One-Pot Synthesis of Cationic Gold Nanoparticles by Differential Reduction
-
Ralph A. Sperling


Abstract
The size-controlled synthesis of cationic particles by differential reduction of HAuCl4 precursor in the presence of NaBH4 and 1-aminoundecane-12-thiol (AUT) is reported. The number of seed particles is determined by the fraction of the initially Au precursor reduced by NaBH4 present in the reaction mixture, which are then grown larger by the AUT, acting as both weak reducing agent and stabilizing surfactant. By this methodology, size controlled synthesis is achieved in a two-step one-pot synthesis at room temperature.
1 Introduction
In recent years, the tunable chemical and physical properties of gold nanoparticles (Au NPs) have been widely exploited because of the improvements of both their direct [1], [2], [3] and seeded-growth [4], [5], [6], [7] production, which yield discrete sizes of the nanomaterials with controlled diameters. The basic components needed for Au NP synthesis include a metal precursor, a stabilizing agent (also called surfactant), a reducer and a solvent system. Due to the high electropositive character of gold, a large number of reducing agents including citrate [8], [9], borohydride [5], [10], ascorbic acid [11], [12], tannic acid [13], Na2S [14] and borane [15] among others, have been used for the reduction of Au+3-salts to form Au NPs in both aqueous and non-aqueous media. In addition, other reducers such as aminoacids [16] or proteins [17] have also been used for Au NP production. Besides, in many approaches, the surfactant in solution, which has the primary role to prevent the aggregation of NPs by providing an electrostatic or steric repulsion between them [18] simultaneously used as the reducing agent. State examples are the case of the Au NPs synthetized with sodium citrate [9], or oleylamine [19], or for the nucleation of Au on CoPt3 NPs with dodecylamine, playing a dual role [20]. This dual role of the surfactant is often required, to control the nucleation and growth steps occurring at all stages of the particle formation, and has a direct influence in the size and size distribution of the resulting NPs.
A common approach for controlling the size of Au NPs over a broad dispersity of diameters is seeded growth, in which small Au particles are prepared first and later used as nucleation centers for the preparation of larger particles. Providing a controlled number of preformed seeds and a growth condition that inhibits any secondary nucleation [7], the particle size can be controlled simply by varying the ratio of seed to metal precursor added [7], [13], [21], [22]. However, difficulty in finding a suitable growth condition that promotes uniform growth and inhibits additional nucleation during the growth stage limits the application of such methods. Despite the large number of studies highlighting the importance of kinetically control the growth of NPs, to our knowledge our group was the first to explore the differential reducing performance of two competing agents, initially in the case of the synthesis of Pt nanocrystals in organic solvents [21] and later in the size-controlled production of noble metal (Au and Ag) NPs in aqueos solvents [13], [22]. The results showed that it was possible to precisely accelerate the reduction of a fraction of the precursor molecules at a desired moment during the nucleation-growth process.
Beyond size, surface charge plays a critical role in determining the interactions of NPs with their surrounding environments. This is particular important in biological scenarios, where surface interactions [23] ultimately determine the formation of the protein corona [24], [25], the intracellular uptake and localization of the NPs [26], and in turn, their biological functions [27]. Although their relevance, the production of cationic Au NP is still limited [28], due in part to the difficulties in obtaining cationic particles from citrate-stabilized Au NPs upon surface ligand exchange [29]. Thus, the direct displacement of the negative citrate by positively charged molecules causes multiple electrostatic bridging, leading to irreversible agglomeration of the conjugates [29]. Alternatively, most direct synthetic approaches involves the reduction of an aqueous solution of Au3+ precursor in the presence of cationic ligands [26], [30], [31], [32], or following minor modifications of the well-known Brust-Schiffrin two-phase methodology [10], [33], which is generally limited to Au NPs of very small sizes (from 1.5 to 6 nm).
In this context, we take herein advantage of the recent improvements on the kinetic control of seeded-growth synthesis of metal NPs (Au and Ag) to explore the possibility to produce cationic Au NPs with controlled sizes between 10 nm and 30 nm by the combined use of a mixture of two competing reducing agents, a mild in excess, 1-aminoundecane-12-thiol (AUT), and a strong one, NaBH4, in stoichiometric deficiency, in order to induce a differential reduction of the HAuCl4 precursor solution [34]. Thus, by this methodology, the fraction of Au+3 precursor reduced by NaBH4 determines the number of seeds, which are then grown and stabilized by the AUT present in the reaction, which reduces the remaining precursor catalyzed by the presence of Au seeds [7]. As a result, the size-control of cationic Au NPs is achieved in a two-step one-pot synthesis at room temperature.
2 Results and discussion
Upon addition of the mixture of both reducers to an aqueous solution of HAuCl4, the fast reduction of Au3+ atoms by NaBH4 induces an initial burst nucleation of small nuclei which results in a decrease in the precursor concentration, leaving the remaining Au3+ atoms for the further growth of the particles. In this growth stage, AUT acts as both a stabilizing surfactant and a weak reducing agent, specially in the presence of the seed particles produced by the stronger reducing agent NaBH4. Following this idea, the fraction of the initially Au+3-precursor reduced by NaBH4 determines the number of seeds in a matter of seconds, and these are then grown by the AUT present in the reaction mixture, which slowly reduces the remaining Au+3-precursor catalyzed by the pre-formed seeds in a matter of few hours (Figure 1). As a result, it is possible to control the number and final size of the Au NPs by systematically adjusting the relative ratio of both reducers.
