once daily for 8 weeks in either 6- or 12-month-old animals, an age that represents early phase AD, rescued the cognitive decline of 12-month-old APP/PS1 mice. Synapse numbers in the cortex and LTP in the hippocampus were also protected. Importantly, the amyloid plaque load in the cortex was reduced. The chronic inflammation response in the brain was also reduced in the cortex (Faivre and Hölscher, 2013b), see Figure 2. In a follow-up study, D-Ala2GIP was tested in 19-month-old APP/PS1 mice or littermate controls to find out if the drug may have protective effects even at an advanced stage of AD. Mice were injected for 21 days at 25 nmol/kg i.p. once daily. Interestingly, the age-related reduction of synaptic numbers in the DG and cortex was prevented in wild-type control mice as well as in the APP/PS1 mice. D-Ala2GIP facilitated synaptic plasticity in APP/PS1 and WT mice and reduced the number of amyloid plaques and activated microglia (chronic inflammation) in the cortex (Faivre and Hölscher, 2013a). In another study, i.c.v. infusion of β-amyloid (1-40) in mice produced impairments in a water maze test, and the infusion of GIP i.c.v. prevented the amyloid-induced impairment in spatial learning (Figueiredo et al., 2010).
The results show that GIP analogues not only have protective but also have regenerative properties in the brain of aged wild-type mice, and on key biomarkers found in AD in APP/PS1 mice. A longitudinal study of the brains of these mice showed a much enhanced oxidative stress level in the APP/PS1 mice, which was reduced by D-Ala2GIP (Duffy and Hölscher, 2013a,b). This suggests that novel GIP analogues may have beneficial effects in non-demented aged people and perhaps even in patients with AD even when the disease is further progressed.
Novel dual GIP/GLP-1 incretin agonists as new treatments for AD
GLP-1 is a sister incretin that also showed good effects in treating the key symptoms in preclinical tests of AD (Hölscher, 2014b) and several GLP-1 mimetics are on the market as treatments for type 2 diabetes (Campbell and Drucker, 2013). First results in clinical trials have been published: the GLP-1 receptor agonist exendin-4 improved cognitive performance in a pilot trial in patients with Parkinson’s disease (Aviles-Olmos et al., 2013, 2014). In a pilot study in patients with AD, the GLP-1 analogue liraglutide protected brain activity and energy metabolism from the decline that is associated with AD disease progression (Gejl et al., 2015). GLP-1 and GIP work hand in hand in a synergistic way (Hansotia and Drucker, 2005). Based on the success of GLP-1 analogues and the additional effects that GIP analogues show when administered simultaneously in mouse models of diabetes (Gault et al., 2011), synthetic peptides that activate GLP-1 and GIP receptors simultaneously have been developed as potential treatments in diabetes. In a series of peptide engineering projects to optimize the physiological properties, protease resistance and balanced activity at the two receptors, very effective dual agonists have been developed (Finan et al., 2013). All peptides showed good efficacy in rodent models of diabetes, showing superiority to liraglutide or a GIP analogue. One of these dual agonist peptides (DA1) had been tested in primates, and the efficacy in controlling blood sugar levels is superior to liraglutide. A second dual agonist peptide (DA2) that has a more balanced activity spectrum on the GLP-1 and GIP receptors (but with lower receptor affinity) has been tested in patients with diabetes. The drug was well tolerated and showed high potency with lower side effects compared to liraglutide (Finan et al., 2013).
One side effect of GLP-1 mimetics is that it reduces appetite in obese people. While this is of great advantage in treating overweight people, it may not be ideal for the treatment of patients with AD.
Interestingly, GIP analogues work in the opposite way. Studies have shown that GIP analogues induce weight gain and promote fat cell maturation and lipid uptake (Chan et al., 1985). Therefore, the GIP/GLP-1 dual agonists have shown a much more balanced effect on weight, which may be of advantage when treating patients who are normal weight or below (Finan et al., 2013).
