Addressing the challenges of standalone multi-core simulations in molecular dynamics
-
R.O. Ocaya
 und
J.J. Terblans
und
J.J. Terblans

Abstract
Computational modelling in material science involves mathematical abstractions of force fields between particles with the aim to postulate, develop and understand materials by simulation. The aggregated pairwise interactions of the material’s particles lead to a deduction of its macroscopic behaviours. For practically meaningful macroscopic scales, a large amount of data are generated, leading to vast execution times. Simulation times of hours, days or weeks for moderately sized problems are not uncommon. The reduction of simulation times, improved result accuracy and the associated software and hardware engineering challenges are the main motivations for many of the ongoing researches in the computational sciences. This contribution is concerned mainly with simulations that can be done on a “standalone” computer based on Message Passing Interfaces (MPI), parallel code running on hardware platforms with wide specifications, such as single/multi- processor, multi-core machines with minimal reconfiguration for upward scaling of computational power. The widely available, documented and standardized MPI library provides this functionality through the MPI_Comm_size (), MPI_Comm_rank () and MPI_Reduce () functions. A survey of the literature shows that relatively little is written with respect to the efficient extraction of the inherent computational power in a cluster. In this work, we discuss the main avenues available to tap into this extra power without compromising computational accuracy. We also present methods to overcome the high inertia encountered in single-node-based computational molecular dynamics. We begin by surveying the current state of the art and discuss what it takes to achieve parallelism, efficiency and enhanced computational accuracy through program threads and message passing interfaces. Several code illustrations are given. The pros and cons of writing raw code as opposed to using heuristic, third-party code are also discussed. The growing trend towards graphical processor units and virtual computing clouds for high-performance computing is also discussed. Finally, we present the comparative results of vacancy formation energy calculations using our own parallelized standalone code called Verlet–Stormer velocity (VSV) operating on 30,000 copper atoms. The code is based on the Sutton–Chen implementation of the Finnis–Sinclair pairwise embedded atom potential. A link to the code is also given.
1 Introduction
The last few decades have seen a rapid rise of computational modelling as a powerful tool to develop materials by postulation, inference and tuning in what can only be described as a semi-closed loop approach. The advantage of this approach is that properties that may not be directly ascertained, such as homogeneous nucleation [1], or are costly to investigate empirically can be evaluated [2, 3, 4, 5, 6]. A molecular simulation begins with choosing the best force field that describes the molecular configuration and ends with the iterative calculation of the phase space under specified boundary conditions that denote the external influences. Such conditions convey the applied forces, thermostats [7], pressure, etc. The choice of mathematical model, which in effect denotes the force field, affects the simulation data structures and therefore the design of the software. This in turn affects what can ultimately be parallelized. In our own work, we focus on the Sutton–Chen implementation of the Finnis–Sinclair embedded atom model potential [3, 7, 8, 9]. Force fields describe the interactions of particles in the configuration in terms of normalized dimensionless perturbations. The general assumption is that the internal force field in the molecular configuration is usually much stronger in comparison with the external perturbation. The required phase can be determined deterministically by molecular dynamics (MD), or stochastically through Monte-Carlo approaches [7, 10]. Ultimately, the results of such computation must be compared with the experiment to ascertain its accuracy or even to gauge the efficacy of the model. This leads either to a tuning of the model if at the model development stage or to a tuning of the material properties by parameter adjustment if the model has been tested and is accepted previously. There are generally two simulation approaches used, namely MD and ab-initio methods [11, 12, 13, 14, 15, 16, 17, 18, 19, 20]. The basic computational modelling approach takes the aggregated, macroscopic properties arising as the consequence of their isolated, pairwise and interactive atomistic dynamics and considering the cumulative effects of these atomistic interactions as aggregations by some method that incrementally integrates their equations of motion. The recurrent idea in both approaches is to first derive a mathematical model that best describes the system, through which a state trajectory can be found. The mathematical model and its effectiveness and range depend on a number of factors that ultimately decide the extent of success of the simulation. Therefore, for a given system, it is imperative to derive a model that is as realistic as possible. However, this particular goal must take into consideration the level of accuracy required and the computational resources available. For atomic and molecular systems of macroscopic significance, the number of particles in the system can be extremely large, and studying their pairwise interactions will generate vast amounts of data and consume equally vast amounts of system time and resources. Using a faster processor does not necessarily alleviate the problem immediately, in spite of any mathematical simplifications such as specification of a cut-off distance for the interactions, simplification of the force field and so on that may have been devised. As interest in the computational study of materials grew, the issue of computational speed and accuracy quickly took centre stage and highlighted the shortcomings of the typical computer for these purposes. Compounding the situation is the fact that conformity to Moore’s law [21, 22] is no longer assured using the device interconnect-size concept on which the law is based. Moore’s law predicts a doubling of computational power approximately every 2 years as a consequence of the falling distances of the component interconnections on a semiconductor wafer. In 1971, the interconnect distance was around 10 μm and currently stands around 10 nm. It is expected to reach 5 nm around 2020. This distance affects the fastest speed that a charge conveying bit information can travel between any two devices on the same chip. Operating speed, however, is not simply a consequence of the interconnect distance. Other engineering considerations must be made, such as how heat dissipation will be handled. Also, as the physics shows, this distance is the realm of the lattice parameter (around 10 times the 0.361 nm lattice parameter of copper, for instance) where quantum mechanical effects manifest significantly. Consequently, processor speeds have more or less stabilized over the last 5 years at clocking frequencies of around 3 GHz. Clearly, to keep up with the demands of increasing computational power, there had to be a shift in the thinking. Today, vast increases in computational power are therefore not so much due to unilateral improvements in processor technology, but due to ingeniously interconnected multi-core, multiprocessor arrays. The development of computational frameworks and efficient algorithms appears to be driving innovations in atomistic MD simulations [7, 8, 9]. This is unwittingly a consequence of advancements in computer gaming, where the demand for high-performance computer graphics for more realistic rendering of ever larger and often networked games is ever greater and far outweighs other applications in the public domain. Thus, better models and computational algorithms are increasingly possible that exploit these features and are supported by ongoing developments in internet and computer technology (ICT). The rise of high-performance, supercomputing clusters [6] as low-cost supercomputers may perhaps be considered a secondary revolution in computation. This is because micron-scale, microprocessor interconnects [23] are now falling as fast as the demands of processing power require. However, in spite of these trends, standalone computation is still of considerable importance because it underscores the role of the individual node computer which is an integral component of any cluster, particularly if its inherent power could be unlocked through careful software design. The latter remains the basic computational element, and several such distributed nodes collectively execute chunks of tasks assigned by a task manager, which is also responsible for collating the results of the processing and making it available to the requester. Unfortunately, apart from the nature of interconnects between processors or cores themselves, the design of the node can easily become the weakest link in a computational arrangement and can lead to overheads whose cumulative effects substantially diminish the overall system performance. By standalone, one does not mean that the system is necessarily single processor or single core, but only that the computing node does not feature as part of a distributed system. Standalone can therefore mean any arrangement on a physically isolated, single machine that can support anywhere from single-core to multiple single-core processors to multiple processor multi-core computers. Such arrangements are today quite ubiquitous as standard features on desktop computers. For instance, the i3, i5 and i7 classes of x86-based machines common on desktops today are single processors with two to four cores and can support hyper-threading, such as Turbo Boost and K-technology [24]. The K-technology allows the overclocking of the CPU but for computational purposes illustrates the law of diminishing returns since it can limit the CPU power