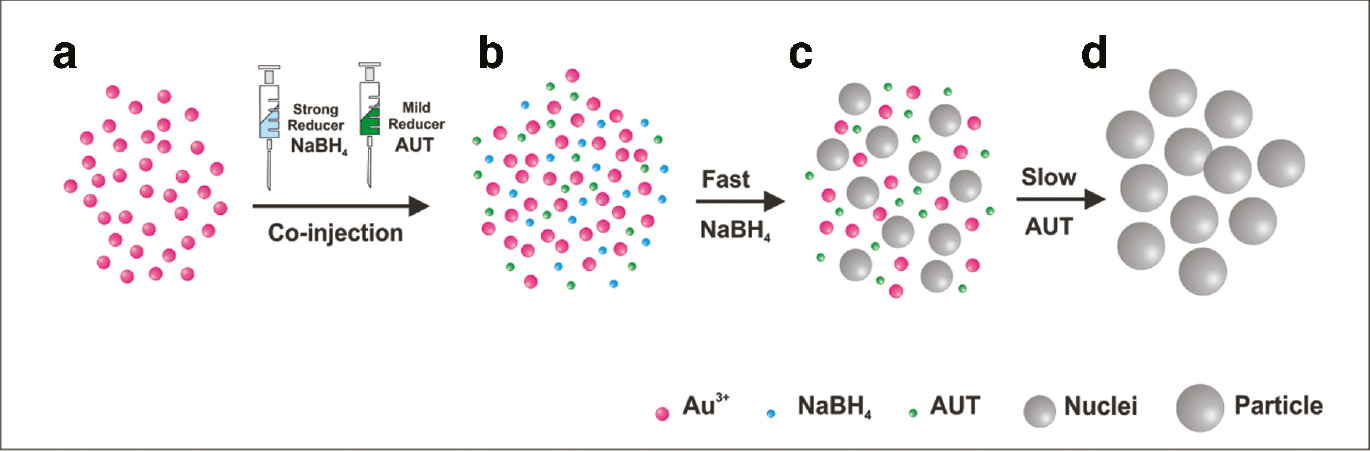
Reaction scheme of the formation mechanism of Au nanoparticles via a differential reduction synthetic approach. Upon addition of the mixture of both reducers to an aqueous solution of HAuCl4 (a, b) the fast reduction of Au3+ atoms by NaBH4 induces an initial burst nucleation of small nuclei which results in a dramatic decrease in the precursor concentration (c). Then, in the growth stage, AUT acts as both a stabilizing surfactant and a weak reducing agent, which slowly reduces the remaining Au+3-precursor catalyzed by the pre-formed seeds in few hours (d). As a result, it is possible to control the final size of the particles by systematically adjusting the relative ratio of both reducers.
Results obtained under identical reaction conditions, fixing the molar Au to AUT ratio at 1.5 (Au:AUT=1:0.66), but with systematically increased concentrations of NaBH4 are shown in Figure 2. The stoichiometric ratio Au:AUT of 1:0.66 was found to be necessary, since for such an amount of Au+3-precursor addition of less AUT resulted in irreversible aggregation, sometimes even before the addition of NaBH4, while under excess of AUT (Au:AUT=1:1.5) nucleation was dramatically reduced due to the formation of stable AUT-Au+3 complexes. These results suggest that the formation, evolution and stability of intermediate Au species depend on the concentration of AUT present in solution. Thus, if the concentration of AUT is lower than a critical value, these intermediate Au species are not stable and tend to aggregate, limiting the size control of the final Au particles. On the contrary, an excess of AUT stabilizes these intermediates, so that they do not evolve into well-defined individual particles.

Morphological (TEM) characterization of Au NPs synthesized with decreased volume fraction of a stock solution of NaBH4 in a fixed molar ratio of the other reagents Au:AUT:NaBH4 1:0.66:X at room temperature. a) X=0.11, b) X=0.053, c) X=0.026, d) X=0.0132, e) X=0.0066, f) X=0.0033, g) X=0.0016, h) X=0.0008, i) X=0.0004, j) X=0.0002.
Representative transmission electron microscopy (TEM) images (Figure 2) and particle size distributions (Figure 3) show how the Au NP size can be tuned from 10 to 30 nm by decreasing the amount of NaBH4 added. Thus, monomodal size distributions with mean diameters between 10.3±2.8 nm (SD~27 %) and 19.7±2.6 nm (SD~13 %) were synthesized with Au:AUT:NaBH4 ratios between 1:0.66:0.053 and 1:0.66:0.0016, respectively (Figures 2b–g and 3b–g).
