Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
-
Kazushi Mashima
 ,
Yukiko Hayashi
,
Yukiko Hayashi

Abstract
Acylation is one of the most abundant organic transformations of alcohols (esterification) and amines (amidation). Because of the greater nucleophilicity of the amino group compared to the hydroxyl group and the stability of amides compared to esters, N-acylation occurs predominantly in organic synthetic reactions. We reported that the μ-oxo-tetranuclear zinc cluster Zn4(OCOCF3)6O efficiently catalyzes highly chemoselective acylation of hydroxyl groups in the presence of primary and secondary alkyl amino groups to afford the corresponding esters in high yields. Not only zinc carboxylate complexes but also various carboxylate complexes of first-row late transition metals, such as Mn, Fe, Co, and Cu, become catalysts for such the hydroxy group-selective acylation in the presence of amines. Among these carboxylate compounds, we found that the combination of an octanuclear cobalt carboxylate cluster [Co4(OCOR)6O]2 (R = CF3, CH3, and tBu) with nitrogen-containing ligands such as 2,2′-bipyridine show sufficient catalytic activity toward O-selective transesterification. Notably, an alkoxide-bridged dinuclear complex, Co2(OCOtBu)2(bpy)2(μ2-OCH2-C6H4-4-CH3)2, was successfully isolated as a key intermediate that proceeds with Michaelis–Menten behavior through an ordered ternary complex mechanism similar to dinuclear metallo-enzymes, suggesting that the formation of alkoxides, followed by coordination of the ester, is responsible for the unique O-selective acylation.
Introduction
Esters and amides are ubiquitous functional groups in natural and synthetic organic compounds [1]. These functional groups are typically synthesized by acylation of the corresponding alcohol and amine with a stoichiometric amount of acylating reagents such as acid halides and acid anhydrides, although stoichiometric amounts of base are required to capture acidic co-products such as hydrochloric acid and carboxylic acids (Fig. 1a). Another standard method of synthesizing esters and amides is a condensation reaction of any carboxylic acid with an appropriate alcohol and amine under suitable condensation reagents, but the carboxylic acids have some disadvantages, such as low solubility in common organic solvents, and acidity, which leads to the decomposition of acid-sensitive functionalities (Fig. 1b). Transesterification and amidation starting from lower esters such as methyl esters are easy to handle due to their high solubility and stability, thereby providing a rational synthetic protocol with respect to atom-economy and environmental concerns (Fig. 1c).
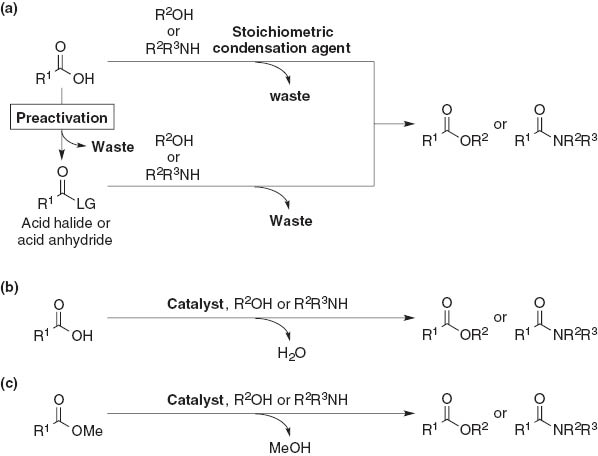
Esterifications and amidations: (a) general method for synthesis of esters and amides from carboxylic acids; (b) catalytic condensation of carboxylic acids with alcohols or amines; (c) catalytic transesterification or ester-amide exchange reaction.
When alcohol and amine coexist, a common reaction occurs in which the nucleophilic amine selectively attacks the carbonyl group of ester to give the corresponding amide.