to only one core. Only applications written with one core in mind will likely reap any speedup benefit from such overclocking. The i7-Extreme can have up to eight cores and offer a unique processing opportunity if properly coded for the extra capability. Typically, most desktop applications do not use that extra feature and therefore do not observe the extra speedup. Additionally, on the same main board supporting the processor, there can be a high-definition hardware graphics processing unit (GPU). This multi-core CPU/GPU combination is the “typical” standalone configuration being referred to here. In our own work, the simulations are done on a Dell Optiplex 3010, which hosts a third-generation i5-3470 (3.2 GHz) with four cores and L3 cache (6 MB), has no hyper-threading but has K-technology (not activated to the maximum 3.6 GHz in this work). The system comes preinstalled with 8 GB (DDR3–1.6 GHz) of SDRAM. The GPU is AMD Radeon HD 7470 with 1 GB DDR3 SDRAM and interfaces the CPU with a PCI Express, 16-bit bus. Our results presented below are based on this hardware specification. The external storage, which is not part of the benchmark performance and mentioned here for completeness, is a 500 GB hard drive. Its function is merely to hold initial particle data and boundary conditions and to store the ultimate output of a simulation. Once the simulations start, no intermediate file access is done, all intermediate calculations being done in a SDRAM scratch pad. In the typical computer, the random access memory (RAM) is shared between cores or processors through a common, managed memory area called a “heap.” There have also been dramatic developments in physical storage devices associated with the modern desktop to address the demands of traditional multimedia data files, if not for the more demanding storage of large gaming applications and data. For computational applications, physical storage media may be used as swap space and intermediate storage, although in the interest of speed they are generally relegated to input and ultimate data output. These considerations put a heavy demand, and therefore stipulation of type and layout, on the RAM requirements of such a system for successful computational modelling application. As a benefit of falling costs, computing clusters are becoming more common in medium-sized research facilities and universities. However, they are still generally still beyond the reach of many smaller organizations and individual researchers for a number of reasons. These reasons may revolve around an inability to justify their costs by any sort of sustainable research output, external third-party usage, running costs such as steady electrical power requirements which are likely to be proportional to the size of the system, availability of skilled manpower for configuration and maintenance, programming expertise for task scheduling, interpretation and result integration and so on. The starting inertia in computational research therefore tends to be high for the same reasons. As the popularity of computation continues to rise, the bulk of research output remains largely experimental, leaving computation largely unexplored.
As mentioned above, large MD simulations are today being run regularly for large particle systems with ever-increasing spatiotemporal scales. New innovations or extensions to existing systems are constantly being devised for increased computational performance. Many of these developments revolve around identification of code aspects that can be further parallelized or optimized. Parallelization starts typically with a decomposition, where the problem is divided into smaller “chunks” which are then assigned to a specific processor or core. There are three decomposition methods that are widely used to parallelize MD simulations. These are particle, force and domain decomposition using a spatial basis [25, 26, 27]. The particle number tends to be large in practically meaningful simulations, and none of these decompositions is yet very effective, due to memory consumption, a large number of processors and communication partners. Spatial decomposition approaches, based on physical subdomains, are generally superior [28]. In recent years, under the technological limitations imposed by Moore’s law, increases in computational power were achieved in two ways, namely increasing the number of cores in a processor and the vector size in single-instruction, multiple data instruction set (SIMD) computers. It has become almost de facto in this quest that the only avenue available to increase computational power is by an upward scaling of the number of processors and cores per processor in a chip. For a given configuration of processors, other performance enhancements can be devised though improved algorithms. It is clear that tapping into the full power of such a system will require a highly scalable parallel code. Unfortunately, the secondary issue of power dissipation and age degradation failure (e. g. due to any of the billions of transistors failing because of heat subjection) has to be considered carefully [29, 30]. Domain decomposition can present problems for inhomogeneous systems. The cell-task method [31] can overcome some problems, particularly if used as part of a hybrid decomposition scheme. For instance, by the use of a hybrid algorithm, Mangiardi and Meyer [29] devise a domain decomposition and thread-based hybrid parallelization technique based on large vectorization SIMD processors such as the AVX, AVX-2 and Xeon-Phi processors, comprising several thousands of cores. Their hybrid technique involves modelling with short-ranged forces for both homogeneous and inhomogeneous collections consisting of up to tens of millions of atoms. The hybrid algorithm achieves parallelization by using a task-based approach on smaller sub-domains obtained from domain decomposition. Each sub-domain is handled by a team or “pool” of threads running on multiple cores. The flow of execution of tasks cannot access the same particle simultaneously, thereby averting the need for lockout code to prevent such access of a particle by different tasks. At present, several parallelized MD programs are in existence, for instance [32, 33, 34, 35, 36] and others. However, such programs employ rigid parallelization techniques, and completely new versions will be needed to implement improved parallel algorithms that better exploit advancements in computer architectures [29]. In light of this proliferation, a smaller code that is easier to revise with algorithmic and architectural advancements has clear advantages, and is arguably necessary if advances in architectures are to be exploited fully in future [29, 37]. The effort required for such revisions can be small. There are some issues with hybrid methods, such as synchronization of data structure access which can lead to false sharing of these records because all processors constantly amend the data structures of neighbour cells [38]. These irregular memory access patterns pose significant challenges for parallelization and performance optimization. Additional challenges are associated with the hardware limitations that effectively limit active core performance due to chip configuration, such as memory speed, volume, interchip communication bandwidths, power management, etc. In 2010, Peng et al. [25, 26, 27] proposed three incremental optimizations for an emergent platform, the Godson-T multi-core architecture [39], which was formally released in late 2010. They presented behavioural, instruction-level simulations of a chip that was still in development and aimed at petaflops supercomputing. Their work addressed the foregoing issues of power efficiency, performance, programming and parallelism and platform-operating system portability (particularly to Linux and BSD; portability to WindowsCE is reported) by exploiting polymorphic parallelism. Its main contributions to high-performance computing (HPC) is improved interconnection data communications (both internal and external), fine-grained division of threads, improved thread synchronization and locality awareness. Its unique architecture achieves transfer bandwidths of up to 512 Gb/s for internal register transfers and 51.2 Gb/s for off-chip transfers. It implements 64, 64-bit cores and several levels of memory on chip, specifically 16 address-interleaved 128 kB L2 cache, 32 kB scratch pad memory and four external DDR3 memory controllers. The actual chip was shipped in the optimization strategy employed, termed cell-centred addressing that sequentially maps neighbour 3D cells to 2D cores by a scalar transformation vector which are then searched for migrating atoms in O (1) time. Such mappings are readily solved by classical algorithms but what makes the Godson-T and its progenies unique is the presence of on-chip hardware that implements a locality-aware parallel algorithm for data reuse and latency and congestion avoidance. Such issues typically arise from core to core communications when shared data structures are accessed in L2 cache or off-chip memory during the two-body force calculations. In this contribution, we present a short review of the current state of MD simulations with the aim of achieving speedups on a standalone computer. The discussion is expected to permit a reasonably easy starting point for standalone computational MD for the reader, with the view to readily scale such computers to larger clusters by virtue of interconnection. The work is intended to address single machines that support thread and message passing by virtue of the processor and memory architecture, by using any selection of proliferated tools which are freely available for such purposes. We discuss the rationales for the chosen approach of our own ongoing work in the hope that the generally high initial inertia could be overcome by the interested reader. Finally, we present some of our results thus far and point to an online repository where the evolving code is being maintained. We begin by outlining some standalone architectural configurations that are decidedly parallel or can be made so with relative ease.
