Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
-
 ,
,

Abstract
The rhenium-catalyzed reaction of terminal alkynes with imines gives N-alkylideneallylamine derivatives. A diphenylmethyl group as the substituent on the imine nitrogen gave the best result. Deuterium labeling experiments revealed that the regioselective addition of both the hydrogen and the N-alkylideneaminoalkyl group to the terminal alkynes also proceeded stereoselectively. While alkynes bearing primary and secondary alkyl-, vinyl-, and aryl groups were applicable to the catalytic reaction, tertiary alkyl- and silyl-substituted alkynes gave propargylamines. The C–C bond-forming step via the nucleophilic attack of the alkynyl β-carbon on the imine carbon leading to the formation of a vinylidene rhenium species appears to be involved in the catalytic cycle.
Introduction
Transition-metal alkynyl complexes have been widely studied not only in the area of organic synthesis [1] but also in materials science [2]. These complexes are typically prepared by the treatment of terminal alkynes with a stoichiometric amount of a strong base such as organolithium and Grignard reagents and the subsequent addition of transition-metal halides. In some cases, transition-metal halides are directly reacted with terminal alkynes in the presence of a base, including tertiary amines and inorganic carbonates, to afford a complex. The oxidative addition of an alkyne C(sp)–H bond to transition metals is also possible. Alkynylmetal complexes can react with a variety of electrophiles at the alkynyl α-carbon to give new acetylenic compounds (path a in Fig. 1). For example, imines have been widely studied as an electrophile in the synthesis of propargylamines, which was recently developed as a catalytic reaction [3]. The reaction proceeds through the nucleophilic addition of the alkynyl α-carbon to the imine carbon, leading to the formation of propargylaminometal species. On the other hand, electrophiles can also add to the alkynyl β-carbon to give vinylidene metal complexes (path b) [4, 5]. Some catalytic reactions involving the addition of electrophiles to the alkynyl β-carbon in the catalytic cycle have recently been reported, as shown in Scheme 1. The first catalytic reaction involving a β-addition was the rhodium-catalyzed cyclization of 3-haloalkyl-1,6-enynes to give fused bicyclic compounds reported by Lee (eq. 1) [6]. The catalytic cycle appears to involve the formation of an alkynylrhodium species followed by the intramolecular nucleophilic substitution of alkyl halides by the alkynyl β-carbon. In 2012, Aue and Zhang demonstrated the cyclization of 1,5-diynes catalyzed by a Au(I) complex, as shown in eq. 2 [7]. The proposed reaction mechanism involves the intramolecular nucleophilic attack of the alkynyl β-carbon to gold-coordinated alkynes to give vinylidene gold complexes. Hashmi also independently studied similar inter- and intramolecular reactions [8]. After our report of the present catalytic reaction [9], the rhodium-catalyzed intermolecular [2 + 2]-cyclization of terminal alkynes with α,β-unsaturated electron-deficient alkenes via the conjugate addition of the alkynyl β-carbon was reported by Kakiuchi (eq. 3) [10].
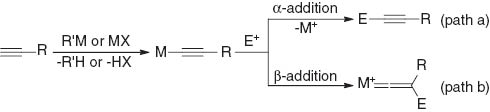
Reaction of alkynylmetal complexes with electrophiles.
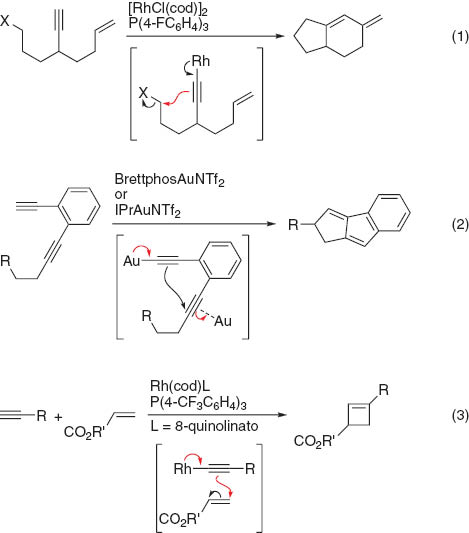
Examples of the catalytic reaction via the β-addition.