Accordingly, under standard acylating conditions, acylation of alcohol in the presence of a free amino group requires the selective protection of a free amine, followed by esterification of a hydroxyl group, and then deprotection of the amine moiety (Fig. 2a). Thus, although selective direct O-esterification over N-amidation of aminoalcohols is a challenging task in synthetic organic chemistry, it is worth developing to minimize the reaction process and reduce chemical waste (Fig. 2b) [2]. Herein, we concisely review chemoselective acylation of the hydroxy group in the presence of amino groups, and report our recent achievements on O-selective esterification of aminoalcohols catalyzed by zinc and cobalt clusters.
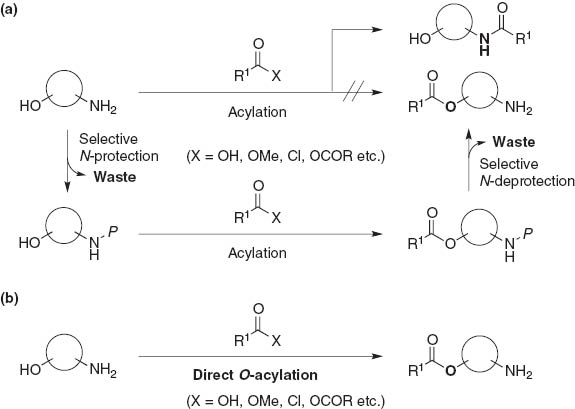
(a) Common strategy for esterification of aminoalcohols; (b) direct esterification of amino alcohols.
Chemoselective acylation of alcohols in the presence of amines
Pre-protonation of alkyl amino groups by highly acidic reagents such as hydrochloric acid and trifluoroacetic acid is a straightforward route for the esterification of aliphatic amino alcohols (eq. 1). A unique example was reported by Takata and Kihara et al. who used rotaxane compounds for a O-selective acylation followed by N-acylation to lock the rotaxane structure (Scheme 1) [3]. Copper ions were coordinated in a chelating fashion to amino acid functionalities so that the hydroxyl group was esterified, and then removed by hydrogen sulfide [4] or 8-hydroxyquinoline (Scheme 2) [5].
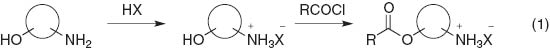
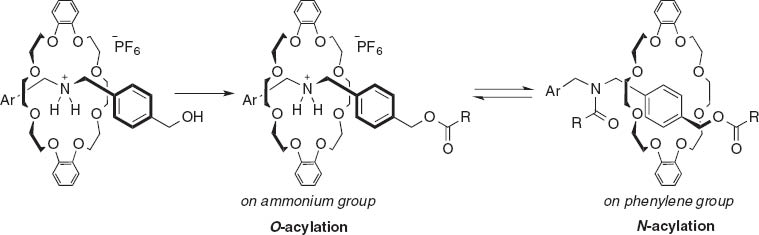
O-selective acylation of rotaxane compounds.
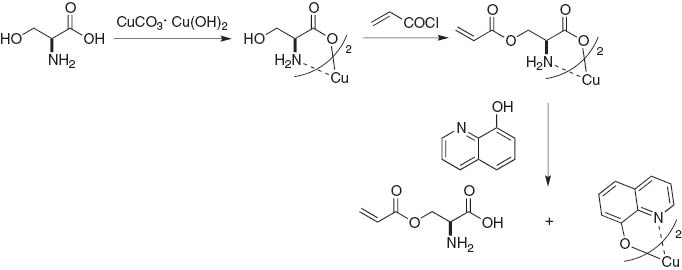
O-selective acylation of Cu-chelated amino acid derivatives.
Enhancement of the nucleophilicity of the hydroxy group is another strategy used for the chemoselective acylation of aromatic substrates. Selective O-acylation of phenols in the presence of aromatic amino groups is accomplished by acylating reagents bearing a specific leaving group, such as N-methyl-N-nitroso [6] and 2-bipyridinyl groups [7] in which a proton of the hydroxy group is accepted via a concerted proton transfer, as schematically depicted in Fig. 3.