2 Standalone architectures
The processor memory architectures of the traditional computer fall into one of two broad categories. In the first category, instruction fetch-execution cycles are repeated from the reset vector up until the last instruction is executed. The instructions and data are stored contiguously in the same, indistinguishable memory blocks. The instruction and data fetches happen along the same paths. In the second category, the instructions are generally stored in different memory locations and the paths to them are different. This sort of architecture is generally faster since simultaneous fetches and pre-fetches are possible. Several enhancements have been developed to support this behaviour, such as pipelining or super pipelining, which involve look-ahead methods and ways to avoid the problems that arise when branches are encountered in the linearly pre-fetched sequence of instructions and data. Such methods include branch prediction, cache memory for the frequently used data thereby saving on fetch times, and so on. A complete discussion of these techniques is beyond the present scope.
2.1 Classifications of parallelization paradigms
Parallel problems fall into two broad categories, embarrassingly parallel problems and serial problems. The placement of a problem depends on the extent and readiness to which it can be parallelized. A problem is said to be “embarrassingly parallel” if it is readily separated into identifiable, unique tasks that may then be executed separately. For such problems, the execution paths of the different threads are independent in the sense of the results of one thread not depending on any other thread. On the other hand, a “serial” problem is one that cannot be split into independent sub-problems and the results of one computation drive the next. Therefore, such a problem cannot easily be distributed across independent processing units without requiring interprocess communication. However, some problems can contain both elements and must be coded as combinations of embarrassingly parallel and serial. Most parallelization of interest occurs at the level of the iterations where the force fields are applied. Only these loops are considered for performance benchmarking. The other parts of the typical program are the user and output interfaces of a program. These parts of the code are concerned with loading particle structures into memory and can implement iterations that allow the particles in the configuration to relax to an equilibrium decided by the (new) boundary condition (called “setup” time), such as applied temperature. These parts of the code can take inordinately large multiples of the simulation time step, but typically occur only once per simulation and are therefore not considered in performance benchmarking [29].
3 Getting started with standalone computation
3.1 To code or not to code
For the innumerable material science problems that could be solved by MD techniques, there may or may not already exist a software, either specific for the class of the problem on hand or with an innate ability to be tuned for the problem. Many available packages are heuristic or “black box” in nature and cannot be dissected readily; others target a specific class of problems (through the available potentials and force fields), which introduce inflexibility towards new problems. Also, as mentioned elsewhere in this article, advancements in computer architecture and improved parallelization efficient and/or more accurate algorithms for the same software make it advantageous to develop software with high modularity in mind to keep up with developments in the field. Naturally, one then asks whether embarking on writing one’s own code is a reasonable venture. To address this question meaningfully, careful considerations of the problem to be solved must be made, such as availability or access to similar software, the complexity of the problem if no software precedent exists, the technical resources available in terms of development time, target hardware and know-how, to highlight a few. An illustration of this scenario relates to density functional theory (DFT) calculations [40], for which numerous free software exist. However, different DFT software have different strengths and weaknesses. Many of them demonstrate applicability to the newer class of molecular and atomic aggregations found in the emerging field of nanotechnology, but perform poorly in many others. Next, the question would be whether the software is user adaptable to a new problem, such as ability to resolve small atom arrays of a totally different particle configuration and boundary conditions. For an academic undertaking, writing own software has obvious advantages and can lead to a software that is small, fast and potentially extendable to new classes of problems, particularly if the software is designed with modularity for code reuse in mind. Such codes can be understood and readily evolved by more developers with respect to parallelization efficiency and accuracy.
3.2 General tools
Standalone techniques that are considered to be successful are those that are highly scalable to implement larger systems while assuring/improving the desired levels of accuracy. Here, we define scalability as the ease and ability to increment the system size and computational power by physically adding similar systems to an existing system while adjusting only a small set of reconfiguration parameters. The closest parallel to this is the addition of a RAM module to upgrade system memory. Today, many interconnected systems are said to be “hybrid” since they are comprised of interconnections of subsystems that are heterogeneous with respect to hardware and operating system, sometimes referred to as a “farm”. Interconnectivity is an obvious, fundamental tenet of the internet, where hardware compliance with controlled communication protocols and standards assures conformity, rather than the actual hardware and operating system in use. For this reason, the computing farm could readily support Apple or x86-based hardware, running Linux, BSD or Windows operating systems, all of them cooperating to reinforce the overall computational power. If one takes cognisance of the lower starting inertia and the need for simplicity for a better understood system, then the scaled system should ideally be homogeneous to inherently possess a plug-and-play character.
3.3 Parallelizable tools
Naturally, the question that can be asked at the onset is, how does one begin to exploit the full potential of a standalone computer having P processors and C cores? Second, is this set {P, C} available by default by virtue of the unit being on? Third, how does one benchmark the performance of this set? Interestingly, as suggested above, these questions are still largely unanswered. There are some inroads to the answers, and several studies have been conducted to attempt to correlate the performance of the system as a function of the size in terms of the set {P, C}. Peng et al. [25] measure the speedup on
4 Parallel processing paradigms in the C-language
4.1 Threads and message passing
The C-language is native to Unix-based systems, but its high portability has made it standardized and widely available across several platforms. Although there are newer contenders to parallelization, but the C-language is still widely used for parallelization for three main reasons. First, complex data structures that closely resemble collections of structured particles in a system can be defined, referenced and manipulated within memory with considerable ease. The basic data structure typically consists of a particle type object with directly accessible attributes such as mass, position and velocity. Entire collections of such particles, which represent particle configurations within a domain, can be manipulated just as readily as its individual particles through structure access functions. Second, the use of pointers within the language introduces a flexibility and enabling modularity. In this way, individual particles or entire collections of particles can be passed on between different sections the code with ease. An instance where this flexibility is called for is during particle migration. This requires that a particle is added or deleted from a collection. Third, the language is fast and has a number of excellent heap memory management functions. It is native and highly popular on Unix-based systems. The main idea behind thread processing as exploited in parallel programming involves a single problem process giving rise or “spawning” multiple, dynamically managed sub-programs called “threads.” The threads run concurrently and independently and are structured such that they can access shared memory. Shared memory access can involve some interthread communication, requiring careful management of all spawned threads, from the moment each is spawned to the moment it terminates. The process of killing a thread releases its resources for reassignment by the manager. A critical role of the manager is to implement careful synchronization of threads to avoid thread collisions and racing conditions by assuring a mutual exclusivity of memory access during a focus by a particular thread. The concept of threads is well-established in the C-programming language and is conveyed by a number of paradigms. For instance, the POSIX threads standard, or p-threads, makes use of a standardized library header “pthread.h,” which contains the necessary definitions of constants, functions and types. The code in Figure 1 spawns three threads using this approach. Other popular parallelization techniques available to C-programs include the multiprocessing application programming interfaces (APIs), of which the open multiprocessing API (OpenMP) is a popular implementation, and various message passing interfaces (MPIs). The MPIs are libraries that define code and routines which run on a diverse cross-section of platforms including Linux and Windows. The most commonly referenced and freely available MPIs are OpenMPI (not to be confused with OpenMP), MPI, MPICH2 and LAM-MPI. Each of these MPIs may be considered a cross between p-threads and OpenMP.