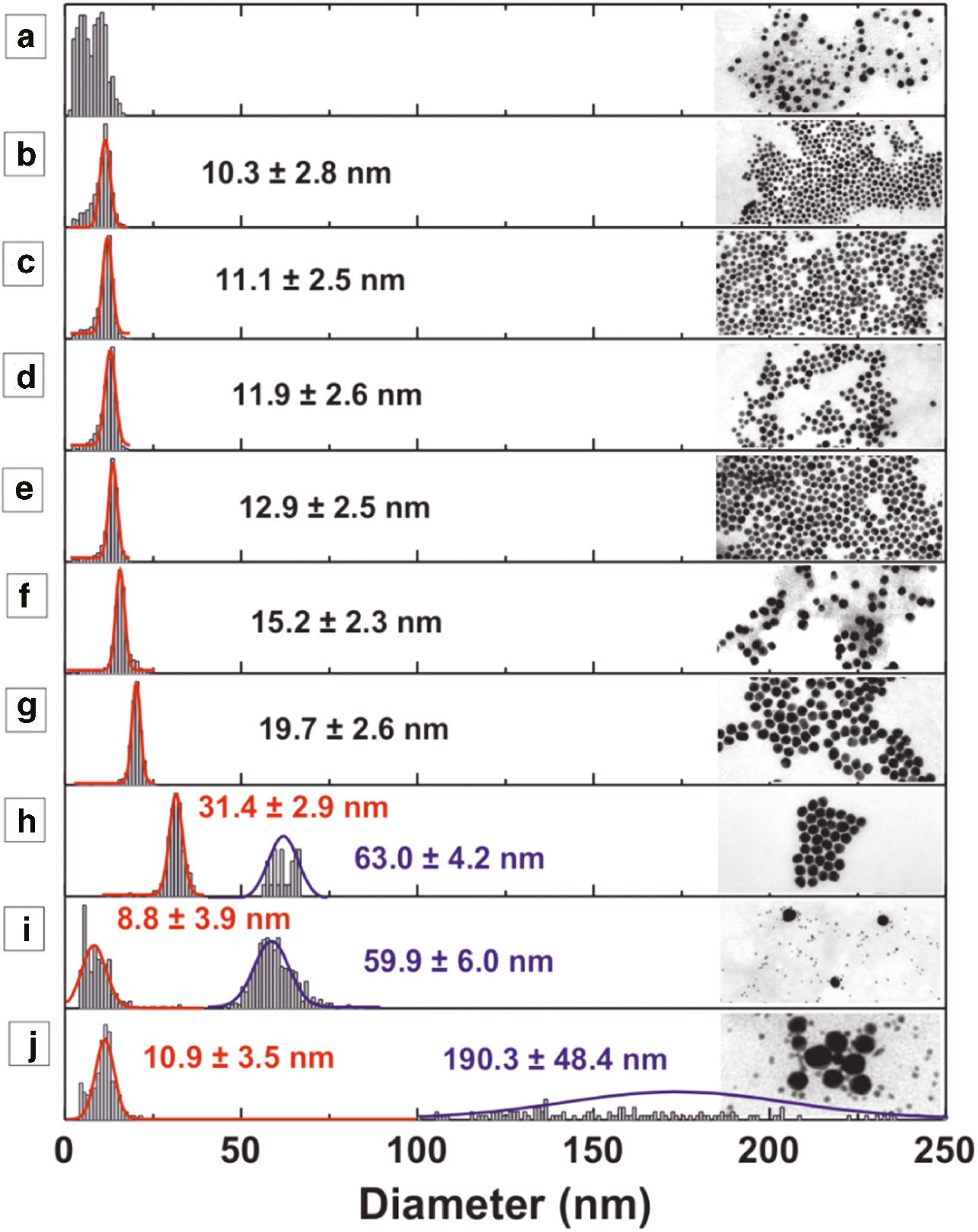
Size distributions measurements of Au NPs synthesized with decreased volume fraction of a stock solution of NaBH4 in a fixed molar ratio of the other reagents Au:AUT:NaBH4 1:0.66:X at room temperature. The molar ratios of the three components are the same as in Figure 2. In plots h, i and j the histograms of the two populations were normalized and evaluated individually because of the very low counts of the large fraction.
Interestingly, a controlled growth of the particles without significatively new nucleation and with narrow size distributions is achieved. This narrowing of the size distribution during Au NP’s growth seen from sample B to sample G (Figure 3b–g) has been observed previously in other nanocrystal systems, which was called size focusing growth regime [35].
Remarkably, in the absence of sufficient NaBH4 (Au:NaBH4<1:0.0008) the balance between the number of Au seeds formed and remaining Au precursor in solution do not favor the controlled growth of NPs, resulting in the secondary nucleation of a new Au population [7]. Therefore, when slow reduction of the Au+3-salt by AUT occurs, nucleation of Au NPs is observed several days, which resulted in bimodal size distributions with a population of small particles (~10–30 nm) together with much larger Au NPs (between 60 and 200 nm), attributed to the slow growth of the initial particles (Figures 2h–j and 3h–j). This polydispersity is a consequence of the large differences existing between the growth histories of each NP. The nuclei produced first by the NaBH4 have a much longer time for growth, resulting into larger particles. In the meanwhile additional nuclei are produced by the AUT as a consequence of the large concentration of Au+3-precursor remaining in the reaction mixture, so the resulting size distributions splits into two separate populations. By contrast, addition of an excess of NaBH4 (Au:NaBH4>1:0.11) resulted in a broad distribution of small particles (Figures 2a and 3a). In most of the cases, the small clusters generated by an excess of NaBH4 were not stable and caused the immediate aggregation (and further precipitation) of the samples.