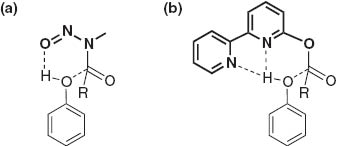
Possible transition state in acylation of phenol with specific acyl donor. (a) N-methyl-N-nitroso group; (b) 2-bipyridinyl group.
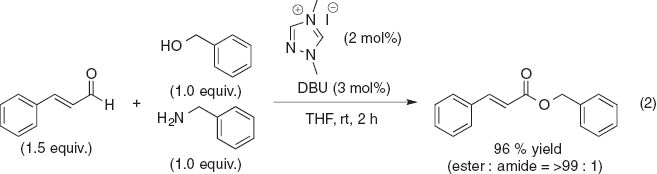
Studer et al. developed N-heterocyclic carbene (NHC), which mediates the chemoselective acylation of alcohols in the presence of amines (eq. 2) [8]. The DFT calculations on NHC–H2NMe and NHC–HOMe indicate that the stronger hydrogen bonding energy for NHC–HOMe than that for NHC–H2NMe elongates the O–H bond.
The 1,2- and 1,3-amino alcohols are unique in that they act in intramolecular hydrogen bonding of the hydroxy group with the amino group in nonpolar solvent, and as non-hydrogen-bonded molecules in polar solvent (Fig. 4a). Such hydrogen bonding enhances the nucleophilicity of the oxygen atom, while attenuating the nucleophilicity of the nitrogen, which results in an inversion of reactivity toward the acylation. Maeng and Funk demonstrated chemoselective O-esterification for the synthesis of cytotoxin fasicularin (Fig. 4b) [9]. Using the same synthetic strategy, Piarulli and Gennari et al. conducted O-selective acylation of (R)-N-benzylserine methyl ester using HATU to afford the corresponding ester compound (Fig. 4c) [10].
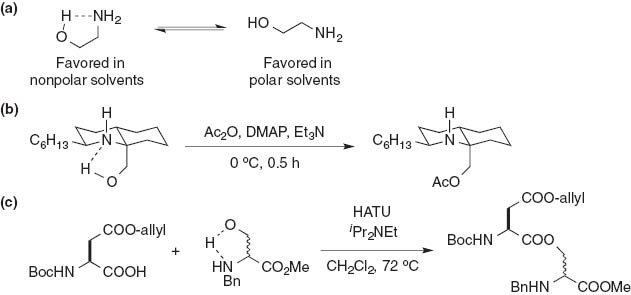
Intramolecular hydrogen bonding of the hydroxy group with the amino group in 1,2-amino alcohols. (a) The lone pair of electrons on the nitrogen atom is used to form the intramolecular hydrogen bond with the hydrogen atom on the hydroxy group; (b) and (c) examples of chemoselective O-acylations of 1,2-amino alcohols under basic conditions.
Some enzymes act to fix the transition state conformation in favor of O-acylation. Lipase catalyzes the selective O-acylation of the serine moiety of a dipeptide, (S)-Phe-(S)-Ser(OH)-NH-β-Naph, even in the presence of highly nucleophilic α-NH2 group of Phe, to give the O-acetylated product, (S)-Phe-(S)-Ser(OAc)-NH-β-Naph, in 98 % yield[11]. Molecular modeling studies suggest that such O-selectivity is caused by interference with the hydrogen transfer in the transition state [12].