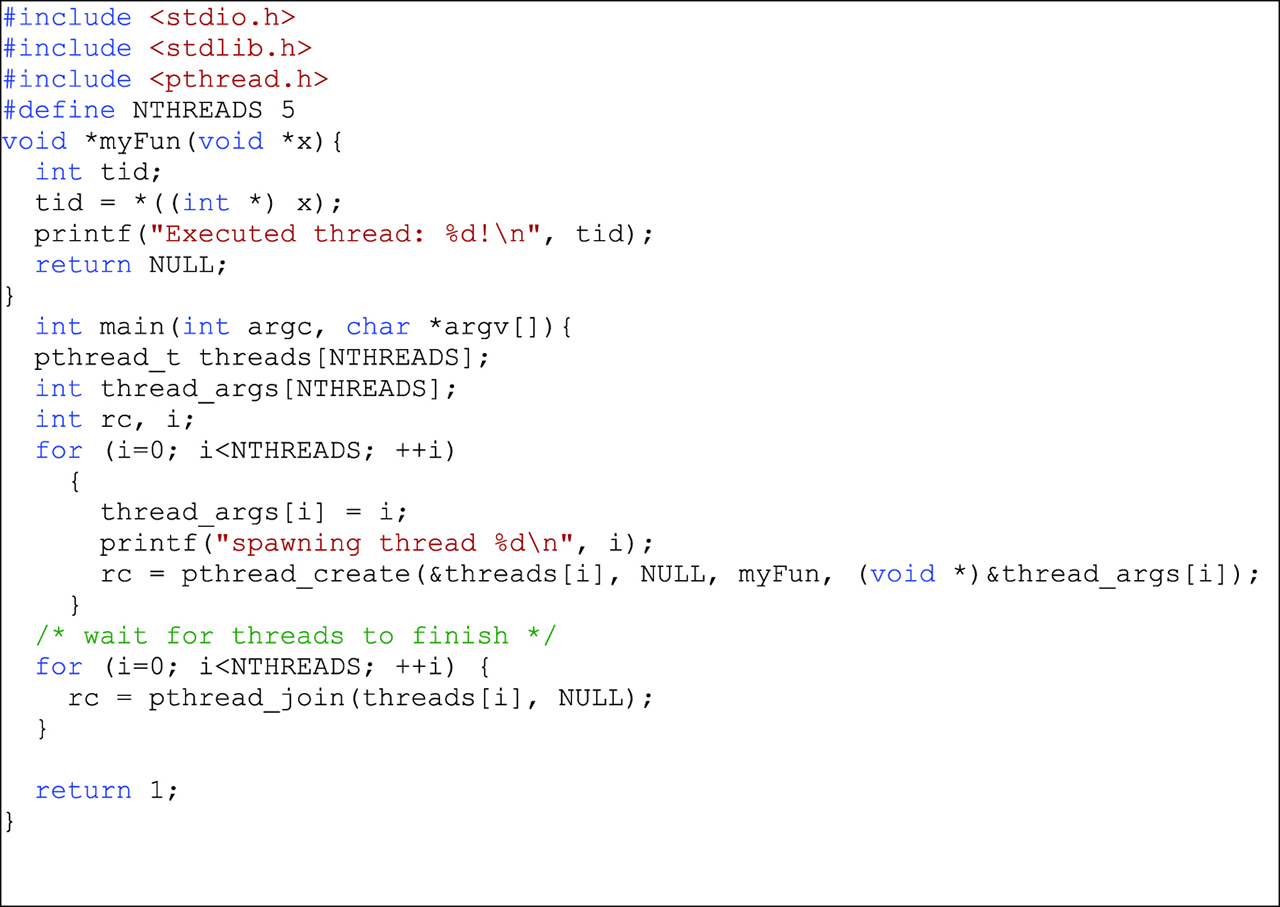
An example of p-thread code spawning three threads.
4.2 Open multiprocessing programming
A significant amount of time in complex, repetitive calculations is spent executing loop statements. These iterations lend themselves readily to parallelization using OpenMP by the inclusion of the compiler directive #pragma followed by the code to be parallelized. An example is shown in Figure 2. The same directive can be used to specify the aspects of critical code, i. e. code that is common to all the threads, as shown in Figure 3. The critical code can, for instance, manage a variable that is global relative to the other parallelized code sections. This approach is implicitly serial and has the virtue of eliminating the problems caused by collisions where code sections try to access the same variable simultaneously.

An example of the OpenMP implementation for parallelization.
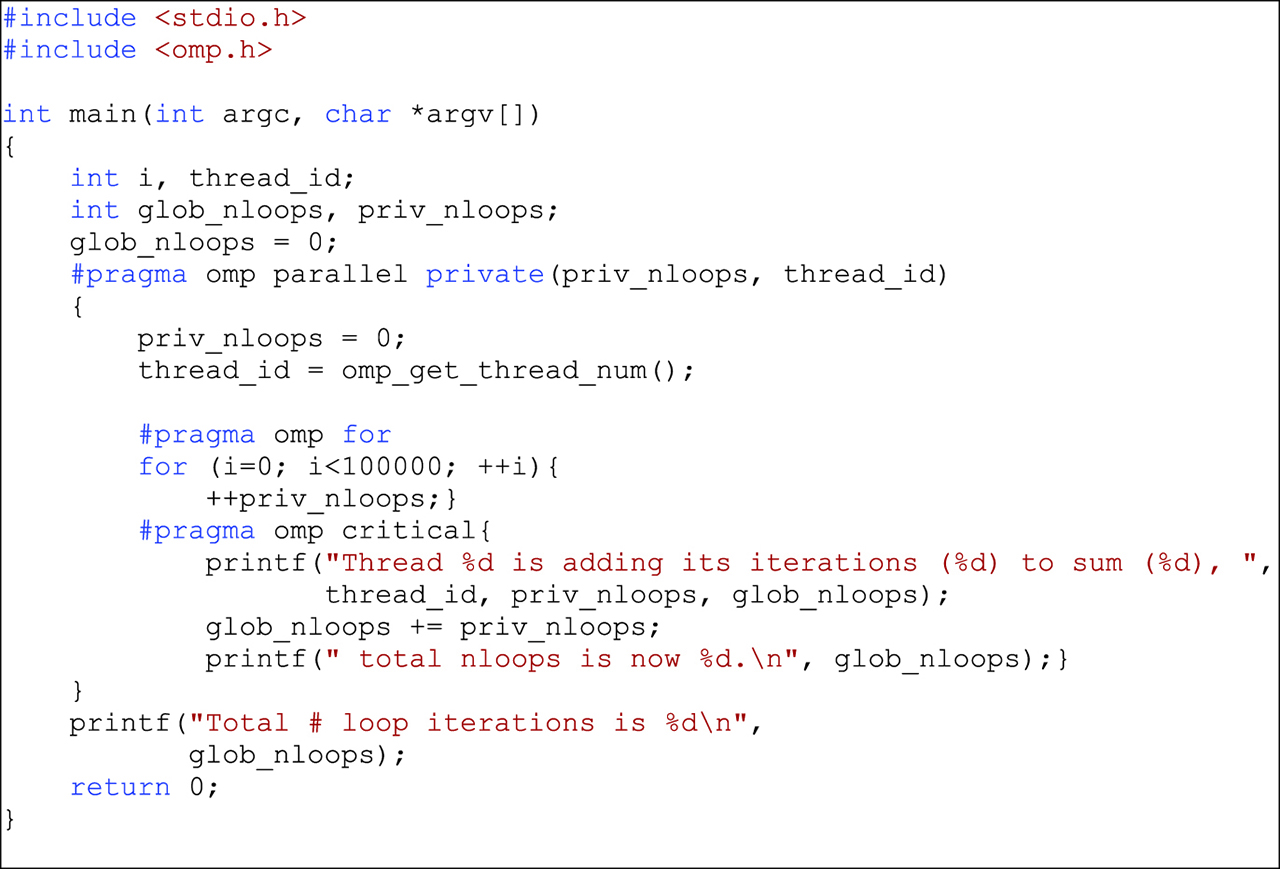
An example of ordinary OpenMP and critical OpenMP processes.