The analysis of the polydispersity of the samples yields also to some interesting insights. On the one side, it is clear that as the NPs grow, the SD decreases. This is tipical of the size focusing regime during standard NC seeded growth [7], indicating that in the one-pot reaction, there is indeed an spontaneous seeded-growth process where fewer larger NPs grow at expenses of many smaller ones. Besides, in the bimodal distributions, it can be observed that the polydispersity of the large sized NP population is significantly broader than the small ones. Polydispersity is normally due to multiple nucleation during a broad time, or irregular growth [22], [36], [37]. Besides, considering that the degree of polidispersity increases as NP are getting larger, it is feasable to argue that the polydispersity could be due to the irregular growth of the population of NPs that were nucleated at the initial stages of the reaction.
It is interesting to observe that an excess of either NaBH4 or AUT leads to similar results in terms of the formation of a broad population of small particles (Figure 3a and j). Unexpectantly, the particles reduced by an excess of a weak AUT are not much larger than those obtained when using and excess of the strong NaBH4. Although one could argue that the presence of a mild reducer would result into larger particles, this was not observed in our case. These results suggest that the number of nuclei is not only determined by the strength of the reducing agent, and illustrates the importance of having the adequate combination and ratios of reducers to control the monomodal formation of collections of NPs with controlled size in a synthesis. Consequently, the combined used (and action) of both reducers is needed to tune the final size of the cationic Au NPs.
The particles were further characterized by UV-vis absorption spectroscopy, in which the effect of systematically decreasing the amount of NaBH4 resulted in a red-shift of the surface plasmon resonance (SPR) peak from 523 to 541 nm (Figure 4a). Considering that the SPR absorption peak is indicative of the particle size [38], [39], this shift confirmed a gradual increase of the Au NPs’ diameter. The trend of size variation measured by dynamic light scattering (DLS) (Figure 4b) correlated well with the optical behavior observed by UV-Vis spectroscopy (Figure 4c) and mean diameter observed by TEM (Figure 3), confirming the dependence of resulting particle size to the relative concentration of both reducers used. However, a further comparison of absolute diameters measured with DLS to the data obtained from TEM was difficult because of the different nature of measurements (i.e. inorganic core diameter of single particles from TEM vs hydrodynamic diameter from an ensemble by DLS). It is worth noting that while DLS is sensitive even to small size changes in case of monodisperse samples, bimodal size distributions could not be resolved neither by the general purpose nor the “multiple narrow peak” algorithm of the Malvern DTS software. Besides, the cationic surfactant AUT renders the resultant Au NPs positively charged, which was confirmed by zeta potential measurements (Figure 4d).
