Activation of stable σ-bonds for organic synthesis
-
Tamejiro Hiyama


Abstract
Since the conference on Organometallic Chemisrty Directed Towards Organic Synthesis (OMCOS) started in 1981, the author has been involved in the invention of novel synthetic reactions for C–C bond formation, taking advantage of organometallic reagents and catalysis. Herein is described a guideline story of how it is done.
Introduction
At the time when the conference on Organometallic Chemistry Directed Towards Organis Synthsisi (OMCOS) started in 1981, Prof. Hitosi Nozaki, my senior collaborator, and I discussed the possibility of generation of a system that involves carbocation and anion before making C–C bonds, namely, the reaction of allylic (or cyclopropylmethyl) acetates with triorganoaluminum. For example [1], the following reactions (Scheme 1) show cyclopropylmethyl cations and alanates are produced and then coupling of these takes place to give alkyation products. Thus, I became interested in the new transformation through heterolysis of C–C bond into carbocationic and carbanionic fragments. This simple retrosynthetic analysis made me examine reaction mechanisms in the simplest way.
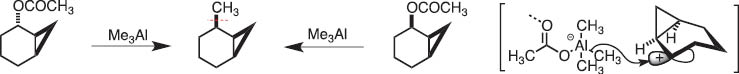
Methylation of norcarenyl acetate with trimethylaluminum.
I immediately proposed that a system derived from hexamethyldisilane and fluoride anion should give silyl anions and silyl cations. This idea turned out to be true and we could invent new reactions, which proceed through insertion of aldehyde carbonyls and 1,3-dienes between Si–Si bonds (Scheme 2) [2].
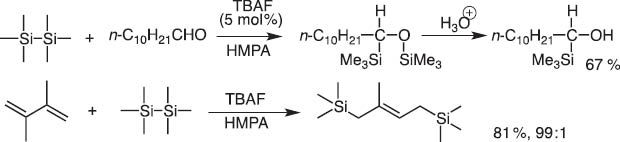
TBAF-catalyzed addition of hexamethyldisilane to aldehyde and 1,3-diene.
We soon extended the concept of the disilane/fluoride ion reagent system to hydrosilane/fluoride ion and disclosed that pentacoordinate silicates derived from hydrosilanes reduce carbonyls highly stereoselectively (Schemes 3 and 4) [3].
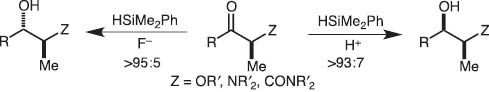
Stereodivergent reduction of ketones with hydrosilane mediated by fluoride ion or proton.
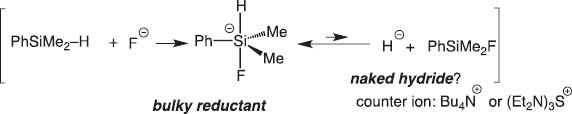
Reaction of fluoride ion with hydrosilane.
Silicon-based cross-coupling
Further extension of the concept organosilane/fluoride ion to transmetalation to palladium catalysts led to the discovery of silicon-based cross-coupling reaction (Scheme 5) [4]. We worried about the simple nucleophilic attack of the fluoride ion on the palladium complex. Luckily, this did not take place. The protocol required the use of fluoride nucleophilic activator and the presence of one to three heteroatoms like fluorine or oxygen on silicon to assist formation of penta-coordinate silicates responsible for the transmetalation to palladium catalyst through a 4-membered cyclic transition state [5].
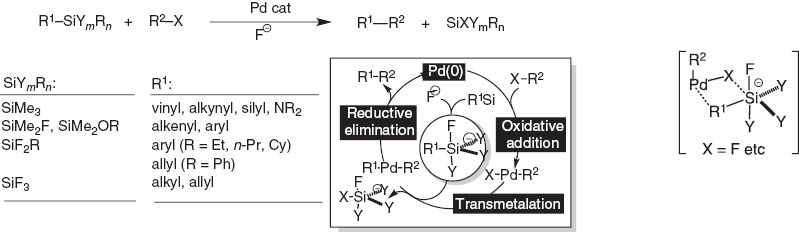
Summary and mechanism of silicon-based cross-coupling reaction.