Conclusion
Long-acting GIP analogues initially developed to treat type 2 diabetes show good neuroprotective effects in a range of key hallmarks of neurodegeneration and AD. Similar effects have been reported for long-acting GLP-1 analogues (Hölscher, 2014b). Novel GLP-1/GIP dual agonists that appear to be superior to single agonists show great promise to perhaps combine the effects of GIP and GLP-1. However, clinical trials testing the effects in patients with AD will have to be initiated to test what the actual benefits will be.
References
Abe, K. and Kimura, H. (1996). Amyloid beta toxicity consists of a Ca2+-independent early phase and a Ca2+-dependent late phase. J. Neurochem. 67, 2074–2078.10.1046/j.1471-4159.1996.67052074.xSearch in Google Scholar
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G.M., Cooper, N.R., Eikelenboom, P., Emmerling, M., Fiebich, B.L., et al. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421.10.1016/S0197-4580(00)00124-XSearch in Google Scholar
Arias, C., Arrieta, I., and Tapia, R. (1995). Beta-amyloid peptide fragment 25-35 potentiates the calcium-dependent release of excitatory amino acids from depolarized hippocampal slices. J. Neurosci. Res. 41, 561–566.10.1002/jnr.490410416Search in Google Scholar PubMed
Aviles-Olmos, I., Dickson, J., Kefalopoulou, Z., Djamshidian, A., Ell, P., Soderlund, T., Whitton, P., Wyse, R., Isaacs, T., Lees, A., et al. (2013). Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Invest. 123, 2730–2736.10.1172/JCI68295Search in Google Scholar PubMed PubMed Central
Aviles-Olmos, I., Dickson, J., Kefalopoulou, Z., Djamshidian, A., Kahan, J., Fmedsci, P.E., Whitton, P., Wyse, R., Isaacs, T., Lees, A., et al. (2014). Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J. Parkinsons Dis. 4, 337–344.10.3233/JPD-140364Search in Google Scholar PubMed
Baggio, L.L. and Drucker, D.J. (2007). Biology of incretins: GLP-1 and GIP. Gastroenterology 132, 2131–2157.10.1053/j.gastro.2007.03.054Search in Google Scholar PubMed
Biessels, G.J., Bravenboer, B., and Gispen, W.H. (2004). Glucose, insulin and the brain: modulation of cognition and synaptic plasticity in health and disease: a preface. Eur. J. Pharmacol. 490, 1–4.10.1016/j.ejphar.2004.02.057Search in Google Scholar PubMed
Biessels, G.J., De Leeuw, F.E., Lindeboom, J., Barkhof, F., and Scheltens, P. (2006). Increased cortical atrophy in patients with Alzheimer’s disease and type 2 diabetes mellitus. J. Neurol. Neurosurg. Psychiatry 77, 304–307.10.1136/jnnp.2005.069583Search in Google Scholar PubMed PubMed Central
Bomfim, T.R., Forny-Germano, L., Sathler, L.B., Brito-Moreira, J., Houzel, J.C., Decker, H., Silverman, M.A., Kazi, H., Melo, H.M., McClean, P.L., et al. (2012). An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβoligomers. J. Clin. Invest. 122, 1339–1353.10.1172/JCI57256Search in Google Scholar PubMed PubMed Central
Buhren, B.A., Gasis, M., Thorens, B., Muller, H.W., and Bosse, F. (2009). Glucose-dependent insulinotropic polypeptide (GIP) and its receptor (GIPR): cellular localization, lesion-affected expression, and impaired regenerative axonal growth. J. Neurosci. Res. 87, 1858–1870.10.1002/jnr.22001Search in Google Scholar PubMed
Campbell, J.E. and Drucker, D.J. (2013). Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 17, 819–837.10.1016/j.cmet.2013.04.008Search in Google Scholar
Carro, E. and Torres-Aleman, I. (2004). Insulin-like growth factor I and Alzheimer’s disease: therapeutic prospects? Expert Rev. Neurother. 4, 79–86.10.1586/14737175.4.1.79Search in Google Scholar