Metal-catalyzed O-selective acylation
Yttrium catalysts
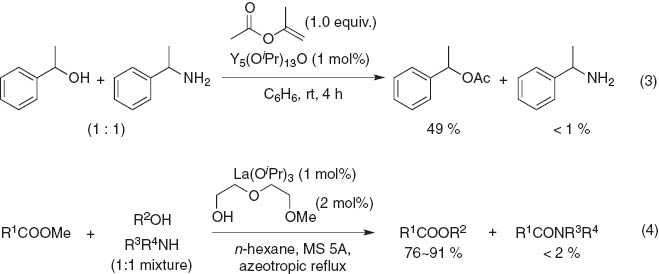
Lin and RajanBabu reported yttrium-catalyzed selective O-acylation over N-amidation using Y5(OiPr)13O as a catalyst to promote the selective O-acetylation of amino alcohols without the formation of the amide by activated acylation reagents such as vinyl or isopropenyl acetate [13]. When a stoichiometric mixture of 1-phenylethanol and 1-phenylethanamine is treated with the yttrium catalyst and isopropenyl acetate, the alcohol quantitatively converts to O-acetate and only a trace amount of the N-acetyl derivative is detected (eq. 3). Recently, Ishihara et al. also reported that a La(OiPr)3–2-(2-methoxyethoxy)ethanol catalyst system catalyzes the chemoselective transesterification of methyl esters in the presence of amines (eq. 4) [14].
Zinc catalysts
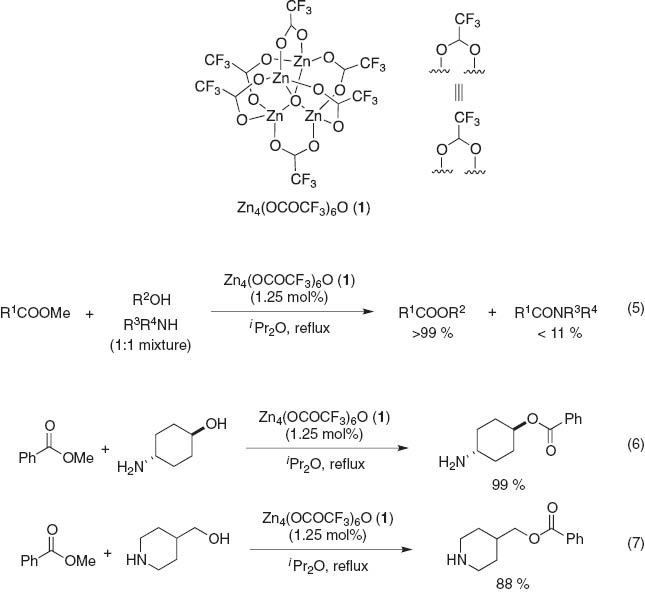
We reported that the zinc cluster Zn4(OCOCF3)6O (1) [15] effectively catalyzes the hydroxy group-selective acylation with unactivated methyl esters in the presence of primary and secondary aliphatic amino groups (eq. 5) [15b]. The present method provides a new protocol to directly convert amino alcohols to the corresponding amino esters without the use of protecting groups, outlined in Fig. 2a. Representative examples are the transesterification of methyl benzoate with amino alcohols such as trans-4-aminohexan-1-ol and 4-piperidinemethanol to give the corresponding amino esters without any amides (eqs. 6 and 7), while ethanolamine and cis-4-aminohexane-1-ol predominantly produce the corresponding amides via intramolecular O,N-rearrangement of the initially generated amino esters. In addition, under refluxing conditions in excess amounts of EtOAc, the alcohols are acetylated in high yield while hydroxyl group selectivity is maintained [15d].