These limitations made the cross-coupling with halo- or alkyoxysilane reagents somewhat less accessible, and we and others developed numerous modifications. Nevertheless, the protocol is recently shown applicable to C–N coupling under mild reaction conditions useful for the synthesis of triarylamines in a straightforward manner under conditions milder than those of Hartwig–Buchwald coupling (Scheme 6) [6]. Polycondensation to give polycarbazole is also achieved.
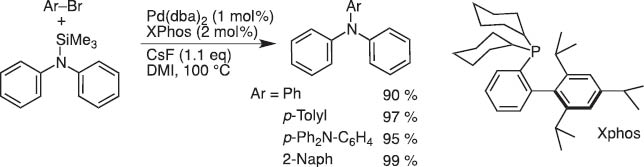
C–N coupling with N-silylamine.
In 2004, we invented dimethyl(o-hydroxymethylphenyl)silane (HOMSi) reagents (abbreviated Si that are stable and tolerate chromatography on silica). Protected Si reagents are cross-coupling inactive; however, they become active upon orthogonal hydroxyl deprotection (Scheme 7). After the reaction, the silicon moiety in HOMSi reagents are converted to a cyclic silyl ether which is readily recovered quantitatively and transformed back to HOMSi reagents (Scheme 8) [7]. This new version of silicon-based cross-coupling reaction is shown to be applicable to polycondensation polymerization to give organic light-emitting materials under mild conditions [8].
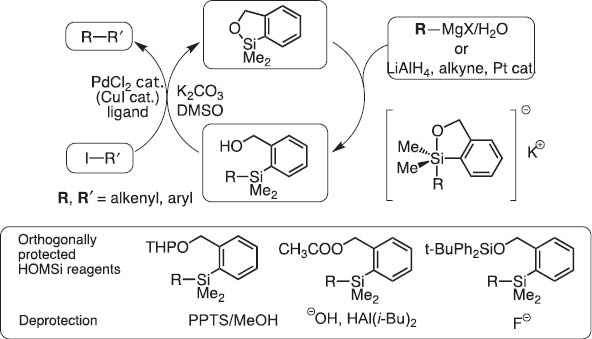
Homsi reagents for silicon-based cross-coupling reaction.
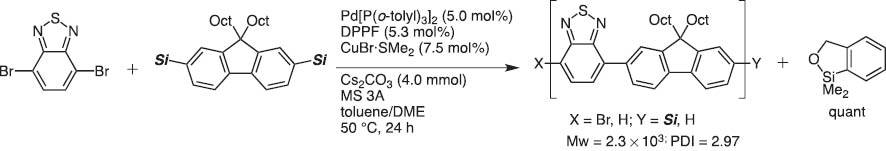
Polymer synthesis with bisHOMSi reagent.
Conjugate addition of Homsi reagents
Transmetalation from Si to Rh also proceeds smoothly to effect conjugate addition of HOMSi reagents (Scheme 9). In collaboration with Prof. Tamio Hayashi, we could show the high synthetic potential in asymmetric conjugate addition to α,β-unsaturated carbonyl substrates [9].
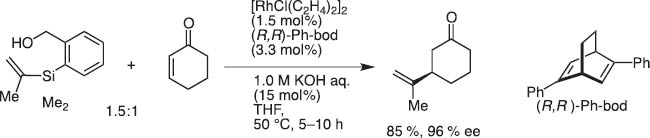
Rhodium-catalyzed conjugate addition of HOMSi reagent.
Carbostannylation
When I returned to Kyoto University as a full professor in 1997, Dr. Eiji Shirakawa was studying the mechanism of the Kosugi–Migita–Stille coupling and proposed a new catalytic cycle which should involve oxidative addition of Pd(0) to a C–Sn bond followed by ligand exchange with organic iodides. I could not immediately believe his new mechanism and thus suggested, if the mechanism were correct, unsaturated bonds might insert between either of the C–Pd–Sn bonds to give, after reductive elimination, cis-adducts of both C and Sn. To our delight, this working hypothesis turned out to be correct and thus we could invent the catalytic carbostannylation reaction of alkenes and dienes (Schemes 10 and 11) [10].

Carbostannylation of alkynes, 1,3-dienes, and allenes.
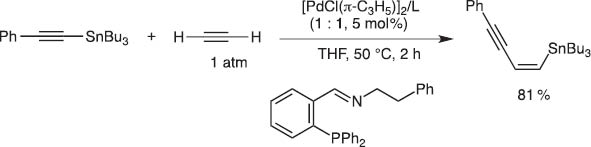
Carbostannylation of acetylene with alkynylstannane.