We report herein a new class of C–C bond formation reaction, namely, the rhenium-catalyzed reaction of terminal alkynes with imines to give N-alkylideneallylamine derivatives. The present reaction permits the regioselective preparation of N-protected 1,2-disubstituted allylamines. In mechanistic aspects, to the best of our knowledge, this is the first catalytic reaction that appears to involve the nucleophilic attack of the alkynyl β-carbon atom on an imine carbon to produce a vinylidene rhenium complex in the catalytic cycle (Scheme 2).
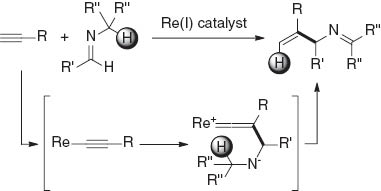
Rhenium-catalyzed reaction of terminal alkynes with imines leading to N-alkylideneallylamines.
Screening of the reaction conditions
In an initial experiment, 1-octyne was reacted with 1,3,5-trisbenzyl-hexahydro-1,3,5-triazine, with the latter having undergone thermolysis to give N-benzylimine [11], in the presence of ReBr(CO)5 to afford N-benzylidene-2-methylidene-1-amine in 26 % yield (Scheme 3). No evidence of the formation of the corresponding propargylamine was observed, unlike the catalytic alkynylation of N-phenyl imines using ReBr(CO)2(THF)3, reported by Kuninobu and Takai [12]. A series of imines derived from formaldehyde were next examined. The results revealed that the substituent on the imine was important in terms of the efficiency of the reaction. Whereas no reaction occurred when N-butyl-substituted imine was used, N-diphenylmethyl imine gave the best result yield (66 %). N-Phenyl imine, which has no hydrogen that can migrate, reacted with 1-octyne to afford the propargylamine in low yield along with the unreacted alkyne being recovered.
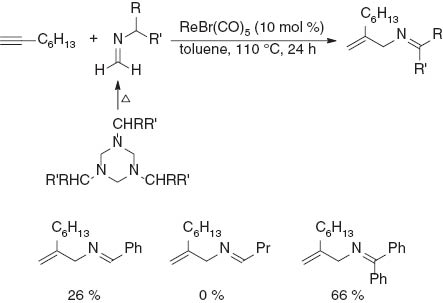
Screening of imines.
We next examined the use of various phosphines as an additive in the ReBr(CO)5-catalyzed reaction of 1-octyne with N-diphenylmethylimine derived from formaldehyde (Scheme 4). When phosphines bearing electron-withdrawing groups on the phenyl ring such as P(4-CF3C6H4)5 and P(C6F5)3 were used, the product yield was improved to 77 %. The product was isolated by chromatography on a NH2-modified silica-gel column in 73 % yield in the reaction using P(C6F5)3. However, the addition of P(OPh)3 retarded the reaction, and a moderate amount of unreacted 1-octyne remained. The screening of rhenium complexes with revealed that Re(I) complexes such as ReCl(CO)5 (70 % yield) and ReBr(CO)3(thf)2 (71 % yield) resulted in a catalytic activity comparable to that for ReBr(CO)5, but Re2(CO)10, as a Re(0) complex, showed no catalytic activity. Complexes such as CpRuCl(PPh3)2, RhCl(PPh3)3, and AuCl(PPh3), which are frequently used in the catalytic reactions involving vinylidene metal complexes in the catalytic cycle, were also ineffective in the present reaction.
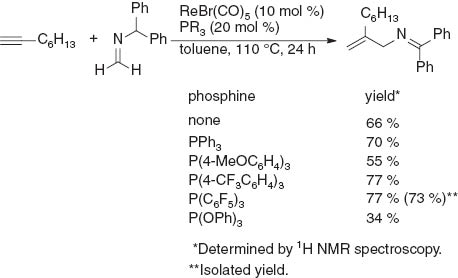
Screening of phosphines.
Scope and limitations
The scope of the present regioselective reaction of N-methylene-1,1-diphenylmethanamine with a variety of terminal alkynes was investigated (Scheme 5). Cyclohexylacetylene and 3-pentyloct-1-yne reacted to give the corresponding allylamines in 72 and 66 % isolated yields, respectively. Functional groups including an ester, a nitrile, THP, a siloxy group, and an alkene were tolerated in the reaction. Phenylacetylene was also applicable to the reaction, but when the reaction was carried out at 110 °C, the product yield was 58 % yield along with oligomerization products derived from the starting alkyne. The yield was improved to 67 % when the reaction temperature was lowered to 80 °C. No evidence of the formation of propargylamines was found in any of the runs. However, when terminal alkynes bearing sterically demanding groups, such as tert-butyl and trimethylsilyl groups, were reacted under the standard reaction conditions, propargylamines were produced as the sole products in 70 and 87 % yields, respectively. No reactions occurred when internal acetylenes such as 4-octyne and diphenylacetylene were tested.
