Synthesis, biological activity and modeling study of some thiopyrimidine derivatives and their platinum(II) and ruthenium(III) metal complexes
-
Najim A. Al-Masoudi
 ,
Azhar Abbas
,
Azhar Abbas

Abstract
A new series of 6-amino-5-(aryldiazenyl)-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one derivatives 17–27 and the N1-methyl-azothiopyrimidine analog 28 were synthesized from the pyrimidine derivatives 4 and 5, respectively, via diazotization reaction, with various amines. The platinum(II) metal complexes [bis(4-amino-N3-methyl-6-oxo-2-thioxo-pyrimidin-1-yl)]Pt(II) (29) and [(6-amino-5-(4-R-phenyl)diazenyl)-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one)]Pt(II)Cl2 derivatives 30–33 were prepared from the treatment of 5 and the azo analogs 17 and 21–23 with PtCl2, respectively. Analogously, 5 and 17 were treated with RuCl3·3H2O to give the Ru(III) complexes 34 and 35, respectively. The newly synthesized compounds were assayed against HIV-1 and HIV-2 in MT-4 cells. The results revealed that 26 and 30 were the only compounds in the series inhibiting HIV replication in cell cultures with an IC50 value of >2.07 and >3.02 μg mL–1, respectively. The molecular modeling interactions of 26 with some amino acids of HIV reverse transcriptase were studied. In addition, the antibacterial activity of 17–35 and 31–33 against Staphylococcus aureus and Escherichia coli was evaluated.
1 Introduction
The pyrimidine molecule is a promising structural moiety for drug design because it forms a component in a number of useful drugs and is associated with many biological and therapeutic activities [1]. Besides the biological significance of pyrimidine, several of its derivatives have been developed as chemotherapeutic agents and have found wide clinical applications such as antiviral [2, 3], antiprotozoal [4], antimicrobial, anti-insecticidal [5–8], anticancer [9–14], antihypertensive [15], antihypoglycemic and hypolipidemic [16], and anti-HIV agents [17–19], in addition to their cardiovascular [20] and diuretic [21, 22] properties. Furthermore, some pyrimidines are used as hypnotic and sedative drugs [23, 24]. Two diarylpyrimidines (DAPY), rilpivirine (1) [25] and etravirine [26, 27], have been classified as non-nucleoside reverse transcriptase inhibitors, whereas bacimethrin (4-amino-5-(hydroxymethyl)-2-methoxypyrimidine) (2) (Fig. 1) is a pyrimidine antibiotic that is active against several staphylococcal bacteria [28].
![Fig. 1: Chemical structures of rilpivirine (1), bacimethrin (2) and [M(pyrimidinyl-pyrazole)Cl2] [M=Pt, Ru(Cl)] complexes (3).](/document/doi/10.1515/znb-2014-0246/asset/graphic/znb-2014-0246_fig1.jpg)
Chemical structures of rilpivirine (1), bacimethrin (2) and [M(pyrimidinyl-pyrazole)Cl2] [M=Pt, Ru(Cl)] complexes (3).
Some platinum and ruthenium complexes play a crucial role in preclinical as well as in clinical studies. Saha and Muherjee [29] have reported the metal complexes of palladium(II) and platinum(II) 3 of (3,5-dimethyl-2′-pyrimidyl)pyrazole as potent anticancer agents (Fig. 1). Furthermore, we have reported the synthesis and anti-HIV activity of a new series of Pt(II), as well as those of other metal complexes with 3-methyl-6,7-diphenyllumazine [30]. In addition, Joshi et al. [31] have discussed the selectivity of anticancer drugs toward cancer cells as one of the main goals of drug optimization, by using the complexes Ru(dppz)2(CppH)](PF6)2 [where CppH = 2-(2-pyridyl)pyrimidine-4-carboxylic acid and dppz = dipyrido[3,2-a:2′,3′-c]phenazine)] as a “photocaging” agent to give a light illumination at 350 nm for detection of cancer cells. This is the first substitutionally inert cytotoxic metal complex to be used as a light-triggered prodrug candidate.
In continuation of our ongoing work on the synthesis of a new anti-HIV agent and our recent antiviral data on new pyrimidine derivatives [32–34], we report here the synthesis of a new series of pyrimidines having arylazo residues, including some of their Pt(II) and Ru(III) metal complexes, and the evaluation of their anti-HIV and antibacterial activities, together with a molecular modeling study.