Carbocyanation
In 2002 Dr. Yoshiaki Nakao and I discussed the synthetic potential of oxidative addition of Ni(0) to the C–CN bond. We immediately ran the reaction in the presence of internal alkynes as an acceptor, as we did for carbostannylation [11]. This indeed worked well, and we could invent the carbocyanation reaction of alkynes and alkenes (Scheme 12). However, the catalytic efficiency was unsatisfactory at the beginning.
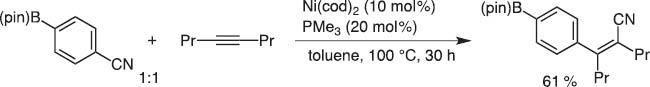
First carbocyanation reaction.
Later we were pleased to find that moderately Lewis acidic organoaluminum, -boron, or -zinc reagents accelerated the Ni(0) catalysis to invent truly catalytic C–CN bond activation [12]. Aryl, allyl, alkenyl, and aryl cyanides, even methyl cyanide, i.e., acetonitrile, were found to undergo such reaction under the Ni(0)/Lewis acid catalysis (Scheme 13).
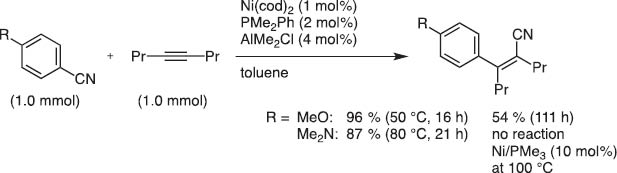
Arylcyanation of 4-octyne with a Ni/Al cocatalyst system.
Hydro(hetero)arylation
By chance, we observed the Ni(0)/Lewis acid system could activate particularly the >+N=C–H bond. This catalyst system led to the discovery of regioselective C–H bond activation of (hetero)aromatic rings and the hydro(hetero)arylation reaction with alkynes and alkenes [13].Typical and excellent examples are C4-selective normal alkylation of pyridines with terminal alkenes [14]and one carbon homologation of terminal alkenes with DMF (Scheme 14) [15]. The bulky tightly bound NHC-ligands separate the Al and Ni centers and Ni inserts the C(4)–H bond highly selectively as illustrated for the C4-selective pyridine n-alkylation with terminal alkenes (Scheme 15).
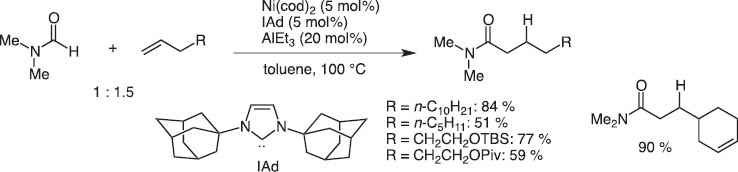
Hydrocarbamoylatin of terminal alkenes with DMF.
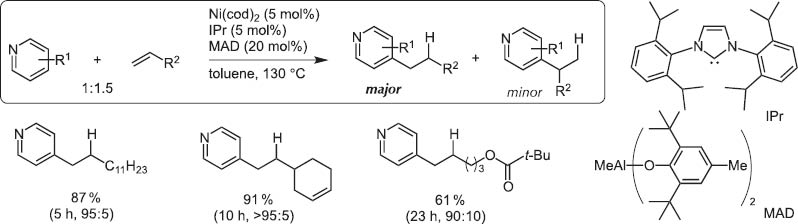
Normal alkylation of pyridines with terminal alkenes.
Hydroarylation of polyfluorobenzonitrile with inner alkynes is possible after arylcyanation (Schemes 16 and 17). Thus, extension of the π-conjugate system is readily achieved by the stepwise activation of first C–CN bond and then C–H bond [16]. The intermediate oxidative adduct of Ni is identified by X-ray crystallographic analysis.