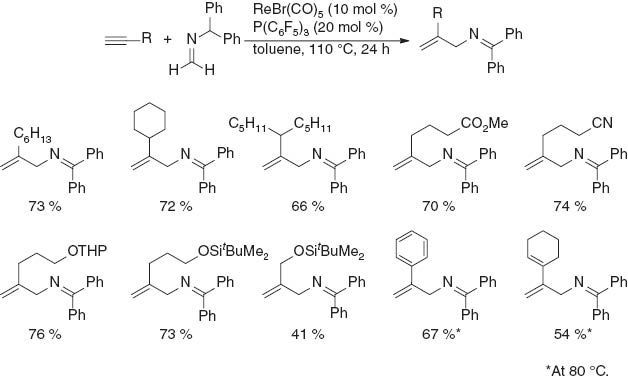
Reaction of various terminal alkynes with N-methylene-1,1-diphenylmethanamine.
The reaction of 1-octyne was expanded to include some C-substituted N-diphenylmethylimines, as shown in Scheme 6. The imine derived from acetaldehyde was treated under the aforementioned reaction conditions to give the 1-methyl-substituted allylamine in 62 % yield, and in 66 % yield when the reaction time was extended to 48 h with 1-octyne being recovered in ca. 10 % yield. Therefore, we reinvestigated the reaction conditions and found that, when the catalyst loading was increased to 15 mol % and the reaction time was extended to 48 h, the product yield was improved to 73 % with complete consumption of the starting alkyne. These modified reaction conditions were also used in the reactions of imines. The reactivity of pentyl-substituted imine was similar to that of the methyl-substituted imine. The reaction of the phenyl-substituted imine resulted in the formation of the desired product along with some unidentified by-products. No conversion was observed when the imine derived from acetone was used in the reaction.
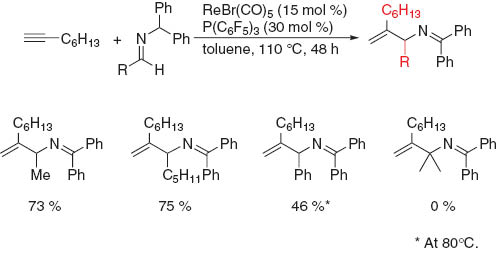
Reaction of 1-octyne with some C-substituted imines.
Mechanistic studies
A plausible reaction mechanism for the reaction is depicted in Scheme 7. The reaction of a rhenium complex I with a terminal alkyne gives the alkynyl rhenium species II with the elimination of HX. The subsequent nucleophilic attack of the β-carbon of the rhenium alkynylide on an imine carbon then occurs to form the vinylidene rhenium species III [13]. A 1,5-shift of the hydrogen atom adjacent to the nitrogen atom to the α-carbon atom of the vinylidene moiety gives the vinyl rhenium species IV. The final protonolysis of IV affords the product with the regeneration of I. Oshima and Yorimitsu reported the stoichiometric reaction of an alkynyliron complex with imines in the presence of BF3‧OEt2 to give α,β-unsaturated iminoiron complexes, which includes the β-addition of an alkynyl ligand to an imine carbon in the reaction mechanism [14]. A related 1,5-hydrogen shift to the vinylidene α-carbon is proposed in the catalytic cyclization of o-alkyl-substituted phenylacetylenes [15] and some stoichiometric reactions [16]. The α-phenyl group on the imine substituent would assist the intramolecular migration of hydrogen in III by stabilizing the partial cationic character of the carbon, to which the hydrogen is attached. Bulky tert-butyl and trimethylsilyl groups would inhibit the nucleophilic attack of the alkynyl β-carbon atom to the imine carbon; therefore, an attack at the α-carbon atom proceeds to afford propargylamines when tert-butylacetylene and trimethylsilylacetylene were used.