Chan, C.B., Pederson, R.A., Buchan, A.M., Tubesing, K.B., and Brown, J.C. (1985). Gastric inhibitory polypeptide and hyperinsulinemia in the Zucker (fa/fa) rat: a developmental study. Int. J. Obes. 9, 137–146.Search in Google Scholar
Darsalia, V., Mansouri, S., Ortsater, H., Olverling, A., Nozadze, N., Kappe, C., Iverfeldt, K., Tracy, L.M., Grankvist, N., Sjoholm, A., et al. (2012). Glucagon-like peptide-1 receptor activation reduces ischaemic brain damage following stroke in Type 2 diabetic rats. Clin. Sci. (Lond.) 122, 473–483.10.1042/CS20110374Search in Google Scholar
Darsalia, V., Ortsäter, H., Olverling, A., Darlöf, E., Wolbert, P., Nyström, T., Klein, T., Sjöholm, Å., and Patrone, C. (2013). The DPP-4 inhibitor linagliptin counteracts stroke in the normal and diabetic mouse brain: a comparison with glimepiride. Diabetes 62, 1289–1296.10.2337/db12-0988Search in Google Scholar
Drucker, D.J. and Nauck, M.A. (2006). The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 368, 1696–1705.10.1016/S0140-6736(06)69705-5Search in Google Scholar
Duffy, A. and Hölscher, C. (2013a). Novel GIP incretin analogues as a potential treatment for Alzheimer disease. Society for Neuroscience Meeting, San Diego, vol. 39, p. 41.23.Search in Google Scholar
Duffy, A.M. and Hölscher, C. (2013b). The incretin analogue D-Ala(2)GIP reduces plaque load, astrogliosis and oxidative stress in an APP/PS1 mouse model of Alzheimer’s disease. Neuroscience 228, 294–300.10.1016/j.neuroscience.2012.10.045Search in Google Scholar PubMed
Ehses, J.A., Casilla, V.R., Doty, T., Pospisilik, J.A., Winter, K.D., Demuth, H.U., Pederson, R.A., and McIntosh, C.H. (2003). Glucose-dependent insulinotropic polypeptide promotes β-(INS-1) cell survival via cyclic adenosine monophosphate-mediated caspase-3 inhibition and regulation of p38 mitogen-activated protein kinase. Endocrinology 144, 4433–4445.10.1210/en.2002-0068Search in Google Scholar PubMed
Faivre, E. and Hölscher, C. (2013a). D-Ala2GIP facilitated synaptic plasticity and reduces plaque load in aged wild type mice and in an Alzheimer’s disease mouse model. J. Alzheimers Dis. 35, 267–283.10.3233/JAD-121888Search in Google Scholar PubMed
Faivre, E. and Hölscher, C. (2013b). Neuroprotective effects of D-Ala2GIP on Alzheimer’s disease biomarkers in an APP/PS1 mouse model. Alzheimers Res. Ther. 5, 20–28.10.1186/alzrt174Search in Google Scholar PubMed PubMed Central
Faivre, E., Gault, V.A., Thorens, B., and Hölscher, C. (2011). Glucose-dependent insulinotropic polypeptide receptor knockout mice are impaired in learning, synaptic plasticity, and neurogenesis. J. Neurophysiol. 105, 1574–1580.10.1152/jn.00866.2010Search in Google Scholar PubMed
Faivre, E., Hamilton, A., and Hölscher, C. (2012). Effects of acute and chronic administration of GIP analogues on cognition, synaptic plasticity and neurogenesis in mice. Eur. J. Pharmacol. 674, 294–306.10.1016/j.ejphar.2011.11.007Search in Google Scholar
Figueiredo, C.P., Pamplona, F.A., Mazzuco, T.L., Aguiar, A.S., Jr., Walz, R., and Prediger, R.D. (2010). Role of the glucose-dependent insulinotropic polypeptide and its receptor in the central nervous system: therapeutic potential in neurological diseases. Behav. Pharmacol. 21, 394–408.10.1097/FBP.0b013e32833c8544Search in Google Scholar
Finan, B., Ma, T., Ottaway, N., Muller, T.D., Habegger, K.M., Heppner, K.M., Kirchner, H., Holland, J., Hembree, J., Raver, C., et al. (2013). Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci. Transl. Med. 5, 209ra151.10.1126/scitranslmed.3007218Search in Google Scholar