The process of combining the outputs of several smaller parallel sections is known as reduction. In OpenMP, there is a separate keyword that is used with the #pragma omp directive to carry out more reduction efficiently because of its importance. The syntax is shown in Figure 4.

Syntax of the compiler directive that invokes output reduction of parallel processes.
There is a multitude of other keywords that are associated with the #pragma omp directive, for the streamlining of various multiprocessing tasks.
Figure 5 shows the specific application of the authors to which parallelization was applied. The code snippet is only a small part of a bigger program and shows the calculation of the total potential energy using the Sutton–Chen implementation of the Finnis–Sinclair potential.
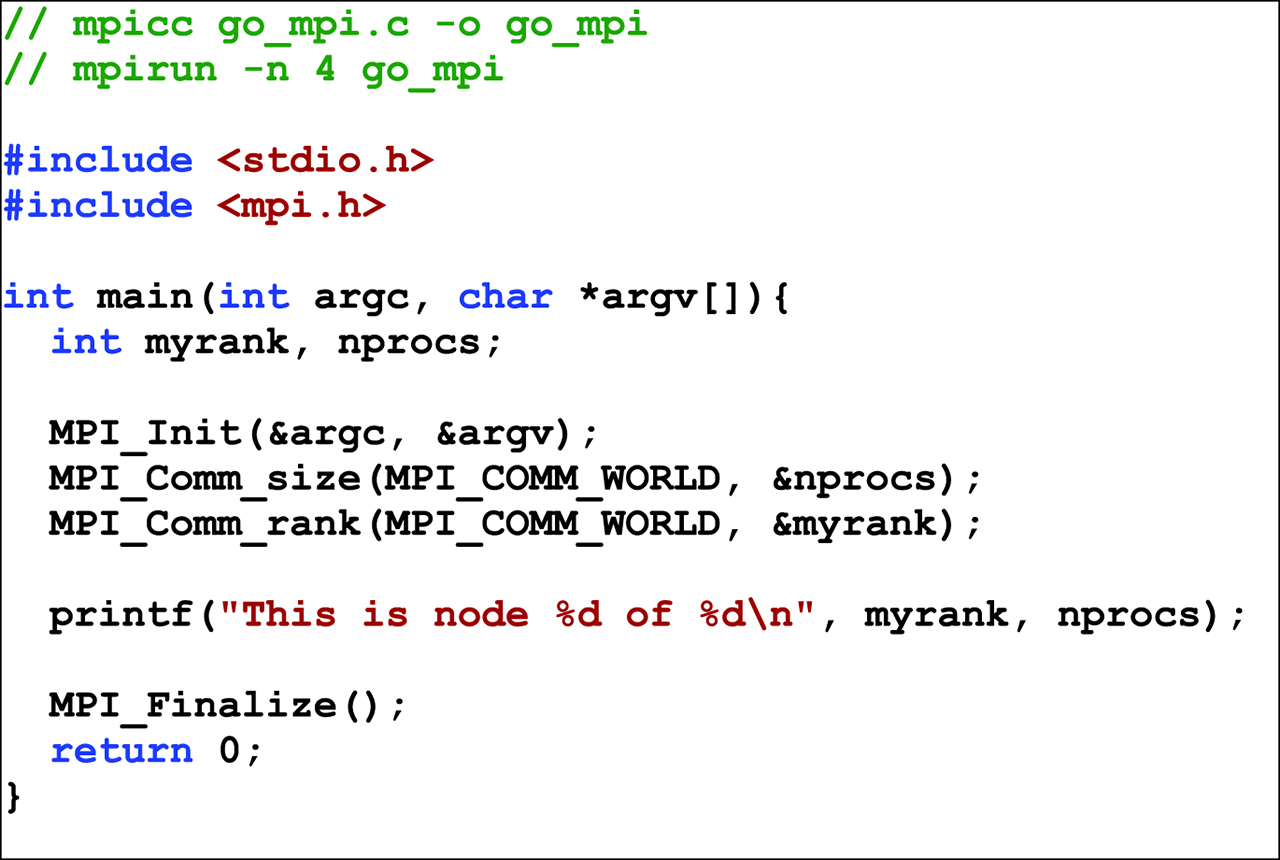
An example of a program parallelized using MPI.
4.3 Message passing interface programming
The MPI has several implementations that support cross-platform multiprocessing. Through this interface, the program assigns a collection of computational nodes resources such as memory and coordinates communication and synchronization with a set of processes running on the nodes with ease. In this approach, any computing node is handled identically. Thus, the interface does not distinguish the elements in the set {P, C}, but ropes each element as a computational resource. This approach is attractive because it operates well across clusters. The scalability is then a matter of increasing the number of such interconnects. Various supercomputers on the planet today are implemented by such arrangements, with some using proprietary interconnects and others with custom, well-guarded interconnects. A typical example of MPI code running on a Linux cluster is shown in Figure 5.
4.4 The GPU approach
GPUs are increasingly being used to implement compute nodes due to their vast processing power as a consequence of sheer speed and their reduced instruction set computing (RISC) architectures and low power consumption [45, 46, 47]. GPUs are specialized at certain operations requiring massive amounts of data at blazing speeds, at relatively low cost. Many GPUs carry multiple cores and are naturally suited to the kinds of calculations that are common in advanced physics and engineering. Multi-GPUs each on a native board known as blades daughter boards can be clustered together on bigger motherboards. Thus, several blades can implement high-performance compute clusters while taking up very little space. Understandably, the power electrical power demands of such clusters need to be carefully considered, and such clusters may be housed specially in localities referred to as render farms. Brown et al. [48, 49] discuss some issues of porting a large MD for CPUs onto parallel hybrid machines based on GPUs. They also use hybrid decomposition on code intended for distributed memory and accelerator cores with shared memory and efficient code porting of short-range force calculations through an accelerated programming model within an existing LAMMPS MD program with a custom library called Geryon. Their technique is based on load balancing of work between the CPU and accelerator cores and can be benefit cases that require the processing power of CPU cores over GPUs. This is still necessary because many GPU algorithms currently cannot achieve peak floating-point performance at present as measured relative to wall-clock time when compared to CPUs. This is a consequence of the specialization of the GPU’s specialization of certain operations and not others. Having some operations run on CPUs can minimize the amount of coding required for functions executed on GPUs in a hybrid system [48, 49, 50, 51, 52]. For instance, Brown et al. [48] show that double-precision performance can be poorer than single or mixed precision in such clusters, indicating that further optimization is required. Other workers [10] argue that increased modularity is advantageous for evolvable scientific software. The goal of writing the software is to assure functionality alongside while carefully considering parallelization efficiency and most importantly, accuracy and reproducibility of results relative to empirical results that may exist.