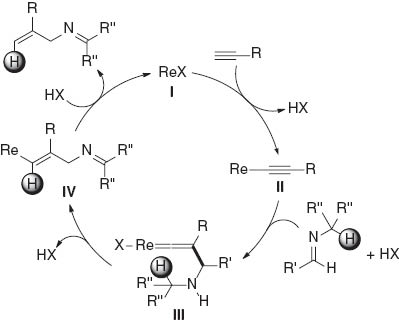
Plausible reaction mechanism.
To gain further insight into the reaction mechanism, deuterium labeling experiments were carried out, as shown in Scheme 8. The reaction of 1-octyne with the imine labeled with deuterium at the α-position of the diphenylmethyl group was examined (eq. 4). As expected, the deuterium completely shifted to the vinylic position in a cis configuration to the amino methyl group in the product. On the other hand, the reaction of 1-deuterio-1-octyne with the imine also gave the trans-labeled product with good stereoselectivity. However, a moderate deuterium ratio of 53 % D was observed, which can be attributed to rapid proton exchange with the trace H2O in the reaction system. Therefore, the intentional addition of D2O (6 mmol) to the reaction mixture improved the H/D ratio to 90 % D (eq. 5). These results are consistent with the proposed reaction mechanism and demonstrate that the present reaction can be used to stereoselectively produce both E- and Z-monodeuterated allylamines. Other mechanisms, for example, via the formation of a metallacycle by the oxidative coupling of terminal alkynes, imines, and rhenium species [17, 18], cannot be ruled out. However, such a mechanism does not account for the regioselectivity of the products observed here.
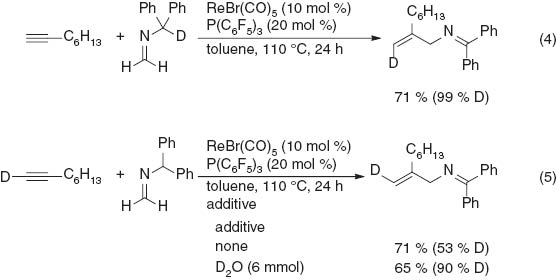
Deuterium labeling experiments.
Conclusions
In summary, the rhenium-catalyzed reaction of terminal alkynes with imines to afford allylamine derivatives is demonstrated. A diphenylmethyl group as the substituent on the imine nitrogen gave the best result. Deuterium labeling experiments revealed that the regioselective addition of hydrogen and the N-alkylideneaminoalkyl group to the terminal alkynes also proceeds stereoselectively. The C–C bond-forming step that occurs via the nucleophilic attack of the alkynyl β-carbon on the imine carbon leading to the formation of a vinylidene rhenium species appears to be involved in the catalytic cycle.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (No. 22550100). Thanks are given to the Instrumental Analysis Center, Faculty of Engineering, Osaka University, for Assistance with HRMS and elemental analyses.
References
[1] Reviews, see: (a) S. Fujimori, T. F. Knöpfel, P. Zarotti, T. Ichikawa, D. Boyall, E. M. Carreira. Bull. Chem. Soc. Jpn. 80, 1635 (2007); (b) B. M. Trost, A. H. Weiss. Adv. Synth. Catal. 351, 963 (2009); (c) C.-J. Li. Acc. Chem. Res.43, 581 (2010).Search in Google Scholar
[2] Recent reviews, see: (a) W.-Y. Wong, P. D. Harvey. Macromol. Rapid Commun.31, 671 (2010); (b) G.-J. Zhou, W.-Y. Wong. Chem. Soc. Rev. 40, 2541 (2011); (c) J. C. Lima, L. Rodríguez. Chem. Soc. Rev. 40, 5442 (2011); (d) A. S. Abd-El-Aziza, E. A. Strohmb. Polymer53, 4879 (2012).Search in Google Scholar
[3] Reviews, see: (a) P. G. Cozzi, R. Hilgraf, N. Zimmermann. Eur. J. Org. Chem. 4095 (2004); (b) C. Wei, Z. Li, C.-J. Li. Synlett5, 1472 (2004); (c) L. Zani, C. Bolm. Chem. Commun. 42, 4263 (2006); (d) W.-J. Yoo, L. Zhao, C.-J. Li. Aldrichim. Acta44, 43 (2011); (e) V. A. Peshkov, O. P. Pereshivko, E. V. Van der Eycken. Chem. Soc. Rev. 41, 3790 (2012). See also refs. [1a–c].Search in Google Scholar
[4] For reviews which contain the description of the addition of electrophiles to alkynyl β-carbons to give vinylidene metal complexes, see: (a) S. G. Davies, J. P. McNally, A. J. Smallridge. Adv. Organomet. Chem. 30, 1 (1990); (b) V. Cadierno, M. P. Gamasa, J. Gimeno. Coord. Chem. Rev. 248, 1627 (2004).Search in Google Scholar