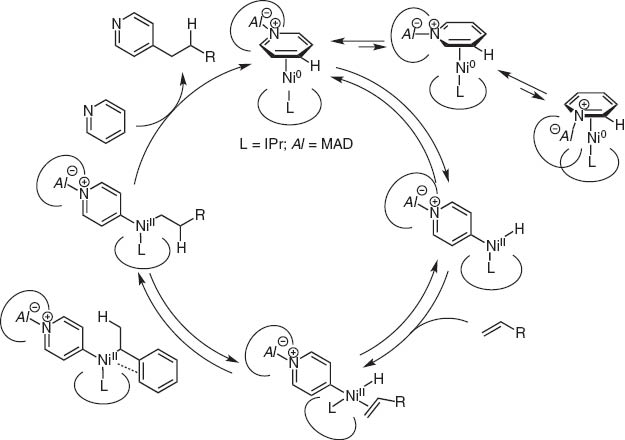
Mechanism of nickel/aluminum catalyzed C4-selective alkylation of pyridines.
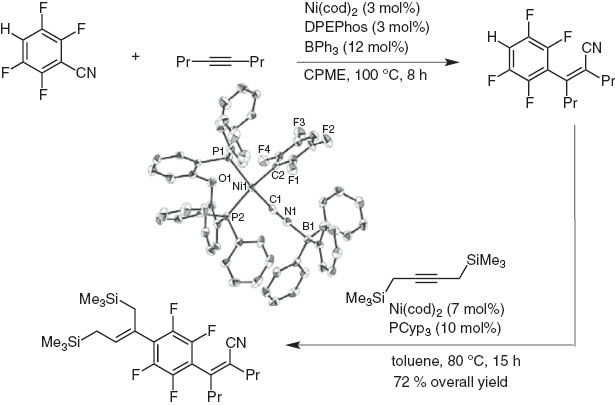
Iterative carbocyanation and hydroarylation of internal alkynes.
C–H activation followed by cycloaddition
Since 2010, Dr. Yasunori Minami and I are studying in Chuo University to cleave either C–O bond in Ph–O–C≡CSi(i-Pr)3 with low-valent metal catalysts. Instead we have observed the ortho-C–H bond activation by a Pd(II)/Zn system, the triple bond apparently working as a directing and accepting group (Scheme 18). In the presence of internal alkynes, a cycloaddition reaction took place to give 2-exo-methylenechromenes [17].
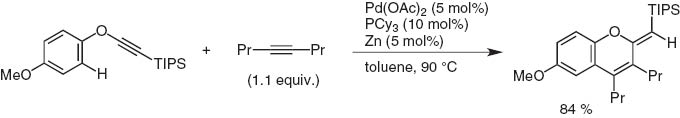
Cycloaddition of palladium-catalyzed aryl ethynyl ether with internal alkyne via ortho-C–H activation.
When mono-substituted propa-1,2-dienes are employed in lieu of internal alkynes, 2,3-bis(methylene)chromanes are produced. Taking advantage of the conjugate exo-methylenes, the Diels–Alder reaction of the cycloadducts with N-phenylmaleimide takes place smoothly to give tetracyclic structures with high stereoselectivity (Scheme 19) [18].
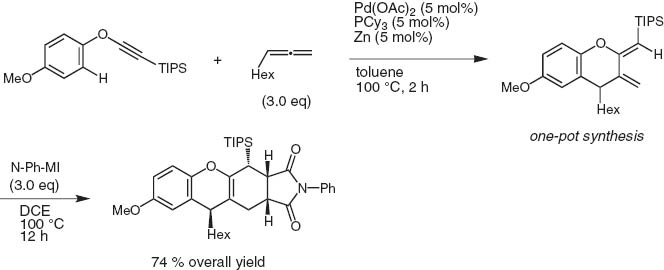
Cycloaddition of palladium-catalyzed aryl ethynyl ether with allene via ortho-C–H activation.
Upon treatment with Pd(II) and Zn catalyst system, o-tolyl TIPS-ethynyl ethers undergo ring closure to give 2-silylmethylene dihydrobenzofurans possibly through benzylic γ-C–H activation (Scheme 20) [19].
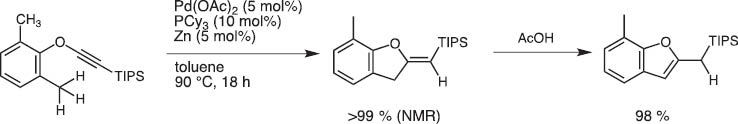
Benzofuran synthesis by palladium-catalyzed cyclization of ethynyl ortho-tolyl ether.