4.5 Cloud virtualization
In very recent years, the concept of the computing Cloud has arose initially as a means of getting large, safe and storage facilities for organization files. This has gradually been progressed to include outright distributed computation and large-data crunching. For instance, financial markets generate vast amounts of time-series data during a trade. The needs of forecasting, fraud detection, internet trade, etc. all require an almost real-time computational element. Depending on the intended functionality and application of the remote user, these powerful new features may be supplied in a limited way free of charge, or at a nominal fee. In any case, this current trend absolves the user from making any major financial outlay in terms of the starting hardware while reaping the benefits of powerful compute facilities and storage. However, security and data privacy and a full grasp of the trappings of this relatively young concept remains a concern for large corporations with obvious interest to safeguard their operations. One recurrent feature of such remote compute facilities is the ability to define virtual environments familiar to the user through virtualization. In essence, several instances of a virtual high-performance computer can be created and loaded by a simple script or graphical interface from a simple less powerful one serving as a terminal from across the planet. The instances can be either homogenous or heterogeneous in terms of the loaded operating system. Examples of organizations with such offerings are Yellow Circle [53], Microsoft Cloud (Azure) [54], Google Cloud platform [55] and others that support free virtualization for individuals in a research university through a secure account. However, the free accounts suffer the limitation in the maximum number of possible virtual instances, a drawback not suffered by paying users. In addition, the technical expertise required to have tasks up and running on such systems with carefully enumerated set {P, C} is essentially nil since server side support exists to tailor applications, particularly for the paying users. In the case of Yellow Circle, for instance, the user can remotely configure the network topology by designing routers, internet protocol addresses, subnets and so on, all through a simple graphical user interface. An image capture of the configuration screen on Yellow Circle with a free account is shown in Figure 6. The tasks of assigning compute nodes, assimilating final output data gathering and task scheduling and general network management are equally straightforward. Consequently, clouds may indeed be the ultimate future of cluster computing. Cloud virtualization can improve the utilization of compute resources while reducing implementations costs. It also introduces a desirable ability to a user to customize their system without many of the technical challenges experienced in traditional grids and cluster configuration. However, at present, virtualization-based cloud has limited performance in its infancy and is yet limited for HPC for a number of reasons. One such limitation can be caused by system virtual machine monitors for supervisory control for the cores and virtual memory peripherals. Methods therefore have to be found to minimize such delays. Ren et al. [56] propose a unique, lightweight supervisory cloud infrastructure called nOSV that serves both HPC and normal applications concurrently. The environment constructs high-performance virtual machines with dedicated resources which operate without supervisory interference, thereby keeping the performance of the traditional Cloud. With their implementations, they report significant performance improvements over other similar virtualization environments.
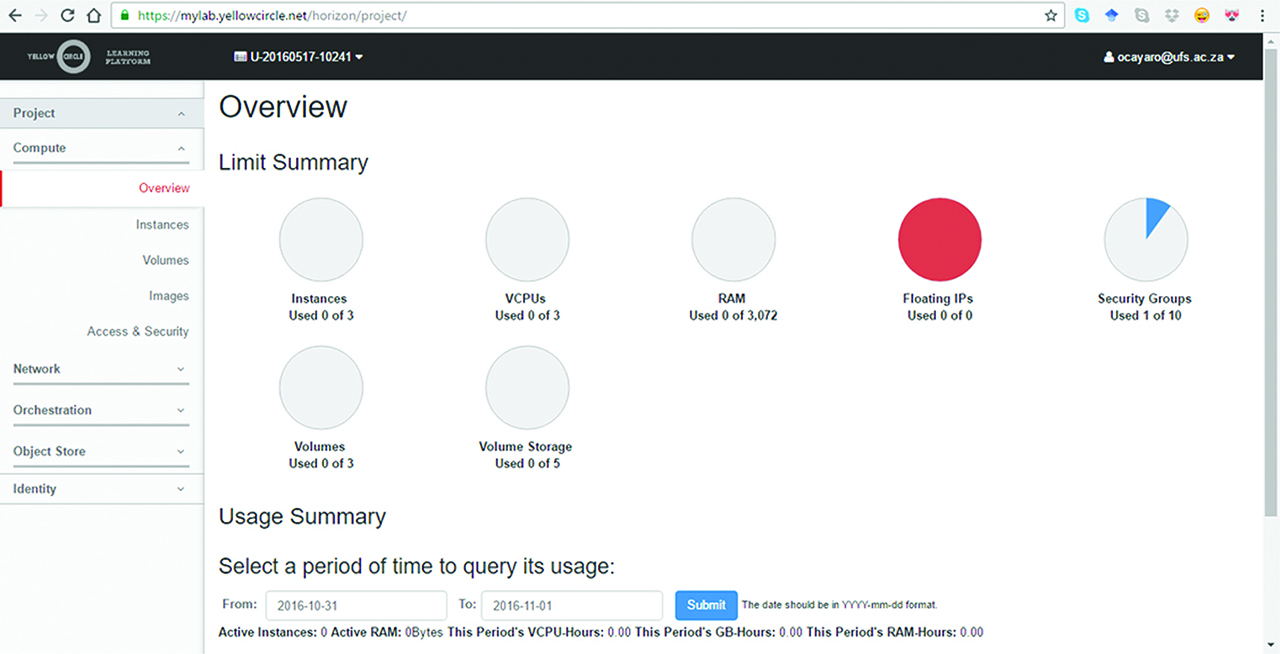
A remote virtualization cloud implemented in Yellow Circle for computational purposes.
5 Summary of results
Our own work pertains to face-centred cubic (fcc) atomic structures of metallic atoms, such as copper atoms. The force fields were derived from the embedded atom model adaptation by Sutton–Chen of the Finnis–Sinclair potential. To illustrate the speedup, we calculated the equilibrium lattice parameter (
Parameter calculations and on 30,000 atom fcc copper array using thread-based parallelization.
| Method | a (nm) | Speedup | ||
|---|---|---|---|---|
| Deterministic | 0.363 | 1.31 | 4.39 | 1.8 |
| Monte-Carlo (30 % acceptance) | 0.374 | 1.66 | 5.84 | 2.2 |
6 Conclusions
This article discusses the considerations to embark on computational research in MD using optimized multi-core computers that can also be operated as a standalone computer, or a node in cluster-based computing. At the onset, a fundamental goal was in the establishment of a computing facility that exploits the features of the single machine, with the secondary aim to extend the functionality to a cluster of such machines, making low-cost HPC possible for the group. The article presents a survey of the current state of the art, on which we base our own ongoing work. We consider what it takes to achieve parallelism and efficiency, providing arguments and pointers to aspects to consider if the need to write own as opposed to using third-party software should arise. Furthermore, we discuss the growing trends in high-performance computation, from the use of GPUs to internet-based virtualization on computing clouds. Finally, we present the summarized results for a sizable array of copper atoms. Both MD and Monte-Carlo methods showed speedup when the iterative steps were parallelized using threads on four cores. A link to a version of the evolving code in a public repository, called the Verlet–Stormer velocity program code [57], is also given for the interested reader. Since threads and MPI are concepts that are standardized and supported in libraries across many platforms, the C-program on which the present work is based could readily be ported to other systems.
Correction Statement
Correction added after ahead-of-print publication on 19 July 2017: The DOI of this article has been corrected to: https://doi.org/10.1515/psr-2016-5100.
The DOI of this article has been used for another publication by mistake. If you intended to access the other publication, please use this link: https://doi.org/10.1515/psr-2016-0100
Acknowledgment
This article is also available in: Ramasami, Computational Sciences. De Gruyter (2017), isbn 978–3–11–046536–5.