Amine additive effects
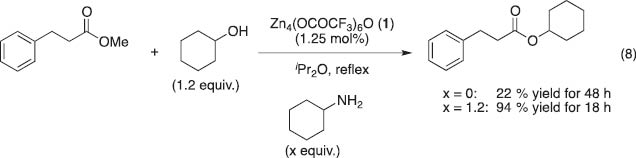
The addition of catalytic amounts of amines and N-heteroaromatics to zinc catalyst 1 has drastic additive effects on the transesterification of alcohols to increase the catalytic activity compared with the original reactivity of 1. A typical example is shown in eq. 8: in the presence of cyclohexylamine, transesterification proceeds smoothly to reach 94 % yield while maintaining the O-selectivity [15h]. DMAP works better than primary amines, facilitating the transesterification of sterically demanding esters and alcohols (Scheme 3). In fact, in the presence of a catalytic amount (20 mol%) of DMAP, transesterification of methyl adamantine-1-carboxylate and 1-butanol under toluene refluxing conditions for 90 h affords 94 % yield, which is greatly improved compared with only a trace amount obtained with no additive and 29 % yield by the addition of cyclohexylamine. We propose that the addition of DMAP stabilizes the clusters with lower nuclearities and enhances catalytic activity for transesterification.
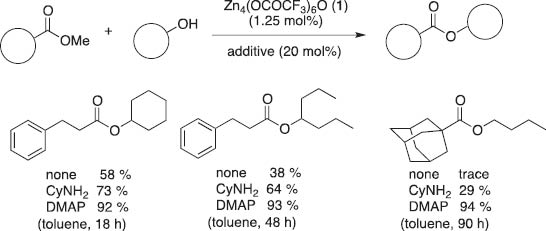
Additive effects of the zinc cluster-catalyzed transesterification.
Cobalt catalysts
Because zinc and yttrium cluster complexes show high hydroxyl group selectivity and the additive of amines and N-heteroaromatic compounds to zinc cluster catalyst 1 improved catalytic activity, we tested the catalytic performance of various metal carboxylates for chemoselective acylation of alcohols over amines [15i]. Scheme 4 shows the results of the test reaction of methyl 3-phenylpropanoate with a 1:1 mixture of n-hexanol (1.2 equiv) and n-hexylamine (1.2 equiv) in the presence of various metal acetates, Mn(OCOCH3)m (5 mol% metal). Acetate complexes of middle and late first-row transition metals in an oxidation state of two show moderate catalytic activity and maintain sufficient chemoselectivity, producing O-acylated product (blue bar) over N-acylated product (red bar). Without the addition of n-hexylamine, the catalytic activity of transesterification is suppressed to almost no reaction or low yield, and the addition of n-hexylamine interacts with carboxylate compounds of Mn(II), Fe(II), Co(II), Cu(II), Cu(I), and Zn(II) to give catalytically active species that catalyze O-selective acylation.
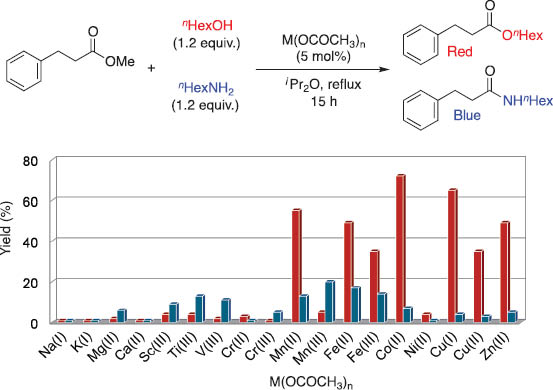
Catalytic activity and chemoselectivity of various metal acetates for acylation of n-hexanol and n-hexylamine with methyl 3-phenylpropionate.
Dinucear cobalt complex
We selected a pivalate-bridged octanuclear cluster, Co8(OCOtBu)12O2 (2)[16], to elucidate O-selective transesterification [15i]. Ligand screening revealed that not only monodentate amines and N-heteroaromatics, but also chelating ligands, including acyclic and cyclic ligands, have suitable additive effects, and finally we selected 2,2′-bipyridine. The reaction of 2 with 2,2′-bipyridine (8 equiv) in the presence of 4-methylbenzyl alcohol (8 equiv) afforded an alkoxide-bridged dinuclear complex Co2(OCOtBu)2(bpy)2(μ2-OCH2-C6H4-4-CH3)2 (3) as a key intermediate (eq. 9), which we characterized by X-ray analysis. Notably, the isolated complex 3 showed hydroxyl group selectivity: competitive reaction of methyl 3-phenylpropanoate with a 1:1 mixture of n-hexanol (1.2 equiv) and n-hexylamine (1.2 equiv) in the presence of 3 (2.5 mol%) produces esters (totally 94 %) and an amide (4 %) (eq. 10).