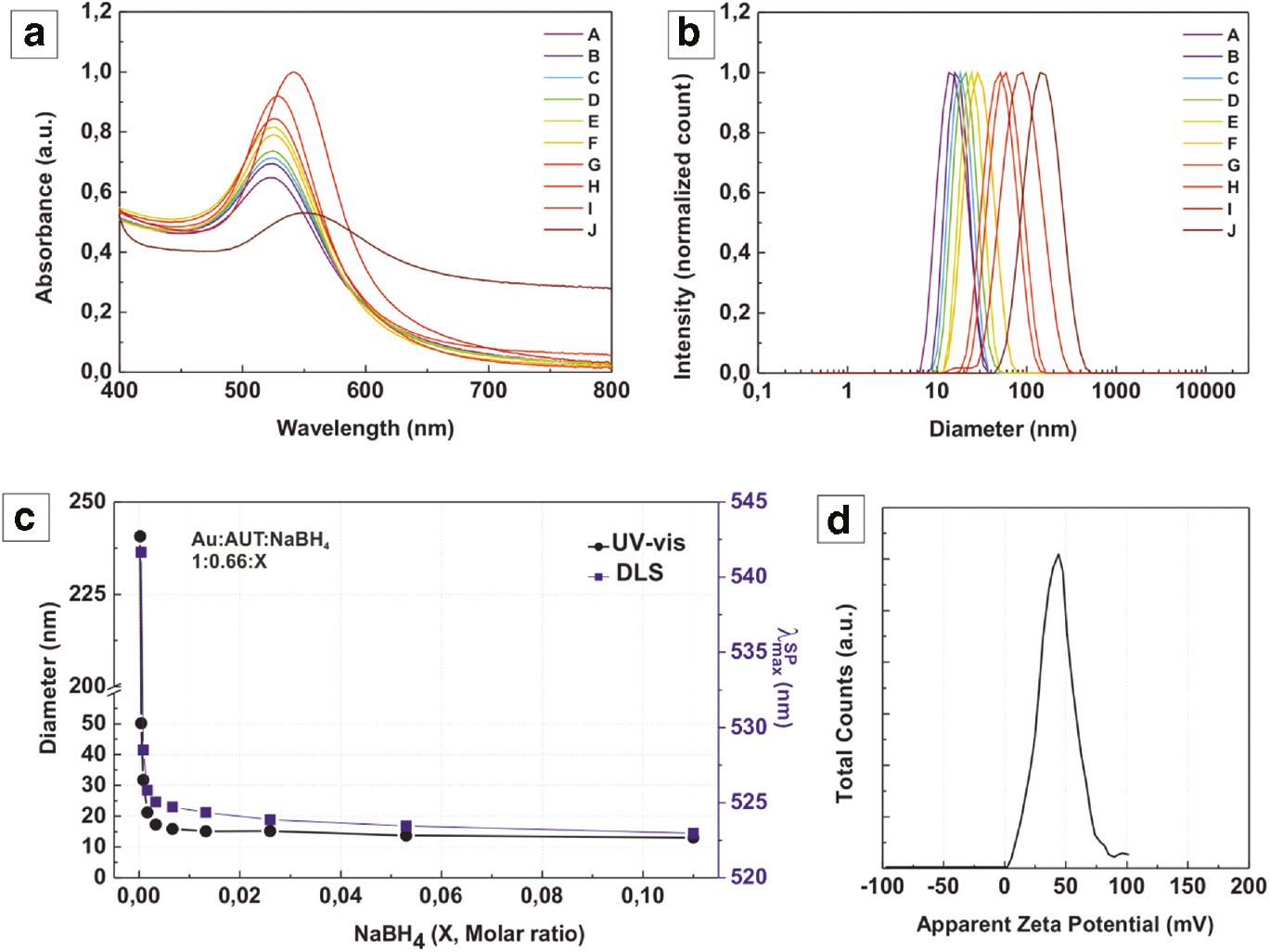
Evolution of the SPR absorbance maximum (a), the volume size by dynamic light scattering (DLS) (b) and a representative zeta potential measurement (d) of Au NPs synthesized with decreasing concentration of NaBH4 in the reaction mixture. The correlation of size variation with the position of SPR band (c) confirms the dependence of resulting particle size to the amount of NaBH4 added.
Closer monitoring of the Au NP growth was also performed by recording a time series of absorbance spectra for two selected reactions (Au:NaBH4=1:0.0033 and 1:0.0008 corresponding to reactions F and H from Figure 1, respectively), and the absorbance maximum, the position of the plasmon peak and the peak width (right half width at half maximum) plotted over time (Figure 5). Analysis of UV-Vis results essentially show similar temporal evolutions, but the addition of larger amounts of NaBH4 resulted into faster reaction rates. As expected, samples at short times have relatively flat absorbance profiles which are translated into low absorption values. Later, as the reaction progresses, the plasmon band intensity increases and blue-shifts smoothly to a constant value which depends on the Au NP size and is characteristic of spherical Au NPs (Figure 5A). This blue-shift in the position of the SPR peak is clearly seen in Figure 5B. The obtained results also reproduce what we have observed when separating the two reduction processes in time using standard seeded growth methods. Exploiting the same mechanism, preformed particles from a first synthesis were grown larger by addition of fresh precursor solution HAuCl4+AUT (Figure 5, red curve).
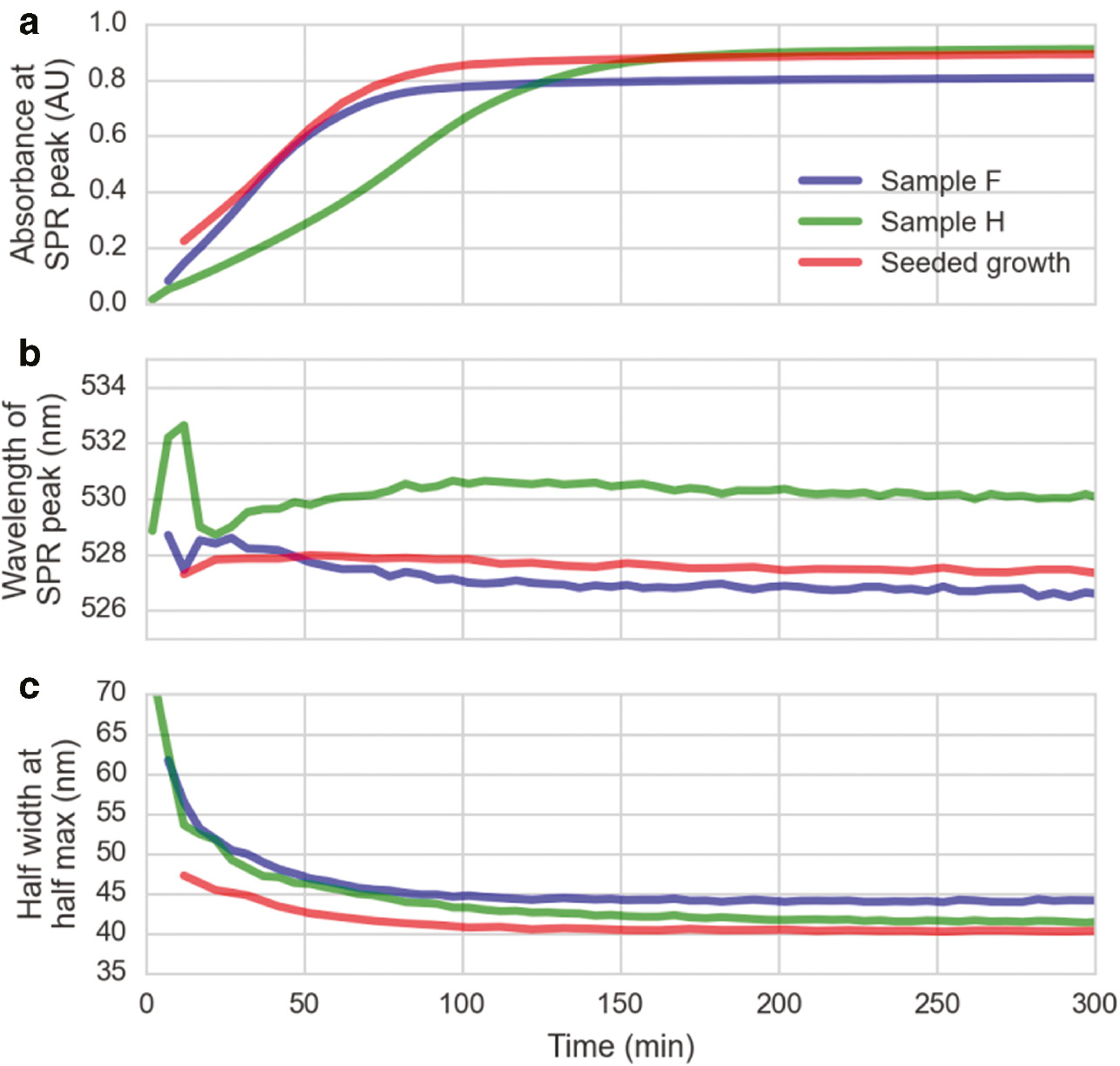
UV-Vis time series. Blue curve: Au:AUT:NaBH4=1:0.66:0.0033 (sample F), green curve: Au:AUT:NaBH4=1:0.66:0.0008 (sample H). After addition of less NaBH4, the absorption increases much slower, indication slower particle growth. Red curve: Seeded growth, addition of 11.1±2.5 nm seed particles to the Au:AUT=1:1.5 solution instead of NaBH4.
3 Conclusions
In conclusion, the synthesis of cationic Au NPs in the presence of two reducing agents has been examined in this work. NaBH4 acts as a strong reducing agent which immediately triggers nucleation of gold clusters, while AUT acts as both a weak reducer and a stabilizing molecule. The fraction of the initial Au+3-precursor reduced by NaBH4 determines the number of seed particles, which are then grown larger by the AUT present in the reaction mixture by the slow reduction of the remaining gold in solution. This fact results in a controlled growth of the particles without significatively new nucleation and with narrow size distributions. The combination of these two reducers does not simply result in an intermediate effect, but they both act sequentially in accordance to their chemical environment. Large initial (NaBH4) yields high concentrations of small Au NPs, while low (NaBH4) results into low concentrations of large Au NPs where the monomodality conditions are lost. Such a control of the number of seeds and the size of NPs achieved in a two-step one-pot synthesis at room temperature is unprecedented.