2 Results and discussion
6-Amino-5-(aryldiazenyl)-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one derivatives 17–27 were prepared in 40–92 % yield via diazotization reaction by treatment of 6-amino-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one (4) with various substituted amines – 4-chloro- (6), 4-bromo- (7), 4-nitro- (8), 4-methyl- (9), 4-sulfonamido- (10) and 4-sulfonylactamido- (11) anilines – as well as with p-aminobenzoic acid (12), p-aminosulfonic acid (13), sulfadiazine (14), sulfamerazine (15) and sulfamethazine (16), in the presence of NaNO2 and HCl at 0–5 °C, then at ambient temperature for 15 h. Under similar conditions, azothiopyrimidine 28 was prepared in 68 % yield from diazotization of 6-amino-N1-methyl-2-thioxo-pyrimidin-4-one (5) (Scheme 1).
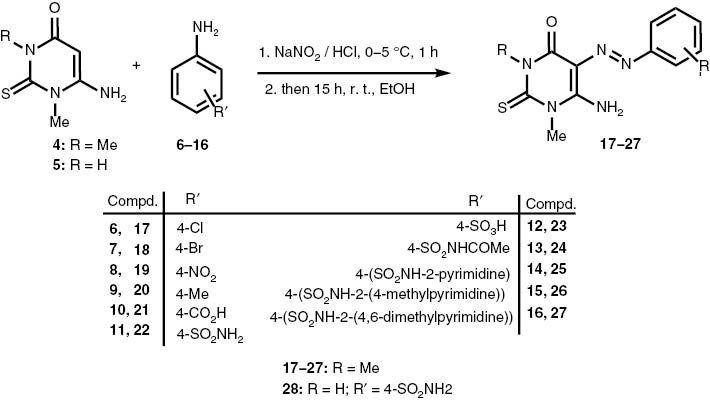
Synthesis of the 6-amino-5-aryl-diazenyl-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one derivatives (17–27) and of the N1-methyl analog (28).
The structures of 17–28 were assigned on the basis of their 1H, 13C NMR and mass spectra. The 1H NMR spectra showed similar patterns for the aromatic and pyrimidine protons and carbon atoms, where the NH2 group of 17, 18, 20 and 23–25 resonated as two separated singlets at the regions δ = 9.14–11.75 ppm and δ = 8.64–9.02 ppm, and this pattern was due to the hydrogen bonding between the NH2 group and the nitrogen atom of the azo residue. Compound 20 was selected for 1H NMR measurement at high temperature (364 K), which revealed the appearance of a broad singlet at δ = 9.99 ppm assigned to the NH2 group, which was exchangeable with D2O, and the disappearance of NH2 protons at δ = 11.64 and 8.65 ppm (92 K). The aromatic, pyrimidine and methyl protons were fully analyzed (see Experimental Section). In the 13C NMR spectra of 17–28, the resonances at the regions δ = 177.2–179.1 and 157.0–151.0 ppm were assigned to C-2 (C=S) and C-4 of the thiopyrimidine ring, whereas the resonances at the regions δ = 75.0–82.7 and 157.1–165.5 ppm were attributed to C-5 and C-6 of the same ring, respectively. The other carbon atoms of the pyrimidine and aromatic substituents were fully assigned (see Experimental Section), whereas all the synthesized compounds were confirmed from their 1H and 13C HSQC NMR spectra [35]. Compound 27 was selected for further NMR experiments. The gradient HMBC [36] NMR spectrum of 27 revealed two 2JC,H couplings: the aromatic carbon atom C-1′ at δ = 143.5 ppm showed a 2JC,H coupling with 2′-H and 6′-H of the same ring at δ = 8.08 ppm, whereas the other one was observed between 5″-H of the pyrimidine ring at δ = 6.67 ppm and two methyl carbon atoms attached to C-4′ and C-6″ of the same ring at δ = 23.0 ppm. Furthermore, C-2 (C=S) of the pyrimidine ring at δ = 177.5 ppm showed two 3JC,H couplings, one with N1-Me protons at δ = 3.69 ppm and the other with N3-Me protons at δ = 3.89 ppm. In addition, a 3JC,H coupling was observed between C-5″ of the pyrimidine scaffold at δ = 113.1 ppm and the two methyl carbon atoms attached to C-4″ and C-6″ of the same ring at δ = 23.0 ppm (Fig. 2).