[5] For recent reviews on the catalytic reactions involving vinylidene metal complexes, see: (a) C. Bruneau, P. H. Dixneuf (Eds.). Metal Vinylidenes and Allenylidenes in Catalysis, pp. 159–332, Wiley-VCH, Weinheim (2008); (b) B. M. Trost, A. McClory. Chem. Asian J.3, 164 (2008).Search in Google Scholar
[6] J. M. Joo, Y. Yuan, C. Lee. J. Am. Chem. Soc.128, 14818 (2006).Search in Google Scholar
[7] L. Ye, Y. Wang, D. H. Aue, L. Zhang. J. Am. Chem. Soc.134, 31 (2012).Search in Google Scholar
[8] (a) A. S. K. Hashmi, I. Braun, M. Rudolph, F. Rominger. Organometallics31, 644 (2012); (b) A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nösel, L. Jongbloed, M. Rudolph, F. Rominger. Adv. Synth. Catal. 354, 555 (2012); (c) A. S. K. Hashmi, I. Braun, P. Nösel, J. Schädlich, M. Wieteck, M. Rudolph, F. Rominger. Angew. Chem., Int. Ed. 51, 4456 (2012); (d) A. S. K. Hashmi, M. Wieteck, I. Braun, M. Rudolph, F. Rominger. Angew. Chem., Int. Ed.51, 10633 (2012); (e) A. S. K. Hashmi, T. Lauterbach, P. Nösel, M. H. Vilhelmsen, M. Rudolph, F. Rominger. Chem.—Eur. J.19, 1058 (2013); (f) M. M. Hansmann, M. Rudolph, F. Rominger, A. S. K. Hashmi. Angew. Chem., Int. Ed.52, 2593 (2013).Search in Google Scholar
[9] Y. Fukumoto, M. Daijo, N. Chatani. J. Am. Chem. Soc.134, 8762 (2012).Search in Google Scholar
[10] K. Sakai, T. Kochi, F. Kakiuchi. Org. Lett.15, 1024 (2013).Search in Google Scholar
[11] For a recent example of the use of cyclic triamines as the precursor of formaldehyde imines, see: T. Ma, X. Fu, C. W. Kee, L. Zong, Y. Pan, K.-W. Huang, C.-H. Tan. J. Am. Chem. Soc.133, 2828 (2011).10.1021/ja1098353Search in Google Scholar PubMed
[12] Y. Kuninobu, Y. Inoue, K. Takai. Chem. Lett. 35, 1376 (2006).Search in Google Scholar
[13] For examples where the formation of a vinylidene rhenium complex is proposed as a key intermediate in catalytic reactions, see: (a) S. S. Yudha, Y. Kuninobu, K. Takai. Org. Lett.9, 5609 (2007); (b) D. Xia, Y. Wang, Z. Du, Q.-Y. Zheng, C. Wang. Org. Lett.14, 588 (2012).Search in Google Scholar
[14] R. Nakaya, S. Yasuda, H. Yorimitsu, K. Oshima. Chem.—Eur. J. 17, 8559 (2011).Search in Google Scholar
[15] For examples of the 1,5-hydrogen shift to the vinylidene β-carbon, see: (a) S. Datta, A. Odedra, R.-S. Liu. J. Am. Chem. Soc. 127, 11606 (2005); (b) G. B. Bajracharya, N. K. Pahadi, I. D. Gridnev, Y. Yamamoto. J. Org. Chem.71, 6204 (2006); (c) A. Odedra, S. Datta, R.-S. Liu. J. Org. Chem. 72, 3289 (2007); (d) M. Tobisu, H. Nakai, N. Chatani. J. Org. Chem.74, 5471 (2009).Search in Google Scholar
[16] (a) J. Ipaktschi, J. Mohsseni-Ala, A. Dümer, C. Loschen, G. Frenking. Organometallics24, 977 (2005); (b) I. Hyder, M. Jiménez-Tenorio, M. C. Puerta, P. Valerga. Organometallics30, 726 (2011).Search in Google Scholar
[17] Nickel-catalyzed alkylative and/or reductive coupling of internal alkynes and imines via the oxidative cyclization was reported, see: (a) S. J. Patel, T. F. Jamison. Angew. Chem., Int. Ed.42, 1364 (2003); (b) S. J. Patel, T .F. Jamison. Angew. Chem., Int. Ed.43, 3941 (2004); (c) Z.-Y. Zhou, S.-F. Zhu, L.-X. Wang, Q.-L. Zhou. J. Am. Chem. Soc. 132, 10955 (2010).Search in Google Scholar
[18] It has been reported that the oxidative coupling of terminal alkynes, imines, and Cp2Zr or (iPrO)2Zr species, followed by hydrolysis, gives allylamines having substituents derived from terminal alkynes on the alkenyl terminal carbon, see: (a) Y. Gao, Y. Yoshida, F. Sato. Synlett8, 1353 (1997); (b) K. Fukuhara, S. Okamoto, F. Sato. Org. Lett. 5, 2145 (2003); (c) C. Dehhez, S. Médégan, F. Hélion, J.-L. Namy, J.-L. Vasse, J. Szymoniak. Org. Lett.8, 2945 (2006); (d) K. Mao, G. Fan, Y. Liu, S. Li, X. You, D. Liu. Beilstein J. Org. Chem.9, 621 (2013).Search in Google Scholar