Freiherr, J., Hallschmid, M., Frey, W.H., 2nd, Brunner, Y.F., Chapman, C.D., Hölscher, C., Craft, S., De Felice, F.G., and Benedict, C. (2013). Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs 27, 505–514.10.1007/s40263-013-0076-8Search in Google Scholar
Freir, D.B. and Herron, C.E. (2003). Inhibition of L-type voltage dependent calcium channels causes impairment of long-term potentiation in the hippocampal CA1 region in vivo. Brain Res. 967, 27–36.10.1016/S0006-8993(02)04190-2Search in Google Scholar
Freir, D.B., Hölscher, C., and Herron, C.E. (2001). Blockade of long-term potentiation by β-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J. Neurophysiol. 85, 708–713.10.1152/jn.2001.85.2.708Search in Google Scholar
Gault, V.A. and Hölscher, C. (2008). Protease-resistant glucose-dependent insulinotropic polypeptide agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. J. Neurophysiol. 99, 1590–1595.10.1152/jn.01161.2007Search in Google Scholar
Gault, V.A., Flatt, P.R., and O’Harte, F.P. (2003). Glucose-dependent insulinotropic polypeptide analogues and their therapeutic potential for the treatment of obesity-diabetes. Biochem. Biophys. Res. Commun. 308, 207–213.10.1016/S0006-291X(03)01361-5Search in Google Scholar
Gault, V.A., Kerr, B.D., Harriott, P., and Flatt, P.R. (2011). Administration of an acylated GLP-1 and GIP preparation provides added beneficial glucose-lowering and insulinotropic actions over single incretins in mice with Type 2 diabetes and obesity. Clin. Sci. (Lond.) 121, 107–117.10.1042/CS20110006Search in Google Scholar PubMed
Gejl, M., Gjedde, A., Egefjord, L., Møller, A., Hansen, S.B., Vang, K., Rodell, A., Brændgaard, H., Gottrup, H., Schacht, A., et al. (2015). No decline of brain glucose metabolism in Alzheimer’s disease patients treated with liraglutide. American Diabetes Association, 75th Annual Meeting, Boston, USA, 5–9 June 2015, abstract 1309-P.Search in Google Scholar
Gilman, C.P., Perry, T., Furukawa, K., Grieg, N.H., Egan, J.M., and Mattson, M.P. (2003). Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J. Neurochem. 87, 1137–1144.10.1046/j.1471-4159.2003.02073.xSearch in Google Scholar PubMed
Gispen, W.H. and Biessels, G.J. (2000). Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 23, 542–549.10.1016/S0166-2236(00)01656-8Search in Google Scholar
Green, B.D., Gault, V.A., Flatt, P.R., Harriott, P., Greer, B., and O’Harte, F.P. (2004). Comparative effects of GLP-1 and GIP on cAMP production, insulin secretion, and in vivo antidiabetic actions following substitution of Ala8/Ala2 with 2-aminobutyric acid. Arch. Biochem. Biophys. 428, 136–143.10.1016/j.abb.2004.05.005Search in Google Scholar
Hamilton, A. and Hölscher, C. (2009). Receptors for the insulin-like peptide GLP-1 are expressed on neurons in the CNS. Neuroreport 20, 1161–1166.10.1097/WNR.0b013e32832fbf14Search in Google Scholar
Hamilton, A., Patterson, S., Porter, D., Gault, V.A., and Hölscher, C. (2011). Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J. Neurosci. Res. 89, 481–489.10.1002/jnr.22565Search in Google Scholar
Hansotia, T. and Drucker, D.J. (2005). GIP and GLP-1 as incretin hormones: lessons from single and double incretin receptor knockout mice. Regul. Pept. 128, 125–134.10.1016/j.regpep.2004.07.019Search in Google Scholar
Harris, M.E., Wang, Y.N., Pedigo, N.W.J., Hensley, K., Butterfield, D.A., and Carney, J.M. (1996). Amyloid ß peptide (25-35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J. Neurochem. 67, 277–286.10.1046/j.1471-4159.1996.67010277.xSearch in Google Scholar