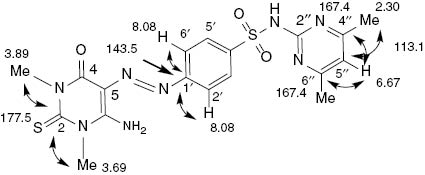
Chemical shifts (δ, ppm) and JC,H correlations in the HMBC NMR spectrum of 27.
Our procedure was modified by preparing some new platinum(II) and ruthenium(III) complexes. Thus, compound 5 was selected as a precursor for the synthesis of the new platinum(II) complex 29 in 48 % yield from the treatment of 5 with PtCl2 in hot MeOH, aiming to examine its antiviral activity (Scheme 2). The structure of 29 was determined from the elemental analysis, 1H, 13C NMR and IR spectra (see Experimental Section). In the 13C NMR spectrum of 5, carbon atoms 2 and 4–6 appeared at δ = 179.7, 164.4, 160.1 and 79.6 ppm, respectively. In the IR spectrum, υ(C=O) and υ(C=S) of the free ligand 5 were observed at 1630 and 1099 cm–1, respectively, which shifted to 1649 and 1134 cm–1 for complex 29. This indicated that the >C=O and >C=S groups of the ligand are involved in the bonding with metal ions. The υ(NH2) observed at 3100 and 3400 cm–1 in the spectrum of free ligand remained constant in the spectrum of the complex. This observation is in agreement with the data reported by Masouda et al. [37] and Parthasarathi et al. [38] concerning the metal complexes of some thiouracil derivatives. The spectrum of 29 also showed a low wavenumber band at 448 cm–1, characteristic for a υ(Pt–O) coordinated bond, as expected [39], whereas a band at 706 cm–1 was probably due to a pyrimidine-ring vibration [40]. These data suggested that the geometrical structure of 29 is a distorted square planar. The chelation via N and S donor atoms and the formation of a four-membered ring at 29 are in agreement with the X-ray data of the related complex Pt(η2-N,S-pymS)(η1-S-pymS)(PPh3)] reported by Lobana et al. [41].
![Scheme 2: Synthesis of the bis(4-amino-N3-methyl-6-oxo-2-thioxo-pyrimidin-1-yl)platinum(II) complex [Pt(ThPyrm)2] (29).](/document/doi/10.1515/znb-2014-0246/asset/graphic/znb-2014-0246_scheme2.jpg)
Synthesis of the bis(4-amino-N3-methyl-6-oxo-2-thioxo-pyrimidin-1-yl)platinum(II) complex [Pt(ThPyrm)2] (29).
The platinum complexes of some synthesized azothiopyrimidine analogs had been explored, aiming to study their stereochemical geometry in addition to evaluating their anti-HIV activity. Thus treatment of the azo ligands 17 and 21–23 with Pt(II)Cl2 in refluxing MeOH gave, after purification, the desired Pt(II) complexes [(AzPyrm-Cl)PtCl2] 30–33 in 55 %, 46 %, 45 % and 58 % yield, respectively (Scheme 3).
![Scheme 3: Synthesis of the bis(4-amino-N1,N3-dimethyl-6-oxo-2-thioxo-pyrimidin-1-yl)Pt(II)Cl2 complexes [(ADiThPyrm)2PtCl2] (29–33).](/document/doi/10.1515/znb-2014-0246/asset/graphic/znb-2014-0246_scheme3.jpg)
Synthesis of the bis(4-amino-N1,N3-dimethyl-6-oxo-2-thioxo-pyrimidin-1-yl)Pt(II)Cl2 complexes [(ADiThPyrm)2PtCl2] (29–33).