4 Materials and methods
4.1 Synthesis
HAuCl4 and NaBH4 were purchased from Sigma-Aldrich, 11-Amino-1-undecanethiol (AUT) as hydrochloride from Asemblon (# 111027) or Sigma-Aldrich (# 674397). Aqueous solutions were prepared with MilliQ water from the reagents as received.
For a typical synthesis, AUT was prepared as a 20 mM stock solution by brief sonication and added to a 1.42 mM solution of HAuCl4 in a plastic tube, the solution was left stirring for at least 30 min. Upon mixing AUT and HAuCl4, the color of the solution immediately changed from pale to a more intense yellow, and faded out again over time. NaBH4 was freshly prepared as 1 mM solution in ice-cold water and added to the precursor solution. Depending of the stoichiometry, the characteristic red color of gold nanoparticles developed within minutes to a few hours. After waiting for at least overnight, the samples were used for further analysis.
For online-monitoring the synthesis progress, a small aliquot of the solution was transferred to a plastic cuvette immediately after the addition of NaBH4 and a series of absorbance spectra were taken over time. The cuvettes were sealed by tape or a layer of silicone oil to prevent evaporation.
4.2 Analysis
UV-vis absorption spectra were acquired with a Shimadzu UV-2401-PC spectrometer. For the measurement of time series, a small amount of synthesis mixture was taken off immediately after the addition of NaBH4, transferred to a microcuvette (Eppendorf UVette), covered with silicone oil or lab tape, and placed into the spectrometer. Spectra were automatically taken every 5 or 10 min. The absorbance and exact position of the plasmon peak was detected fitting a square function to a small interval (±5 nm) around the peak.
TEM grids were prepared by briefly dipping the grids into the nanoparticle solution or by carefully placing a droplet of the sample onto the grid which had been placed on Parafilm and absorbing the liquid after about 5 min. TEM analysis was carried out on a JEOL JEM-1010 or JEM-1400 instrument operating at 80 kV. Digital images were analyzed with the ImageJ software and a custom macro performing smoothing (3×3 or 5×5 median filter), manual global thresholding and automatic particle analysis provided by the ImageJ. The particle size was calculated as circle-equivalent diameter from the area of each individual particle (d=2√(A/π)) and calculating the number averages from the entire population or individual peaks found in the histograms. The macro is available at https://github.com/psa-rs/psa-macro.