Hartley, D.M., Walsh, D.M., Ye, C.P., Diehl, T., Vasquez, S., Vassilev, P.M., Teplow, D.B., and Selkoe, D.J. (1999). Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 19, 8876–8884.10.1523/JNEUROSCI.19-20-08876.1999Search in Google Scholar
Hinke, S.A., Lynn, F., Ehses, J., Pamir, N., Manhart, S., Kuhn-Wache, K., Rosche, F., Demuth, H.U., Pederson, R.A., McIntosh, C.H. (2003). Glucose-dependent insulinotropic polypeptide (GIP): development of DP IV-resistant analogues with therapeutic potential. Adv. Exp. Med. Biol. 524, 293–301.10.1007/0-306-47920-6_35Search in Google Scholar
Holmes, C., Cunningham, C., Zotova, E., Woolford, J., Dean, C., Kerr, S., Culliford, D., and Perry, V.H. (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774.10.1212/WNL.0b013e3181b6bb95Search in Google Scholar
Hölscher, C. (1997). Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 20, 298–303.10.1016/S0166-2236(97)01065-5Search in Google Scholar
Hölscher, C. (2005). Development of βeta-amyloid-induced neurodegeneration in Alzheimer’s disease and novel neuroprotective strategies. Rev. Neurosci. 16, 181–212.10.1515/REVNEURO.2005.16.3.181Search in Google Scholar
Hölscher, C. (2011). Diabetes as a risk factor for Alzheimer’s disease: insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem. Soc. Trans. 39, 891–897.10.1042/BST0390891Search in Google Scholar
Hölscher, C. (2014a). First clinical data of the neuroprotective effects of nasal insulin application in patients with Alzheimer’s disease. Alzheimers Dement. 10, S33–S37.10.1016/j.jalz.2013.12.006Search in Google Scholar
Hölscher, C. (2014b). The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement. 10, S47–S54.10.1016/j.jalz.2013.12.009Search in Google Scholar
Holst, J.J. (2004). Treatment of type 2 diabetes mellitus with agonists of the GLP-1 receptor or DPP-IV inhibitors. Expert Opin. Emerg. Drugs 9, 155–166.10.1517/14728214.9.1.155Search in Google Scholar
Hoyer, S. (2004). Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 490, 115–125.10.1016/j.ejphar.2004.02.049Search in Google Scholar
Hutton, M., Perez, T.J., and Hardy, J. (1998). Genetics of Alzheimer’s disease. Essays Biochem. 33, 117–131.10.1042/bse0330117Search in Google Scholar
Irwin, N., Montgomery, I.A., O’Harte, F.P., Frizelle, P., and Flatt, P.R. (2013). Comparison of the independent and combined metabolic effects of subchronic modulation of CCK and GIP receptor action in obesity-related diabetes. Int. J. Obes. (Lond.) 37, 1058–1063.10.1038/ijo.2012.179Search in Google Scholar
Irwin, N., O’Harte, F.P., Gault, V.A., Green, B.D, Greer, B., Harriott, P., Bailey, C.J., and Flatt, P.R. (2006). GIP(Lys(16)PAL) and GIP(Lys(37)PAL): novel long-acting acylated analogues of glucose-dependent insulinotropic polypeptide with improved antidiabetic potential. J. Med. Chem. 49, 1047–1054.10.1021/jm0509997Search in Google Scholar
Jalonen, T.O., Charniga, C.J., and Wielt, D.B. (1997). β-Amyloid peptide-induced morphological changes coincide with increased K+ and Cl- channel activity in rat cortical astrocytes. Brain Res. 746, 85–97.10.1016/S0006-8993(96)01189-4Search in Google Scholar
Karch, C.M. and Goate, A.M. (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51.10.1016/j.biopsych.2014.05.006Search in Google Scholar PubMed PubMed Central
Kim, S.J., Choi, W.S., Han, J.S., Warnock, G., Fedida, D., and McIntosh, C.H. (2005a). A novel mechanism for the suppression of a voltage-gated potassium channel by glucose-dependent insulinotropic polypeptide: protein kinase A-dependent endocytosis. J. Biol. Chem. 280, 28692–28700.10.1074/jbc.M504913200Search in Google Scholar PubMed