The structures of 30–33 were assigned from the elemental analysis, 1H, 13C NMR and IR spectra, since they showed similar patterns of aromatic and pyrimidine protons. In the 13C NMR spectra, the lower field resonances at δ = 178.2–177.4 ppm were assigned to C-2, whereas the resonances at δ = 154.8–153.2 ppm were attributed to C-4 of the pyrimidine ring, respectively. The resonances at δ = 84.1–77.8 and 158.5–156.7 ppm were assigned to C-5 and C-6 of the same ring, respectively. The aromatic carbon atoms were fully analyzed (see Experimental Section). The IR spectra of the free ligands 17 and 21–23 showed the characteristic υ(C=O) bands in the 1626, 1629, 1630 and 1641 cm–1 region, respectively, which shifted to higher wavenumbers in the spectra of the metal complexes 30–33 (1656, 1675, 1653 and 1657 cm–1, respectively) [41–43]. In addition, the bands at 1414, 1416, 1406 and 1414 cm–1 were due to the azo group υ(N=N), respectively, which shifted to higher wavenumbers of the metal complexes in the 1420, 1421, 1425 and 1427 cm–1 range, respectively, indicating the coordination of C=O with one nitrogen atom of the azo group with platinum(II). Furthermore, the bands of the free ligands of υ(C=S) vibrated at 1091, 1100, 1101 and 1122 cm–1, which almost vibrated at the same wavenumbers for the complexes at 1088, 1104, 1100 and 1120 cm–1, respectively. The wavenumbers at ∼800 cm–1 are characteristic for the pyrimidine ring stretching. The band at 554 cm–1 of complex 32 probably belongs to the Pt–N coordinated bond. In conclusion, the above spectral data suggested that the complexes 30–33 exist in square-planar geometries.
Owing to the biological importance of ruthenium complexes [44, 45], we focused our work on the synthesis of a new derivative of ruthenium(III) pyrimidine complex 34 in 43 % yield by treatment of 5 with RuCl3·3H2O in a 2:1 ratio (Scheme 4). The structure of 34 was confirmed by the elemental analysis, 1H, 13C NMR and IR spectra. In the 13C NMR spectrum of 34, the low field frequency at δ = 182.8 ppm was assigned to C-2 (C=S), whereas C-4–C-6 appeared at δ = 156.6, 82.3 and 162.5 ppm, respectively (see Experimental Section). In the IR spectrum, the C=O at C-6 showed two bands: one band appeared at wavenumber 1646 cm–1, which was higher than that of the starting material 5 (1630 cm–1), indicating a coordination of the C=O group of 34 with Ru(III), whereas the other band vibrated at 1600 cm–1 as a non-coordinated C=O group. Similarly, the C=S group showed two bands as well: one band vibrated at 1124 cm–1, which is a higher wavenumber than that for 5 (1100 cm–1), indicating the coordination of the C=S group at C-2 with Ru, whereas the second band at 1100 cm–1 was attributed to the non-coordinated C=S group. The above data suggested that 34 exists as a distorted compressed octahedron.
![Scheme 4: Synthesis of the bis(4-amino-N1-methyl-6-oxo-2-thioxo-pyrimidine)RuCl·H2O complex [(AMThPyrm)2RuCl·H2O] (34).](/document/doi/10.1515/znb-2014-0246/asset/graphic/znb-2014-0246_scheme4.jpg)
Synthesis of the bis(4-amino-N1-methyl-6-oxo-2-thioxo-pyrimidine)RuCl·H2O complex [(AMThPyrm)2RuCl·H2O] (34).
Next, the treatment of 17 with RuCl3·3H2O (1:1 molar ratio) in refluxing MeOH afforded, after purification, the Ru(III) complex 35 (54 %) (Scheme 5). The 13C NMR spectrum showed two lower field resonances at δ = 177.8 and 163.2 ppm assigned to C-2 and C-6 of the pyrimidine ring, respectively, whereas the resonances at δ = 158.2 and 80.4 ppm were attributed to C-4 and C-5 of the same ring, respectively. The aromatic and methyl carbon atoms were fully assigned (see Experimental Section). The IR spectrum of ligand 17 revealed bands at 1626 and 1414 cm–1 assigned to υ(C=O) and υ(N=N), respectively, which shifted to higher wavenumbers (1649 and 1134 cm–1) in the spectrum of complex 35. These differences in wavenumbers indicate that the >C=O and >N=N groups of 17 are involved in the bonding with metal ions. The υ(C=S) vibration of 17 was observed at 1091 cm–1, which remained constant in the spectrum of the complex at 1088 cm–1. The above spectral data suggest the geometrical structure of Ru(III) complex 35 as a distorted octahedral one.