References
1. Horsch M, Vrabec J, Bernreuther M, Grottel S, Reina G, Wix A, et al. Homogeneous nucleation in supersaturated vapors of methane, ethane, and carbon dioxide predicted by brute force molecular dynamics. J Chem Phys. 2008;128:164510.10.1063/1.2907849Suche in Google Scholar PubMed
2. Mendelev MI, Han S, Srolovitz DJ, Ackland GJ, Sun DY, Asta M. Development of new interatomic potentials appropriate for crystalline and liquid iron. Philosophical Magazine. 2003;83((35)):3977–3994. doi:10.1080/14786430310001613264.Suche in Google Scholar
3. Sutton AP, Chen J. Long-range Finnis–Sinclair potentials. Philos Mag Lett. 1990;61((3)):139–156.10.1080/09500839008206493Suche in Google Scholar
4. Das A, Ghosh MM. MD simulation-based study on the melting and thermal expansion behaviours of nanoparticles under heat load. Computational Mater Sci. 2015;101:88–95. doi:10.1016/j.commatsci.2015.01.008.Suche in Google Scholar
5. Van Der Walt C, Terblans JJ, Swart HC. Molecular dynamics study of the temperature dependence and surface orientation dependence of the calculated vacancy formation energies of Al, Ni, Cu, Pd, Ag, and Pt. Computational Mater Sci. 2014;83:70–77. doi:10.1016/j.commatsci.2013.10.039.Suche in Google Scholar
6. Abraham MJ, Murtolad T, Roland Schulz R, Palla S, Jc J, Hessa B, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015. doi:10.1016/j.softx.2015.06.001.Suche in Google Scholar
7. Ocaya RO, Terblans JJ. to appear in Journal of Physics: Conference Series. Paper presented at CCP2016-28th IUPAP Conference on Computational Physics; Johannesburg, South Africa 2016 July 10–14. 2016July10–14 Temperature specification in atomistic molecular dynamics and its impact on simulation efficacy2017.10.1088/1742-6596/905/1/012031Suche in Google Scholar
8. Ocaya RO, Terblans JJ. C-language package for standalone embedded atom method molecular dynamics simulations of fcc structures. SoftwareX. 2016;5:107–111. doi:10.1016/j.softx.2016.05.005.Suche in Google Scholar
9. Ocaya RO, Terblans JJ. ;to appear in Journal of Physics: Conference Series. Paper presented at CCP2016-28th IUPAP Conference on Computational Physics; Johannesburg, South Africa 2016 July 10–14. 2016July10–14 Coding considerations for standalone molecular dynamics simulations of atomistic structures2017.10.1088/1742-6596/905/1/012018Suche in Google Scholar
10. Buchholz M, H-J B, Vrabec J. Software design for a highly parallel molecular dynamics simulation framework in chemical engineering. J Comput Sci. 2011;2:124–129. doi:10.1016/j.jocs.2011.01.009.Suche in Google Scholar
11. Smirnov BM. Energetics of clusters with a face centered-cubic structure. Zh Eksp Teor Fiz. 1995;107:2080–2091.Suche in Google Scholar
12. Terblans JJ. Calculating the bulk vacancy formation energy (Ev) for a Schottky defect in a perfect Cu(111), Cu(100) and a Cu(110) single crystal. Surf Interface Anal. 2002;33:767–770. doi:10.1002/sia.1451.Suche in Google Scholar
13. Mattsson TR, Mattsson AE. Calculating the vacancy formation energy in metals: Pt, Pd, and Mo. Phys Rev B. 2002;66:214110.10.1103/PhysRevB.66.214110Suche in Google Scholar
14. Sebastian IS, Aldazabal J, Capdevila C, Garcia-Mateo C. Diffusion simulation of CrFe bcc systems at atomic level using a random walk algorithm. Phys Stat Sol (A). 2008;205(6):1337–1342. doi:10.1002/pssa.200778124.Suche in Google Scholar
15. Jian-Min Z, Fei M, Ke-Wei X. Calculation of the surface energy of fcc metals with modified embedded-atom method. Appl Surf Sci. 2004;13(7):34–42. doi:10.1016/j.apsusc.2003.09.050.Suche in Google Scholar
16. Griebel M, Knapek S, Zumbusch G. Numerical simulation in molecular dynamics. Texts in computational science and engineering 5. Berlin: Springer, 2007978-3-540-68094-9 .Suche in Google Scholar
17. Car R, Parrinello M. Unified approach for molecular dynamics and density functional theory. Phys Rev Lett. 1985;55(22):2471–2474.10.1103/PhysRevLett.55.2471Suche in Google Scholar PubMed
18. Car R, Parrinello M, Loubeyre PP, Boccara N. The unified approach for molecular dynamics and density functional theory. In: Loubeyre P.P., Boccara N., editors. Simple molecular systems at very high density. Vol. 186 of NATO ASI Series, Series B, Physics. New York: Plenum Press, 1989:455–476.Suche in Google Scholar
19. Remler DK, Madden PA. Molecular dynamics without effective potentials via the Car–Parrinello approach. Mol Phys. 1990;70(6):921–966.10.1080/00268979000101451Suche in Google Scholar
20. Tuckerman ME. Ab initio molecular dynamics: basic concepts, current trends and novel applications. J Phys: Condens Matter. 2002;14 R1297R1355 Online at stacks.iop.org/JPhysCM/14/R1297.10.1088/0953-8984/14/50/202Suche in Google Scholar
21. Moore GE. Cramming more components onto integrated circuits. Electronics Magazine 1965; 38(8):114–117.10.1109/JPROC.1998.658762Suche in Google Scholar
22. Moore GE. Cramming more components onto integrated circuits. Proc IEEE. 1998;86(1):82–85.10.1109/JPROC.1998.658762Suche in Google Scholar
23. Dongarra J. Paper presented in International Supercomputer Conference. Germany: Heidelberg,. 2004June Survey of present and future supercomputer architectures and their interconnects2225.10.1007/978-3-540-30566-8_1Suche in Google Scholar
24. Intel Corporation. document on the Internet;, January 2016. cited Available from. 2017January14 http://ark.intel.com/products/65703.Suche in Google Scholar
25. Peng L, Tan G, Kalia RK, Nakano A, Vashishta P, Fan D, et al. Preliminary investigation of accelerating molecular dynamics simulation on Godson-T many-core processor. Euro-Par 2010 Parallel Processing Workshops. 2010;6586:349–356.10.1007/978-3-642-21878-1_43Suche in Google Scholar
26. Peng L, Tan G, Kalia RK, Nakano A, Vashishta P, Fan D, et al. Preliminary investigation of accelerating molecular dynamics simulation on godson-T many-core processor. Parallel Processing Workshops, Euro-Par 2010: HeteroPar 2010, HPPC 2010, HiBB 2010, CoreGrid 2010, UCHPC 2010, HPCF 2010, PROPER 2010, CCPI 2010, VHPC 2010, Ischia; Italy; 2010. 2010August 31–September 3 Code 86021.10.1007/978-3-642-21878-1_43Suche in Google Scholar
27. Peng L, Tan G, Kalia RK, Nakano A, Vashishta P, Fan D, et al. Preliminary investigation of accelerating molecular dynamics simulation on Godson-T many-core processor. Vol. 6586 of the Series Lecture Notes in Computer Science Euro-Par. 2010;2010:349–356.10.1007/978-3-642-21878-1_43Suche in Google Scholar
28. Bernreuther M, Bungartz H-J, Hülsemann F, Kowarschik M, Rüde U. Proceedings of the 18th Symposium Simulations Technique (ASIM2005), in Frontiers in Simulation, SCS European Publishing House; 2005;117–123 15.Suche in Google Scholar
29. Mangiardi CM, Meyer R. A hybrid algorithm for parallel molecular dynamics simulations [document on the Internet]. 2016 [cited 2016 October 31]. Available from: https://arxiv.org/abs/1611.00075.Suche in Google Scholar