Kim, S.J., Winter, K., Nian, C., Tsuneoka, M., Koda, Y., and McIntosh, C.H. (2005b). Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic β-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J. Biol. Chem. 280, 22297–22307.10.1074/jbc.M500540200Search in Google Scholar PubMed
Kim, S.J., Nian, C., Widenmaier, S., and McIntosh, C.H. (2008). Glucose-dependent insulinotropic polypeptide-mediated up-regulation of β-cell antiapoptotic Bcl-2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2. Mol. Cell. Biol. 28, 1644–1656.10.1128/MCB.00325-07Search in Google Scholar PubMed PubMed Central
Lee, Y.J., Han, S.B., Nam, S.Y., Oh, K.W., and Hong, J.T. (2010). Inflammation and Alzheimer’s disease. Arch. Pharm. Res. 33, 1539–1556.10.1007/s12272-010-1006-7Search in Google Scholar PubMed
Lee, C.H., Yan, B., Yoo, K.Y., Choi, J.H., Kwon, S.H., Her, S., Sohn, Y., Hwang, I.K., Cho, J.H., Kim, Y.M., et al. (2011). Ischemia-induced changes in glucagon-like peptide-1 receptor and neuroprotective effect of its agonist, exendin-4, in experimental transient cerebral ischemia. J. Neurosci. Res. 89, 1103–1113.10.1002/jnr.22596Search in Google Scholar PubMed
Leech, C. and Habener, J. (1997). Insulinotropic glucagon-like peptide-1-mediated activation of non-selective cation currents in insulinoma cells is mimicked by maitotoxin. J. Biol. Chem. 272, 17987–17993.10.1074/jbc.272.29.17987Search in Google Scholar PubMed
Lennox, R., Porter, D.W., Flatt, P.R., and Gault, V.A. (2013). (Val(8))GLP-1-Glu-PAL: a GLP-1 agonist that improves hippocampal neurogenesis, glucose homeostasis, and β-cell function in high-fat-fed mice. ChemMedChem 8, 595–602.10.1002/cmdc.201200409Search in Google Scholar PubMed
Li, L. and Hölscher, C. (2007). Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res. Rev. 56, 384–402.10.1016/j.brainresrev.2007.09.001Search in Google Scholar PubMed
Maino, B., Ciotti, M.T., Calissano, P., and Cavallaro, S. (2014). Transcriptional analysis of apoptotic cerebellar granule neurons following rescue by gastric inhibitory polypeptide. Int. J. Mol. Sci. 15, 5596–5622.10.3390/ijms15045596Search in Google Scholar PubMed PubMed Central
Martin, C.M., Irwin, N., Flatt, P.R., and Gault, V.A. (2013). A novel acylated form of (d-Ala(2))GIP with improved antidiabetic potential, lacking effect on body fat stores. Biochim. Biophys. Acta 1830, 3407–3413.10.1016/j.bbagen.2013.03.011Search in Google Scholar
McIntosh, C.H., Widenmaier, S., and Kim, S.J. (2009). Glucose-dependent insulinotropic polypeptide (gastric inhibitory polypeptide; GIP). Vitam. Horm. 80, 409–471.10.1016/S0083-6729(08)00615-8Search in Google Scholar
Moloney, A.M., Griffin, R.J., Timmons, S., O’Connor, R., Ravid, R., and O’Neill, C. (2010). Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 31, 224–243.10.1016/j.neurobiolaging.2008.04.002Search in Google Scholar
Nyberg, J., Anderson, M.F., Meister, B., Alborn, A.M., Strom, A.K., Brederlau, A., Illerskog, A.C., Nilsson, O., Kieffer, T.J., Hietala, M.A., et al. (2005). Glucose-dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. J. Neurosci. 25, 1816–1825.10.1523/JNEUROSCI.4920-04.2005Search in Google Scholar
Nyberg, J., Jacobsson, C., Anderson, M.F., and Eriksson, P.S. (2007). Immunohistochemical distribution of glucose-dependent insulinotropic polypeptide in the adult rat brain. J. Neurosci. Res. 85, 2099–2119.10.1002/jnr.21349Search in Google Scholar
Okamoto, N., Hore, S., Akazawa, C., Hayashi, Y., Shigemoto, R., Mizuno, N., and Nakanishi, S. (1994). Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J. Biol. Chem. 269, 1231–1236.10.1016/S0021-9258(17)42247-2Search in Google Scholar