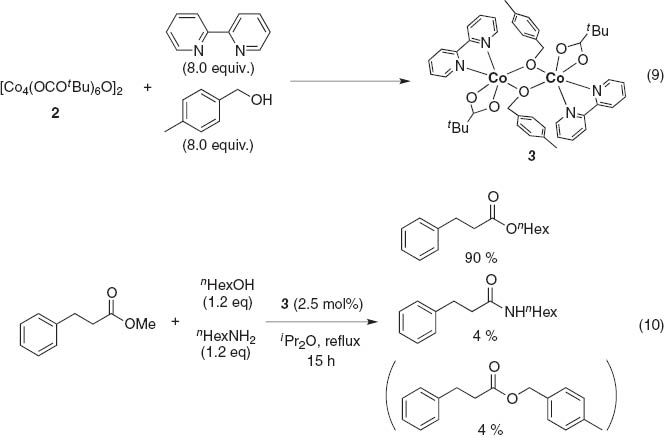
Mechanism
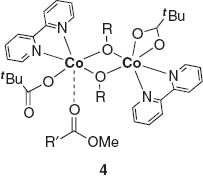
Kinetic studies and DFT calculations revealed Michaelis–Menten behavior of the complex 3 through the ordered ternary complex mechanism similar to dinuclear metallo-enzymes, suggesting the formation of alkoxides followed by coordination of the ester to give the ternary intermediate 4 with η1-coordination of the pivalate moiety and intramolecular nucleophilic attack of the bridged alkoxy moiety to the coordinated ester. In contrast to the Co(II) ion of the active species, Co(III) compounds have very low catalytic activity due to the inertness of a closed-shell d6 Co(III) species for any ligand exchange reaction. We therefore assumed that basic μ-oxo moieties of the cobalt cluster play an important role in the initial deprotonation step of alcohols for producing alkoxo-bridged species.
Conclusion
As summarized in this review, several efficient O-selective acylations in the presence of much more nucleophilic primary and secondary alkyl amino groups have been developed. In general, to reverse the innate reactivity of amino and hydroxyl groups, the protection of more reactive amino groups is necessary, resulting in the formation of more than stoichiometric amounts of chemical waste, which reduces the reaction efficiency. The catalytic O-selective acylation reactions described here clearly indicate that such innate chemoselectivity is reversible in a catalytic manner. A variety of carboxylate complexes of first-row late transition metals, such as Mn, Fe, Co, Cu, and Zn, catalyze the hydroxy group-selective acylation in the presence of amines. Mechanistic studies using Co complexes revealed that the reactions proceeds with Michaelis–Menten behavior through an ordered ternary complex mechanism similar to dinuclear metallo-enzymes, suggesting that the formation of alkoxides followed by coordination of the ester is responsible for the unique O-selective acylation. Because of the high efficiency in terms of atom economy and step economy, these direct catalyses will be a powerful tool for organic synthesis, and further development of new catalyst-controlled chemoselective reactions is needed.
Acknowledgment
This work was supported by CREST from Japan Science and Technology Agency.