Article note
A collection of invited papers based on presentations at the 17th International IUPAC Conference on Organometallic Chemistry Directed Towards Organic Synthesis (OMCOS-17), Fort Collins, Colorado, USA, 28 July–1 August 2013.
©2014 by IUPAC & De Gruyter
Articles in the same Issue
- Masthead
- Masthead
- Conference papers
- International Union of Pure and Applied Chemistry
- Enantioselective palladium(0)-catalyzed C–H arylation strategy for chiral heterocycles
- Organometallic catalysis for applications in radical chemistry and asymmetric synthesis
- Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
- Copper-catalyzed aminoboration and hydroamination of alkenes with electrophilic amination reagents
- Activation of stable σ-bonds for organic synthesis
- Pd-catalyzed reactions of unactivated Csp3−I bonds
- Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
- Nickel-catalyzed oxidative cross-coupling of arylboronic acids with olefins
- Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents
- Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
- Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions
- Beyond C–H and C–O activation: the evolution of components in cross-coupling reactions
- The effect of acceptor-substituted alkynes in gold-catalyzed intermolecular reactions
- Chemoselective silver-catalyzed nitrene insertion reactions
- The strategic generation and interception of palladium-hydrides for use in alkene functionalization reactions
- 3-Acyloxy-1,4-enyne: A new five-carbon synthon for rhodium-catalyzed [5 + 2] cycloadditions
- Cobalt-catalyzed directed alkylation of arenes with primary and secondary alkyl halides
- IUPAC Technical Report
- Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)
Articles in the same Issue
- Masthead
- Masthead
- Conference papers
- International Union of Pure and Applied Chemistry
- Enantioselective palladium(0)-catalyzed C–H arylation strategy for chiral heterocycles
- Organometallic catalysis for applications in radical chemistry and asymmetric synthesis
- Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
- Copper-catalyzed aminoboration and hydroamination of alkenes with electrophilic amination reagents
- Activation of stable σ-bonds for organic synthesis
- Pd-catalyzed reactions of unactivated Csp3−I bonds
- Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
- Nickel-catalyzed oxidative cross-coupling of arylboronic acids with olefins
- Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents
- Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
- Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions
- Beyond C–H and C–O activation: the evolution of components in cross-coupling reactions
- The effect of acceptor-substituted alkynes in gold-catalyzed intermolecular reactions
- Chemoselective silver-catalyzed nitrene insertion reactions
- The strategic generation and interception of palladium-hydrides for use in alkene functionalization reactions
- 3-Acyloxy-1,4-enyne: A new five-carbon synthon for rhodium-catalyzed [5 + 2] cycloadditions
- Cobalt-catalyzed directed alkylation of arenes with primary and secondary alkyl halides
- IUPAC Technical Report
- Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)