Ott, A., Stolk, R.P., van Harskamp, F., Pols, H.A., Hofman, A., and Breteler, M.M. (1999). Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53, 1937–1942.10.1212/WNL.53.9.1937Search in Google Scholar
Pannaccione, A., Secondo, A., Scorziello, A., Sirabella, R., Taglialatela, M., and Annunziato, L. (2004). Molecular mechanisms involved in β-amyloid-induced enhancement of voltage-gated K+ channels during neuronal apoptosis. FENS Meeting, Lisbon, vol. 2, abstract A159.121.Search in Google Scholar
Peila, R., Rodriguez, B.L., and Launer, L.J. (2002). Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 51, 1256–1262.10.2337/diabetes.51.4.1256Search in Google Scholar PubMed
Perry, V.H. (2007). Stress primes microglia to the presence of systemic inflammation: implications for environmental influences on the brain. Brain Behav. Immun. 21, 45–46.10.1016/j.bbi.2006.08.004Search in Google Scholar PubMed
Perry, V.H., Cunningham, C., and Holmes, C. (2007). Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 7, 161–167.10.1038/nri2015Search in Google Scholar PubMed
Porter, D.W., Irwin, N., Flatt, P.R., Hölscher, C., and Gault, V.A. (2010a). Prolonged GIP receptor activation improves cognitive function, hippocampal synaptic plasticity and glucose homeostasis in high-fat fed mice. Eur. J. Pharmacol. 650, 688–693.10.1016/j.ejphar.2010.10.059Search in Google Scholar
Porter, D.W., Kerr, B.D., Flatt, P.R., Hölscher, C., and Gault, V.A. (2010b). Four weeks administration of liraglutide improves memory and learning as well as glycaemic control in mice with high fat dietary-induced obesity and insulin resistance. Diabetes Obes. Metab. 12, 891–899.10.1111/j.1463-1326.2010.01259.xSearch in Google Scholar
Roberts, R.O., Knopman, D.S., Geda, Y.E., Cha, R.H., Pankratz, V.S., Baertlein, L., Boeve, B.F., Tangalos, E.G., Ivnik, R.J., Mielke, M.M., et al. (2014). Association of diabetes with amnestic and nonamnestic mild cognitive impairment. Alzheimers Dement 10, 18–26.10.1016/j.jalz.2013.01.001Search in Google Scholar
Selkoe, D.J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791.10.1126/science.1074069Search in Google Scholar
Stockhorst, U., de Fries, D., Steingrueber, H.J., and Scherbaum, W.A. (2004). Insulin and the CNS: effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration in humans. Physiol. Behav. 83, 47–54.10.1016/S0031-9384(04)00348-8Search in Google Scholar
Suzuki, M., Fujikura, K., Inagaki, N., Seino, S., and Takata, K. (1997). Localization of the ATP-sensitive K+ channel subunit Kir6.2 in mouse pancreas. Diabetes 46, 1440–1444.10.2337/diab.46.9.1440Search in Google Scholar PubMed
Talbot, K. (2014). Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP-1 analogs. Neurodegener. Dis. Manag. 4, 31–40.10.2217/nmt.13.73Search in Google Scholar PubMed PubMed Central
Talbot, K., Wang, H.Y., Kazi, H., Han, L.Y., Bakshi, K.P., Stucky, A., Fuino, R.L., Kawaguchi, K.R., Samoyedny, A.J., Wilson, R.S., et al. (2012). Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338.10.1172/JCI59903Search in Google Scholar PubMed PubMed Central
Tan, T. and Bloom, S. (2013). Gut hormones as therapeutic agents in treatment of diabetes and obesity. Curr. Opin. Pharmacol. 13, 996–1001.10.1016/j.coph.2013.09.005Search in Google Scholar PubMed
Tansey, M.G. and Goldberg, M.S. (2010). Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 37, 510–518.10.1016/j.nbd.2009.11.004Search in Google Scholar PubMed PubMed Central
Teramoto, S., Miyamoto, N., Yatomi, K., Tanaka, Y., Oishi, H., Arai, H., Hattori, N., and Urabe, T. (2011). Exendin-4, a glucagon-like peptide-1 receptor agonist, provides neuroprotection in mice transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 31, 1696–1705.10.1038/jcbfm.2011.51Search in Google Scholar