Similar reaction of biphenyl-2-yl TIPS-ethynyl ether gave 9-oxa-9,10-dihydrophenanthrenes, which are apparently derived from δ-C–H activation (Scheme 21) [20].

9-Oxaphenanthrene synthesis through δ-C–H activation by palladium catalyst.
Through the studies on the topics discussed herein, one will readily learn that the logical examination of the reaction mechanism is a shortcut to invention of novel and useful reactions for organic synthesis.
Acknowledgments
Financial supports by Kyoto University, Chuo University, JSPS, MEXT, and Sumitomo Chemicals and incomparable contributions by my coworkers are deeply appreciated.
References
[1] A. Itoh, S. Ozawa, K. Oshima, S. Sasaki, H. Yamamoto, T. Hiyama, H. Nozaki. Bull. Chem. Soc. Jpn. 53, 2357 (1980).Suche in Google Scholar
[2] T. Hiyama, M. Obayashi, I. Mori, H. Nozaki. J. Org. Chem. 48, 912 (1983).Suche in Google Scholar
[3] (a) M. Fujita, T. Hiyama. J. Am. Chem. Soc. 106, 4629 (1984); (b) M. Fujita, T. Hiyama. J. Am. Chem. Soc. 107, 8294 (1985); (c) M. Fujita, T. Hiyama. Tetrahedron Lett. 28, 2263 (1987); (d) M. Fujita, T. Hiyama. J. Org. Chem.53, 5405 (1988); (e) M. Fujita, T. Hiyama. J. Org. Chem.53, 5415 (1988); (f) M. Fujita, H. Oishi, T. Hiyama. Chem. Lett.15, 837 (1986).Suche in Google Scholar
[4] Y. Hatanaka, T. Hiyama. J. Org. Chem. 53, 918 (1988).Suche in Google Scholar
[5] A. Sugiyama, Y.-y. Ohnishi, M. Nakaoka, Y. Nakao, H. Sato, S. Sakaki, Y. Nakao, T. Hiyama. J. Am. Chem. Soc. 130, 12975 (2008).Suche in Google Scholar
[6] K. Shimizu, Y. Minami, Y. Nakao, K. Ohya, O. Goto, H. Ikehira, T. Hiyama. Chem. Lett. in press.Suche in Google Scholar
[7] (a) Y. Nakao, H. Imanaka, A. K. Sahoo, A. Yada, T. Hiyama. J. Am. Chem. Soc. 127, 6952 (2005); (b) Y. Nakao, A. K. Sahoo, H. Imanaka, A. Yada, T. Hiyama. Pure Appl. Chem. 78, 435 (2006); (c) J. Chen, M. Tanaka, A. K. Sahoo, M. Takeda, A. Yada, Y. Nakao, T. Hiyama. Bull. Chem. Soc. Jpn.83, 554 (2010); (d) Y. Nakao, M. Takeda, T. Matsumoto, T. Hiyama. Angew. Chem., Int. Ed., 49, 4447 (2010); (e) S. Tang, M. Takeda, Y. Nakao, T. Hiyama. Chem. Commun.47, 307 (2011); (f) T. Hiyama. J. Synth. Org. Chem. Jpn.68, 729 (2010); (g) Y. Nakao, T. Hiyama. J. Synth. Org. Chem. Jpn.69, 1221 (2011); (h) Y. Nakao, T. Hiyama. Chem. Soc. Rev. 40, 4893 (2011).Suche in Google Scholar
[8] K. Shimizu, Y. Minami, Y. Nakao, K. Ohya, H. Ikehira, T. Hiyama. Chem. Lett. 42, 45 (2013).Suche in Google Scholar
[9] Y. Nakao, Jinshui Chen, H. Imanaka, T. Hiyama, Y. Ichikawa, W.-L. Duan, R. Shintani, T. Hayashi. J. Am. Chem. Soc. 129, 9137 (2007).Suche in Google Scholar
[10] (a) E. Shirakawa, K. Yamasaki, H. Yoshida, T. Hiyama. J. Am. Chem. Soc. 121, 10221 (1999); (b) E. Shirakawa, T. Hiyama. Bull. Chem. Soc. Jpn.75, 1435 (2002); (c) E. Shirakawa, T. Hiyama. J. Organomet. Chem.653, 114 (2002).Suche in Google Scholar
[11] Y. Nakao, S. Oda, T. Hiyama. J. Am. Chem. Soc.126, 13904 (2004).Suche in Google Scholar
[12] (a) Y. Nakao, A. Yada, S. Ebata, T. Hiyama. J. Am. Chem. Soc.129, 2428 (2007); (b) Y. Nakao. Bull. Chem. Soc. Jpn. 85, 731 (2012).Suche in Google Scholar
[13] Y. Nakao. Chem. Rec.11, 242 (2011).10.1002/tcr.201100023Suche in Google Scholar PubMed
[14] Y. Nakao, Y. Yamada, N. Kashihara, T. Hiyama. J. Am. Chem. Soc. 132, 13666 (2010).Suche in Google Scholar
[15] Y. Miyazaki, Y. Yamada, Y. Nakao, T. Hiyama. Chem. Lett.41, 298 (2012).Suche in Google Scholar
[16] Y. Minami, H. Yoshiyasu, Y. Nakao, T. Hiyama. Angew. Chem., Int. Ed.52, 883 (2013).Suche in Google Scholar