References
[1] For reviews, see: (a) J. Mulzer, in Comprehensive Organic Synthesis B. M. Trost, I. Fleming (Eds.), Vol. 6, Pergamon Press, New York (1992); (b) J. Otera. In Esterification, Wiley-VCH, Weinheim (2003); (c) J. Otera. Chem. Rev. 93, 1449 (1993); (d) R. C. Larock. In Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, New York (1999); (e) M. B. Smith. In Compendium of Organic Synthetic Methods, Vol. 9, p. 100, John Wiley, New York (2001); (f) H. E. Hoydonckx, D. E. De Vos, S. A. Chavan, P. A. Jacobs. Top. Catal. 27, 83 (2004); (g) G. A. Grasa, R. Singh, S. P. Nolan. Synthesis 971 (2004); (h) C. A. G. N. Montalbetti, V. Falque, M. Park. Tetrahedron 61, 10827 (2005).Suche in Google Scholar
[2] For reviews, see: (a) M. Nahmany, A. Melman. Org. Biomol. Chem. 2, 1563 (2004); (b) N. A. Afagh, A. K. Yudin. Angew. Chem., Int. Ed. 49, 262 (2010); (c) T. E. Kristensen, T. Hansen. Eur. J. Org. Chem. 3179 (2010); (d) J. Mahatthananchai, A. M. Dumas, J. W. Bode. Angew. Chem., Int. Ed. 51, 10954 (2012); (e) D. J. Trader, E. E. Carlson. Mol. BioSyst. 8, 2484 (2012).Suche in Google Scholar
[3] Y. Tachibana, H. Kawasaki, N. Kihara, T. Takata. J. Org. Chem. 71, 5093 (2006).Suche in Google Scholar
[4] H. Morawetz, E. J. Sammak. J. Phys. Chem. 61, 1357 (1957).Suche in Google Scholar
[5] S. Nagaoka, A. Shundo, T. Satoh, K. Nagira, R. Kishi, K. Ueno, K. Iio, H. Ihara. Synth. Commun. 35, 2529 (2005).Suche in Google Scholar
[6] B. R. Brown, J. Cocker. J. Chem. Res., Synop. 2, 46 (1984).Suche in Google Scholar
[7] T. Mukaiama, F.-C. Pai, M. Onaka, K. Narasaka. Chem. Lett. 9, 563 (1980).Suche in Google Scholar
[8] S. D. Sarkar, S. Grimme, A. Studer. J. Am. Chem. Soc. 132, 1190 (2010).Suche in Google Scholar
[9] J. H. Maeng, R. L. Funk. Org. Lett. 4, 331 (2002).Suche in Google Scholar
[10] M. Marchini, M. Mingozzi, R. Colombo, C. Gennari, M. Durini, U. Piarulli. Tetrahedron 66, 9528 (2010).10.1016/j.tet.2010.10.007Suche in Google Scholar
[11] L. Gardossi, D. Bianchi, A. M. Klibanov. J. Am. Chem. Soc. 113, 6328 (1991).Suche in Google Scholar
[12] M. Cammenberg, K. Hult, S. Park. Chem. Bio. Chem. 7, 1745 (2006).Suche in Google Scholar
[13] M.-H. Lin, T. V. RajanBabu. Org. Lett. 2, 997 (2000).Suche in Google Scholar
[14] (a) M. Hatano, Y. Furuya, T. Shimmura, K. Moriyama, S. Kamiya, T. Maki, K. Ishihara. Org. Lett. 13, 426 (2011); (b) M. Hatano, S. Kamiya, K. Moriyama, K. Ishihara. Org. Lett. 13, 430 (2011); (c) M. Hatano, S. Kamiya, K. Ishihara. Chem. Commun. 48, 9465 (2012).Suche in Google Scholar
[15] (a) T. Ohshima, T. Iwasaki, K. Mashima. Chem. Commun. 2711 (2006); (b) T. Ohshima, T. Iwasaki, Y. Maegawa, A. Yoshiyama, K. Mashima. J. Am. Chem. Soc. 130, 2944 (2008); (c) T. Iwasaki, Y. Maegawa, Y. Hayashi, T. Ohshima, K. Mashima. J. Org. Chem. 73, 5147 (2008); (d) T. Iwasaki, Y. Maegawa, Y. Hayashi, T. Ohshima, K. Mashima. Synlett 1659 (2009); (e) T. Iwasaki, K. Agura, Y. Maegawa, Y. Hayashi, T. Ohshima, K. Mashima. Chem.—Eur. J. 16, 11567 (2010); (f) Y. Hayashi, T. Ohshima, Y. Fujii, Y. Matsushima, K. Mashima. Catal. Sci. Technol. 1, 230 (2011); (g) Y. Maegawa, K. Agura, Y. Hayashi, T. Ohshima, K. Mashima. Synlett 23, 137 (2012); (h) Y. Maegawa, T. Ohshima, Y. Hayashi, K. Agura, T. Iwasaki, K. Mashima. ACS Catal. 1, 1178 (2011); (i) Y. Hayashi, S. Santoro, Y. Azuma, F. Himo, T. Ohshima, K. Mashima. J. Am. Chem. Soc. 135, 6192 (2013).Suche in Google Scholar