Trudeau, F., Gagnon, S., and Massicotte, G. (2004). Hippocampal synaptic plasticity and glutamate receptor regulation: influences of diabetes mellitus. Eur. J. Pharmacol. 490, 177–186.10.1016/j.ejphar.2004.02.055Search in Google Scholar
Trümper, A., Trümper, K., and Horsch, D. (2002). Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in β(INS-1)-cells. J. Endocrinol. 174, 233–246.10.1677/joe.0.1740233Search in Google Scholar
van Dam, P.S. and Aleman, A. (2004). Insulin-like growth factor-I, cognition and brain aging. Eur. J. Pharmacol. 490, 87–95.10.1016/j.ejphar.2004.02.047Search in Google Scholar
Vaz, A.R., Silva, S.L., Barateiro, A., Fernandes, A., Falcao, A.S., Brito, M.A., and Brites, D. (2011). Pro-inflammatory cytokines intensify the activation of NO/NOS, JNK1/2 and caspase cascades in immature neurons exposed to elevated levels of unconjugated bilirubin. Exp. Neurol. 229, 381–390.10.1016/j.expneurol.2011.03.004Search in Google Scholar
Wang, Q., Walsh, D.M., Rowan, M.J., Selkoe, D.J., and Anwyl, R. (2004). Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci. 24, 3370–3378.10.1523/JNEUROSCI.1633-03.2004Search in Google Scholar
Wheeler, D.B., Randall, A., and Tsien, R.W. (1994). Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264, 107–111.10.1126/science.7832825Search in Google Scholar
Winder, D.G. and Conn, P.J. (1993). Activation of metabotropic glutamate receptors increases cAMP accumulation in hippocampus by potentiating responses to endogenous adenosine. J. Neurosci. 13, 38–44.10.1523/JNEUROSCI.13-01-00038.1993Search in Google Scholar
Yip, R.G. and Wolfe, M.M. (2000). GIP biology and fat metabolism. Life Sci. 66, 91–103.10.1016/S0024-3205(99)00314-8Search in Google Scholar
Zhao, J., O'Connor, T., and Vassar, R. (2011). The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation 8, 150.10.1186/1742-2094-8-150Search in Google Scholar PubMed PubMed Central
©2016 by De Gruyter
Articles in the same Issue
- Frontmatter
- Role of iso-receptors in receptor-receptor interactions with a focus on dopamine iso-receptor complexes
- Different patterns of 5-HT receptor and transporter dysfunction in neuropsychiatric disorders – a comparative analysis of in vivo imaging findings
- Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease
- Permanent dynamic transporter-mediated turnover of glutamate across the plasma membrane of presynaptic nerve terminals: arguments in favor and against
- CXCL12/CXCR4 axis: an emerging neuromodulator in pathological pain
- Traumatic brain injury: a risk factor for neurodegenerative diseases
- Excitatory and inhibitory conversive experiences: neurobiological features involving positive and negative conversion symptoms
Articles in the same Issue
- Frontmatter
- Role of iso-receptors in receptor-receptor interactions with a focus on dopamine iso-receptor complexes
- Different patterns of 5-HT receptor and transporter dysfunction in neuropsychiatric disorders – a comparative analysis of in vivo imaging findings
- Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease
- Permanent dynamic transporter-mediated turnover of glutamate across the plasma membrane of presynaptic nerve terminals: arguments in favor and against
- CXCL12/CXCR4 axis: an emerging neuromodulator in pathological pain
- Traumatic brain injury: a risk factor for neurodegenerative diseases
- Excitatory and inhibitory conversive experiences: neurobiological features involving positive and negative conversion symptoms