[17] Y. Minami, Y. Shiraishi, K. Yamada, T. Hiyama. J. Am. Chem. Soc.134, 6124 (2012).Suche in Google Scholar
[18] Y. Minami, K. Kanda, T. Hiyama. Chem. Lett. 43, 181 (2014).Suche in Google Scholar
[19] Y. Minami, K. Yamada, T. Hiyama. Angew. Chem., Int. Ed. 52, 10611 (2013).Suche in Google Scholar
[20] Y. Minami, T. Anami, T. Hiyama. Unpublished.Suche in Google Scholar
Article note
A collection of invited papers based on presentations at the 17th International IUPAC Conference on Organometallic Chemistry Directed Towards Organic Synthesis (OMCOS-17), Fort Collins, Colorado, USA, 28 July–1 August 2013.
©2014 by IUPAC & De Gruyter
Artikel in diesem Heft
- Masthead
- Masthead
- Conference papers
- International Union of Pure and Applied Chemistry
- Enantioselective palladium(0)-catalyzed C–H arylation strategy for chiral heterocycles
- Organometallic catalysis for applications in radical chemistry and asymmetric synthesis
- Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
- Copper-catalyzed aminoboration and hydroamination of alkenes with electrophilic amination reagents
- Activation of stable σ-bonds for organic synthesis
- Pd-catalyzed reactions of unactivated Csp3−I bonds
- Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
- Nickel-catalyzed oxidative cross-coupling of arylboronic acids with olefins
- Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents
- Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
- Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions
- Beyond C–H and C–O activation: the evolution of components in cross-coupling reactions
- The effect of acceptor-substituted alkynes in gold-catalyzed intermolecular reactions
- Chemoselective silver-catalyzed nitrene insertion reactions
- The strategic generation and interception of palladium-hydrides for use in alkene functionalization reactions
- 3-Acyloxy-1,4-enyne: A new five-carbon synthon for rhodium-catalyzed [5 + 2] cycloadditions
- Cobalt-catalyzed directed alkylation of arenes with primary and secondary alkyl halides
- IUPAC Technical Report
- Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)
Artikel in diesem Heft
- Masthead
- Masthead
- Conference papers
- International Union of Pure and Applied Chemistry
- Enantioselective palladium(0)-catalyzed C–H arylation strategy for chiral heterocycles
- Organometallic catalysis for applications in radical chemistry and asymmetric synthesis
- Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
- Copper-catalyzed aminoboration and hydroamination of alkenes with electrophilic amination reagents
- Activation of stable σ-bonds for organic synthesis
- Pd-catalyzed reactions of unactivated Csp3−I bonds
- Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
- Nickel-catalyzed oxidative cross-coupling of arylboronic acids with olefins
- Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents
- Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
- Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions
- Beyond C–H and C–O activation: the evolution of components in cross-coupling reactions
- The effect of acceptor-substituted alkynes in gold-catalyzed intermolecular reactions
- Chemoselective silver-catalyzed nitrene insertion reactions
- The strategic generation and interception of palladium-hydrides for use in alkene functionalization reactions
- 3-Acyloxy-1,4-enyne: A new five-carbon synthon for rhodium-catalyzed [5 + 2] cycloadditions
- Cobalt-catalyzed directed alkylation of arenes with primary and secondary alkyl halides
- IUPAC Technical Report
- Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)