![Scheme 5: Synthesis of the [6-amino-5-((4-chlrophenyl)diazenyl)-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one)Ru(III)Cl3·H2O complex [(AzPyrm-Cl)RuCl3·H2O] (34).](/document/doi/10.1515/znb-2014-0246/asset/graphic/znb-2014-0246_scheme5.jpg)
Synthesis of the [6-amino-5-((4-chlrophenyl)diazenyl)-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one)Ru(III)Cl3·H2O complex [(AzPyrm-Cl)RuCl3·H2O] (34).
2.1 In vitro anti-HIV activity
Compounds 17–35 were tested for their in vitro anti-HIV-1 (strain IIIB) and anti-HIV-2 (strain ROD) activity in human T-lymphocyte (MT-4) cells based on an MTT assay [46]. The results are summarized in Table 1, where the data for nevirapine (BOE/BIRG587) [47] and azidothymidine (DDN/AZT) [48] are included for comparison. Compounds 26 and 30 were found to be the only compounds from the series inhibiting HIV-1 and HIV-2 replication in a cell culture, which showed an IC50 value of >2.07, and >3.02 μg mL–1, respectively, whereas the analogs 22, 25, 27, 29 and 35 exhibited moderate activity against both virus types, with an IC50 value of >9.56, 8.11, 5.23, 8.06 and 10.01 μg mL–1, respectively.
In vitro anti-HIV-1a and anti-HIV-2b activity of the new pyrimidines [Pt(II) and Ru(III) complexes 17–35].
| Compound | Virus strain | EC50 (μg mL–1)c | CC50 (μg mL–1)d | SIe |
|---|---|---|---|---|
| 17 | IIIB | >28.83 | 28.83 | <1 |
| ROD | >28.83 | 28.83 | <1 | |
| 18 | IIIB | >36.21 | 36.21 | <1 |
| ROD | >36.21 | 36.21 | <1 | |
| 19 | IIIB | >65.34 | 65.34 | <1 |
| ROD | >65.34 | 65.34 | <1 | |
| 20 | IIIB | >95.77 | 95.77 | <1 |
| ROD | >92.68 | 95.77 | <1 | |
| 21 | IIIB | >87.22 | 87.22 | <1 |
| ROD | >87.22 | 87.22 | <1 | |
| 22 | IIIB | >9.56 | 9.56 | <1 |
| ROD | >9.56 | 9.56 | <1 | |
| 23 | IIIB | >37.56 | 37.56 | <1 |
| ROD | >37.56 | 37.56 | <1 | |
| 24 | IIIB | >15.45 | 15.45 | <1 |
| ROD | >15.45 | 15.45 | <1 | |
| 25 | IIIB | >8.11 | 8.11 | <1 |
| ROD | >8.11 | 8.11 | <1 | |
| 26 | IIIB | >2.07 | 2.07 | <1 |
| ROD | >2.07 | 2.07 | <1 | |
| 27 | IIIB | >5.23 | 5.23 | <1 |
| ROD | >5.23 | 5.23 | <1 | |
| 28 | IIIB | >8.06 | 8.06 | <1 |
| ROD | >8.06 | 8.06 | <1 | |
| 29 | IIIB | >54.23 | 54.23 | <1 |
| ROD | >54.23 | 54.23 | <1 | |
| 30 | IIIB | >3.02 | 3.02 | <1 |
| ROD | >3.02 | 3.02 | <1 | |
| 31 | IIIB | >21.25 | 21.25 | <1 |
| ROD | >21.25 | 21.25 | <1 | |
| 32 | IIIB | >39.34 | 39.34 | <1 |
| ROD | >39.34 | 39.34 | <1 | |
| 33 | IIIB | >31.22 | 31.22 | <1 |
| ROD | >31.22 | 31.22 | <1 | |
| 34 | IIIB | >29.34 | 29.34 | <1 |
| ROD | >29.34 | 29.34 | <1 | |
| 35 | IIIB | >10.01 | 10.01 | <1 |
| ROD | >10.01 | 10.01 | <1 | |
| AZT | IIIB | 0.0022 | >25 | >11 363 |
| ROD | 0.00094 | >25 | >26 596 | |
| Nevirapine | IIIB | 0.050 | >4.00 | >80 |
| ROD | >4.00 | >4.00 | >1 |
aAnti-HIV-1 activity measured with strain IIIB.
bAnti-HIV-2 activity measured with strain ROD.
cCompound concentration required to achieve 50 % protection of the MT-4 cells from the HIV-1- and HIV-2-induced cytopathogenic effect.
dCompound concentration that reduces the viability of mock-infected MT-4 cells by 50 %.
eSI: selectivity index (CC50/EC50).