[16] A. A. Sidorov, I. G. Fomina, M. O. Ponina, G. G. Aleksandrov, S. E. Nefedov, I. L. Eremenko, I. I. Moiseev. Russ. Chem. Bull., Int. Ed. 49, 958 (2000).Suche in Google Scholar
Article note
A collection of invited papers based on presentations at the 17th International IUPAC Conference on Organometallic Chemistry Directed Towards Organic Synthesis (OMCOS-17), Fort Collins, Colorado, USA, 28 July–1 August 2013.
©2014 by IUPAC & De Gruyter
Artikel in diesem Heft
- Masthead
- Masthead
- Conference papers
- International Union of Pure and Applied Chemistry
- Enantioselective palladium(0)-catalyzed C–H arylation strategy for chiral heterocycles
- Organometallic catalysis for applications in radical chemistry and asymmetric synthesis
- Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
- Copper-catalyzed aminoboration and hydroamination of alkenes with electrophilic amination reagents
- Activation of stable σ-bonds for organic synthesis
- Pd-catalyzed reactions of unactivated Csp3−I bonds
- Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
- Nickel-catalyzed oxidative cross-coupling of arylboronic acids with olefins
- Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents
- Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
- Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions
- Beyond C–H and C–O activation: the evolution of components in cross-coupling reactions
- The effect of acceptor-substituted alkynes in gold-catalyzed intermolecular reactions
- Chemoselective silver-catalyzed nitrene insertion reactions
- The strategic generation and interception of palladium-hydrides for use in alkene functionalization reactions
- 3-Acyloxy-1,4-enyne: A new five-carbon synthon for rhodium-catalyzed [5 + 2] cycloadditions
- Cobalt-catalyzed directed alkylation of arenes with primary and secondary alkyl halides
- IUPAC Technical Report
- Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)
Artikel in diesem Heft
- Masthead
- Masthead
- Conference papers
- International Union of Pure and Applied Chemistry
- Enantioselective palladium(0)-catalyzed C–H arylation strategy for chiral heterocycles
- Organometallic catalysis for applications in radical chemistry and asymmetric synthesis
- Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
- Copper-catalyzed aminoboration and hydroamination of alkenes with electrophilic amination reagents
- Activation of stable σ-bonds for organic synthesis
- Pd-catalyzed reactions of unactivated Csp3−I bonds
- Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
- Nickel-catalyzed oxidative cross-coupling of arylboronic acids with olefins
- Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents
- Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
- Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions
- Beyond C–H and C–O activation: the evolution of components in cross-coupling reactions
- The effect of acceptor-substituted alkynes in gold-catalyzed intermolecular reactions
- Chemoselective silver-catalyzed nitrene insertion reactions
- The strategic generation and interception of palladium-hydrides for use in alkene functionalization reactions
- 3-Acyloxy-1,4-enyne: A new five-carbon synthon for rhodium-catalyzed [5 + 2] cycloadditions
- Cobalt-catalyzed directed alkylation of arenes with primary and secondary alkyl halides
- IUPAC Technical Report
- Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)