Dynamic light scattering and zeta potential measurements were carried out with the stock solutions using a Malvern Zetesizer and disposable cuvettes. Data was analyzed with the Malvern DTS software using the general purpose algorithm for DLS and the Smochulowski model for obtaining zeta potential values.
Acknowledgments
We acknowledge financial support from the Spanish Ministerio de Ciencia e Innovación (MICINN) (MAT2012-33330) (MAT2015-70725-R) and from the Catalan Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) (2014-SGR-612). Financial support from the FutureNanoNeeds (FP7-NMP-2013-LARGE-7) Project financed by the European Community under the FP7 Capacities Programme is gratefully acknowledged. N.G.B. acknowledges financial support by MINECO through the Ramon y Cajal program (RYC-2012-10991) and by the European Commission Seventh Framework Programme (FP7) through the Marie Curie Career Integration Grant (322153-MINE).
References
1. I. Ojea-Jimenez, N. G. Bastus, V. Puntes, J. Phys. Chem. C 115 (2011) 15752.10.1021/jp2017242Search in Google Scholar
2. F. Schulz, T. Homolka, N. G. Bastús, V. Puntes, H. Weller, T. Vossmeyer, Langmuir 30 (2014) 10779.10.1021/la503209bSearch in Google Scholar PubMed
3. X. Ji, X. Song, J. Li, Y. Bai, W. Yang, X. Peng, J. Am. Chem. Soc. 129 (2007) 13939.10.1021/ja074447kSearch in Google Scholar PubMed
4. L. Cao, T. Zhu, Z. Liu, J. Colloid Interf. Sci. 293 (2006) 69.10.1016/j.jcis.2005.06.012Search in Google Scholar PubMed
5. N. R. Jana, L. Gearheart, C. J. Murphy, Langmuir 17 (2001) 6782.10.1021/la0104323Search in Google Scholar
6. A. A. Volkert, V. Subramaniam, A. J. Haes, Chem. Commun. 47 (2011) 478.10.1039/C0CC02075CSearch in Google Scholar
7. N. G. Bastús, J. Comenge, V. F. Puntes, Langmuir 27 (2011) 11098.10.1021/la201938uSearch in Google Scholar PubMed
8. G. Frens, Nature-Phys. Sci. 241 (1973) 20.10.1038/physci241020a0Search in Google Scholar
9. J. Turkevich, P. C. Stevenson, J. Hillier, Discuss. Faraday Soc. 11 (1951) 55.10.1039/df9511100055Search in Google Scholar
10. M. Brust, M. Walker, D. Bethell, D. J. Schiffrin, R. Whyman, J. Chem. Soc. Chem. Comm. (1994) 801.10.1039/C39940000801Search in Google Scholar
11. D. V. Goia, E. Matijevic, Colloid. Surface. A 146 (1999) 139.10.1016/S0927-7757(98)00790-0Search in Google Scholar
12. N. R. Jana, L. Gearheart, C. J. Murphy, Chem. Mater. 13 (2001) 2313.10.1021/cm000662nSearch in Google Scholar
13. J. Piella, N. G. Bastús, V. Puntes, Chem. Mater. 28 (2016) 1066.10.1021/acs.chemmater.5b04406Search in Google Scholar
14. A. M. Schwartzberg, C. D. Grant, T. van Buuren, J. Z. Zhang, J. Phys. Chem. C 111 (2007) 8892.10.1021/jp067697gSearch in Google Scholar
15. D. H. Rapoport, W. Vogel, H. Colfen, R. Schlogl, J. Phys. Chem. B 101 (1997) 4175.10.1021/jp963301xSearch in Google Scholar
16. Y. Shao, Y. Jin, S. Dong, Chem. Commun. (2004) 1104.10.1039/b315732fSearch in Google Scholar PubMed
17. B. Nivedita, et al. Biomed. Mater. 3 (2008) 034105.10.1088/1748-6041/3/3/034105Search in Google Scholar PubMed
18. N. G. Bastús, E. Casals, S. Vazquez-Campos, V. Puntes, Nanotoxicology 2 (2008) 99.10.1080/17435390802217830Search in Google Scholar
19. C. Shen, C. Hui, T. Yang, C. Xiao, J. Tian, L. Bao, S. Chen, H. Ding, H. Gao, Chem. Mat. 20 (2008) 6939.10.1021/cm800882nSearch in Google Scholar
20. T. Pellegrino, A. Fiore, E. Carlino, C. Giannini, P. D. Cozzoli, G. Ciccarella, M. Respaud, L. Palmirotta, R. Cingolani, L. Manna, J. Am. Chem. Soc. 128 (2006) 6690.10.1021/ja0607741Search in Google Scholar PubMed
21. S. I. Lim, M. Varon, I. Ojea-Jimenez, J. Arbiol, V. Puntes, Chem. Mat. 22 (2010) 4495.10.1021/cm101436pSearch in Google Scholar
22. N. G. Bastús, F. Merkoçi, J. Piella, V. Puntes, Chem. Mater. 26 (2014) 2836.10.1021/cm500316kSearch in Google Scholar
23. A. Verma, F. Stellacci, Small 6 (2010) 12.10.1002/smll.200901158Search in Google Scholar PubMed
24. E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh, V. Puntes, ACS Nano 4 (2010) 3623.10.1021/nn901372tSearch in Google Scholar PubMed