In conclusion, the data in Table 1 suggested that the incorporation of sulfadiazine, sulfamerazine or sulfamethazine into the C-5 of the pyrimidine backbone, via the azo group, would enhance the anti-HIV activity. In addition, the coordination of PtCl2 with the carbonyl oxygen atom (C4=O) and the (C-5-) N atom of the azo group in compound 30 enhanced the anti-HIV activity as well. Therefore, the in vitro screening led to the identification of the new pyrimidines 26 and 30 as new anti-HIV candidates, being promising agents for further structural modification and pharmacological evaluation.
2.2 Molecular modeling analysis
The azothiopyrimidine analog 26, owing to its anti-HIV activity, was selected for the molecular modeling study, which was performed to understand the binding mode of these analogs with the HIV-RT binding pocket (NIBP) (PDB code: 1DTT [49]). The Sybyl-X 1.1 software was used, and the results were visualized with Pymol [50].
The molecular modeling study of compound 26 showed a binding energy score of –8.0, which revealed a selective interaction with the amino acids of the RT HIV enzyme. As shown in Figure 3, the aromatic ring of the sulfonamide group of 26 fitted into an aromatic-rich subpocket surrounded by the aromatic side chains of Tyr179, and a hydrogen bond between the hydroxyl group of Tyr179 and the nitrogen atom of the azo group was observed. The sulfonamide moiety was located in a favorable position of the pocket of RT enzyme, leading to the formation of a hydrogen bond between the oxygen atom of the SO2 group and the NH2 of Lys101, in addition to the hydrogen bond between the NH2 of the sulfonamide group and the oxygen atom of the carboxylic group of Thr132 of the RT enzyme. Overall, the combination of hydrophobic interaction and π stacking appears to govern the binding of 130 with HIV RT.
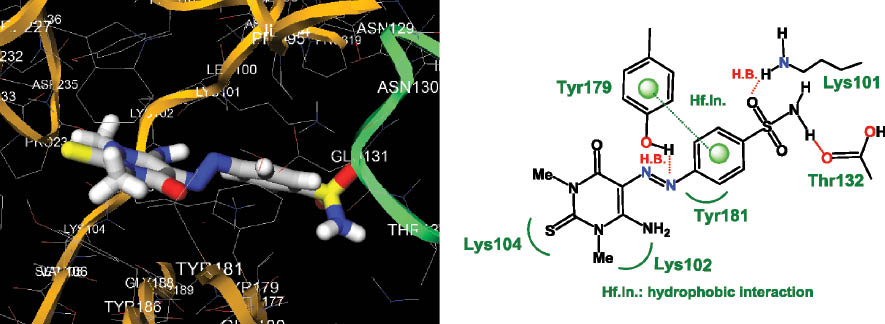
Docked conformation of 26 showing three hydrogen bonds: Lys101 with the oxygen atom of the SO2 group, Thr132 with the NH group of the sulfonamide residue and OH of Tyr179 with the nitrogen atom of the azo group. In addition, a hydrophobic interaction was observed between the phenyl group of the benzenesulfonamido moiety and the aromatic ring of Tyr179 of the reverse transcriptase (RT) enzyme residues.
2.3 In vitro antibacterial activity
The thiopyrimidines 17–35 and 31–33 were screened in vitro for the presence of antibacterial constituents against two strains of bacteria, Staphylococcus aureus and Escherichia coli, by the disc diffusion method [51]. Muller-Hinton agar (MH) was used as the culture media for antibacterial activity. Recommended concentrations of 100, 200 and 300 and 500 μg mL–1 of the test samples in DMSO as a solvent were introduced in the respective method. The bacterial inoculum was uniformly spread using a sterile cotton swab on a sterile Petri dish MH agar. Fifty microliters from 1 mg mL–1 concentration of chemical products was added to each well (7-mm-diameter holes were cut in the agar gel, 20 mm apart from one another). The plates were incubated for 24 h at 36 ± 1 °C under aerobic conditions. After incubation, confluent bacterial growth was observed. Inhibition of the bacterial growth was measured in millimeters using a caliper vernia.