30. Mangiardi CM, Meyer R Molecular-dynamics simulations using spatial decomposition and task-based parallelism. In: Bélair J., Frigaard I., Kunze H., Makarov R., Melnik R., Spiteri R., editors. Mathematical and computational approaches in advancing modern science and engineering. Switzerland: Springer International, 2016:133–140.10.1007/978-3-319-30379-6_13Suche in Google Scholar
31. Meyer R. Efficient parallelization of short-range molecular dynamics simulations on many-core systems. Phys Rev E. 2013;88(5):053309.10.1103/PhysRevE.88.053309Suche in Google Scholar
32. Plimpton S. Fast parallel algorithms for short-range molecular dynamics. J Comput Phys. 1995;117(1):1–19.10.2172/10176421Suche in Google Scholar
33. Todorov IT, Smith W, Trachenko K, Dove MT. DL POLY 3: new dimensions in molecular dynamics simulations via massive parallelism. J Mater Chem. 2006;16(20):1911–1918.10.1039/b517931aSuche in Google Scholar
34. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802.10.1002/jcc.20289Suche in Google Scholar
35. Ackland GJ, D’Mellow K, Daraszewicz SL, Hepburn DJ, Uhrin M, Stratford K. The MOLDY short-range molecular dynamics package. Comput Phys Commun. 2011;182(12):2587–2604.10.1016/j.cpc.2011.07.014Suche in Google Scholar
36. Berendsen HJC, Van Der Spoel D, Van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1):43–56.10.1016/0010-4655(95)00042-ESuche in Google Scholar
37. Needham PJ, Bhuiyan A, Walker RC. Extension of the AMBER molecular dynamics software to Intel’s Many Integrated Core (MIC) architecture. Comput Phys Commun. 2016;201:95–105.10.1016/j.cpc.2015.12.025Suche in Google Scholar
38. Hager G, Wellein G. Introduction to high performance computing for scientists and engineers. Boca Raton, FL: Chapman and Hall, 2011 .10.1201/EBK1439811924Suche in Google Scholar
39. Wang J, Gao X, Li G, Q L, Hu W, Chen Y. Godson-3: a scalable multicore RISC processor with x86 emulation. IEEE Micro. 2009;2(29):17–29. doi:10.1109/MM.2009.30.Suche in Google Scholar
40. Jain A, Shin Y, Persson KA. Computational predictions of energy materials using density functional theory. Nature Reviews Materials. 2016;1. Article number: 15004. doi:10.1038/natrevmats.2015.4.Suche in Google Scholar
41. Pal A, Agarwala A, Raha S, Bhattacharya B. Performance metrics in a hybrid MPI–OpenMP based molecular dynamics simulation with short-range interactions. J Parallel Distrib Comput. 2014;74:2203–2214.10.1016/j.jpdc.2013.12.008Suche in Google Scholar
42. Petitet A, Whaley RC, Dongarra J, Cleary A. HPL - A portable implementation of the high-performance linpack benchmark for distributed-memory computers [document on the internet]. 2016;[cited Available from. 2016 October 22 http://www.netlib.org/benchmark/hpl/.Suche in Google Scholar
43. The Linpack Benchmark [document on the Internet]. October 2016. [cited Available from. 2016 October 22 https://www.top500.org/project/linpack/.Suche in Google Scholar
44. Cleve M. Parallel MATLAB: multiple processors and multiple cores [document on the Internet]. October 2016. [cited Available from 2007 January 01] https://www.mathworks.com/company/newsletters/articles/parallel-matlab-multiple-processors-and-multiple-cores.html.Suche in Google Scholar
45. Govender N, Wilke DN, Kok S, Els R. Development of a convex polyhedral discrete element simulation framework for NVIDIA Kepler based GPUs. J Comput Appl Math. 2014;270:386–400. doi:10.1016/j.cam.2013.12.032.Suche in Google Scholar
46. Govender N, Wilke DN, Kok S, Els R. Collision detection of convex polyhedra on the NVIDIA GPU architecture for the discrete element method. Appl Math Comput. 2015;267:810–829. doi:10.1016/j.amc.2014.10.013.Suche in Google Scholar
47. Govender N, Rajamani RK, Kok S, Wilke DN. Discrete element simulation of mill charge in 3D using the BLAZE-DEM GPU framework. Minerals Eng. 2015;79:152–168. doi:10.1016/j.mineng.2015.05.010.Suche in Google Scholar
48. Brown WM, Wang P, Plimpton SJ, Tharrington AN. Implementing molecular dynamics on hybrid high performance computers - short range forces. Comput Phys Commun. 2011;182(4):898–911. doi:10.1016/j.cpc.2010.12.021.Suche in Google Scholar
49. Brown WM, Kohlmeyer A, Plimpton SJ, Tharrington AN. Implementing molecular dynamics on hybrid high performance computers - Particle-particle particle-mesh. Comput Phys Commun. 2012;183(3):449–459. doi:10.1016/j.cpc.2011.10.012.Suche in Google Scholar
50. Stone JE, Phillips JC, Freddolino PL, Hardy DJ, Trabuco LG, Schulten K. J Comput Chem. 2007;28:2618–2640.10.1002/jcc.20829Suche in Google Scholar PubMed
51. Schmid N, Botschi M, Van GWF. J Comput Chem. 2010;31:1636–1643.10.1002/jcc.21447Suche in Google Scholar
52. Hampton S, Alam SR, Crozier PS, Agarwal PK. Proceedings of the 2010 International Conference on High Performance Computing and Simulation (HPCS 2010), 2010.Suche in Google Scholar
53. Yellow Circle [document on the Internet. October 2016. [cited Available from. 2016October29 https://mylab.yellowcircle.net.Suche in Google Scholar
54. Microsoft Azure [document on the Internet]. 2016;[cited Available from. 2016 October 29 https://azure.microsoft.com/en-us/.Suche in Google Scholar
55. Google Cloud Platform [document on the Internet]. October 2016. [cited Available from. 2016October29 https://cloud.google.com/.Suche in Google Scholar
56. Ren J, Qi Y, Dai Y, Xuan Y, Shi Y. nOSV: a lightweight nested-virtualization VMM for hosting high performance computing on cloud. J Syst Softw. 2017;124:137–152. doi:10.1016/j.jss.2016.11.001.Suche in Google Scholar
57. VSV Software [software on the Internet]. 2016;[cited Available from:. 2016October29 https://github.com/ElsevierSoftwareX/SOFTX-D-15-00054.Suche in Google Scholar
© 2017 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Applications of silver nanoparticles stabilized and/or immobilized by polymer matrixes
- Addressing the challenges of standalone multi-core simulations in molecular dynamics
- Virtually going green: The role of quantum computational chemistry in reducing pollution and toxicity in chemistry
- BioArtificial polymers
- Green analytical chemistry – the use of surfactants as a replacement of organic solvents in spectroscopy
- Membrane contactors for CO2 capture processes – critical review
- Elemental Two-Dimensional Materials Beyond Graphene
Artikel in diesem Heft
- Applications of silver nanoparticles stabilized and/or immobilized by polymer matrixes
- Addressing the challenges of standalone multi-core simulations in molecular dynamics
- Virtually going green: The role of quantum computational chemistry in reducing pollution and toxicity in chemistry
- BioArtificial polymers
- Green analytical chemistry – the use of surfactants as a replacement of organic solvents in spectroscopy
- Membrane contactors for CO2 capture processes – critical review
- Elemental Two-Dimensional Materials Beyond Graphene