25. E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh, V. F. Puntes, Small 7 (2011) 3479.10.1002/smll.201101511Search in Google Scholar PubMed
26. I. Ojea-Jimenez, L. Garcia-Fernandez, J. Lorenzo, V. F. Puntes, ACS Nano 6 (2012) 7692.10.1021/nn3012042Search in Google Scholar PubMed
27. E. C. Cho, J. Xie, P. A. Wurm, Y. Xia, Nano Letters 9 (2009) 1080.10.1021/nl803487rSearch in Google Scholar PubMed
28. J. Hassinen, V. Liljeström, M. A. Kostiainen, R. H. A. Ras, Angew. Chem. Int. Edit. 54 (2015) 7990.10.1002/anie.201503655Search in Google Scholar PubMed
29. I. Ojea-Jimenez, V. Puntes, J. Am. Chem. Soc. 131 (2009) 13320.10.1021/ja902894sSearch in Google Scholar PubMed
30. T. Niidome, K. Nakashima, H. Takahashi, Y. Niidome, Chem. Commun. (2004) 1978.10.1039/b406189fSearch in Google Scholar PubMed
31. P. Li, D. Li, L. Zhang, G. Li, E. Wang, Biomaterials 29 (2008) 3617.10.1016/j.biomaterials.2008.05.020Search in Google Scholar PubMed
32. L. Zhang, X. Sun, Y. Song, X. Jiang, S. Dong, E. Wang, Langmuir 22 (2006) 2838.10.1021/la052822lSearch in Google Scholar PubMed
33. D. V. Leff, L. Brandt, J. R. Heath, Langmuir 12 (1996) 4723.10.1021/la960445uSearch in Google Scholar
34. Y. Xiong, A. R. Siekkinen, J. Wang, Y. Yin, M. J. Kim, Y. Xia, J. Mater. Chem. 17 (2007) 2600.10.1039/b705253gSearch in Google Scholar
35. X. G. Peng, J. Wickham, A. P. Alivisatos, J. Am. Chem. Soc. 120 (1998) 5343.10.1021/ja9805425Search in Google Scholar
36. J. Turkevich, Gold Bull. 18 (1985) 125.10.1007/BF03214694Search in Google Scholar
37. J. Turkevich, Gold Bull. 18 (1985) 86.10.1007/BF03214690Search in Google Scholar
38. S. Link, M. A. El-Sayed, J. Phys. Chem. B 103 (1999) 8410.10.1021/jp9917648Search in Google Scholar
39. N. G. Bastus, J. Piella, V. Puntes, Langmuir 32 (2016) 290.10.1021/acs.langmuir.5b03859Search in Google Scholar PubMed
©2017 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Editorial
- Hierarchical Colloidal Nanostructures – from Fundamentals to Applications
- One-Pot Synthesis of Cationic Gold Nanoparticles by Differential Reduction
- Impact of the Crosslinker’s Molecular Structure on the Aggregation of Gold Nanoparticles
- Modeling the Optical Responses of Noble Metal Nanoparticles Subjected to Physicochemical Transformations in Physiological Environments: Aggregation, Dissolution and Oxidation
- Tetrazole-Stabilized Gold Nanoparticles for Catalytic Applications
- Catalytic Properties of Cryogelated Noble Metal Aerogels
- Graded Shells in Semiconductor Nanocrystals
- Determination of all Dimensions of CdSe Seeded CdS Nanorods Solely via their UV/Vis Spectra
- Ultrafast Transient Absorption and Terahertz Spectroscopy as Tools to Probe Photoexcited States and Dynamics in Colloidal 2D Nanostructures
- Trap-Induced Dispersive Transport and Dielectric Loss in PbS Nanoparticle Films
- Towards Photo-Switchable Transport in Quantum Dot Solids
Articles in the same Issue
- Frontmatter
- Editorial
- Hierarchical Colloidal Nanostructures – from Fundamentals to Applications
- One-Pot Synthesis of Cationic Gold Nanoparticles by Differential Reduction
- Impact of the Crosslinker’s Molecular Structure on the Aggregation of Gold Nanoparticles
- Modeling the Optical Responses of Noble Metal Nanoparticles Subjected to Physicochemical Transformations in Physiological Environments: Aggregation, Dissolution and Oxidation
- Tetrazole-Stabilized Gold Nanoparticles for Catalytic Applications
- Catalytic Properties of Cryogelated Noble Metal Aerogels
- Graded Shells in Semiconductor Nanocrystals
- Determination of all Dimensions of CdSe Seeded CdS Nanorods Solely via their UV/Vis Spectra
- Ultrafast Transient Absorption and Terahertz Spectroscopy as Tools to Probe Photoexcited States and Dynamics in Colloidal 2D Nanostructures
- Trap-Induced Dispersive Transport and Dielectric Loss in PbS Nanoparticle Films
- Towards Photo-Switchable Transport in Quantum Dot Solids