Compound 26, carrying a sulfamerazine scaffold, was the most active candidate in the series, with inhibition zones of 12–13 and 16 mm against S. aureus and E. coli bacteria, at a concentration of 100 μg mL–1, respectively, in comparison with the antibacterial drug sulfamerazine itself [52]. The docking study of 26 showed a binding energy score of –9.2 with the aspartate transaminase (AST) enzyme of E. coli, indicating highly selective interactions with the enzyme pocket, which led to a higher binding energy score.
3 Experimental section
3.1 General
Melting points were uncorrected and were measured on a Büchi melting point apparatus (B-545, Büchi Labortechnik AG, Flawil, Switzerland). Microanalytical data were obtained with a Vario elemental analyzer (Shimadzu, Kyoto, Japan). NMR spectra were recorded on a spectrometer (Bruker, Rheinstetten, Germany) at 400 and 600 MHz for 1H and at 150.91 MHz for 13C, with tetramethylsilane as an internal standard, and on a δ scale in parts per million. Signal assignments for protons were performed by selective proton decoupling or by COSY spectra. Heteronuclear assignments were verified by HSQC, HMBC and DFQ-COSY experiments. Mass spectra (EI, 70 eV, and fast atom bombardment) were recorded on a MAT 8200 spectrometer (Finnegan MAT, Ringoes, NJ, USA). TLC plates (60 F254) were purchased from Merck.
3.2 General procedure for the preparation of 6-amino-5-(aryldiazenyl)-N1, N3-dimethyl-2-thioxo-pyrimidin-4-one derivatives 17–27
A solution of substituted aniline 6–16 (2.90 mmol) in 6 mol/L of HCl (4.0 mL) was cooled to 0–5 °C, and then NaNO2 (58 mg, 0.84 mmol) in water (2.0 mL) was added dropwise with stirring. After the addition was completed, the solution was stirred for another 15 min and checked with iodine-starch paper to destroy the excess of HNO2. The diazonium salt solution was then poured onto a solution of 6-amino-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one (4) (500 mg, 2.90 mmol) or of the N1-methyl analog 5 in EtOH and stirred for 30 min. Potassium acetate (485 mg) was then added, and the mixture was stirred for 16 h at room temperature. The resulting precipitate was filtered, washed with water and dried in a vacuum desiccator over P2O5 to give the desired azothiopyrimidine derivatives.
3.2.1 6-Amino-5-((4-chlorophenyl)diazenyl)- N1,N3-dimethyl-2-thioxo-pyrimidin-4-one (17)
From 4-chloroaniline 6 (450 mg). Yield: 704 mg (82 %), m. p. 226–228 °C (dec.), Rf = 0.9. – IR (KBr, ν, cm–1): 3297 (NH2, br.), 1626 (C=O), 1509 (C=C), 1414 (N=N), 1091 (C=S). – 1H NMR ([D6]DMSO): δ = 9.14, 8.84 (2 × s, 2H, NH2), 7.75 (d, 2H, J5′,6′ = 8.0 Hz, Harom.-3′ + Harom.-5′), 7.55 (d, 2H. J2′,3′ = 8.0 Hz, Harom.-2′ + Harom.-6′), 3.89 (s, 3H, N3-CH3), 3.69 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.4 (C(2)pyrimid.), 165.5 (C(6)pyrimid.), 157.5 (C(4)pyrimid.), 148.4 (C(1′)arom.), 134.5 (C4′)arom.), 129.8 (C(2′)arom. + C(6′)arom.), 123.1 (C(3′)arom. + (C5′)arom.), 77.8 (C(5)pyrimid.), 37.6 (N3-CH3), 36.1 (N1-CH3) ppm. – Anal. for C12H12ClN5OS (309.77): calcd. C 46.53, H 3.09, N, 22.61; found: C 46.33, H 2.94, N 22.39.
3.2.2 6-Amino-5-((4-bromophenyl)diazenyl)- N1,N3-dimethyl-2-thioxo-pyrimidin-4-one (18)
From 4-bromoaniline 7 (502 mg). Yield: 631 mg (61 %), m. p. 205–209 °C, Rf = 0.71. – IR (KBr, ν, cm–1): 3294 (NH2, br.), 1630 (C=O), 1511 (C=C), 1416 (N=N), 1096 (C=S). – 1H NMR ([D6]DMSO): δ = 9.16, 8.83 (2 × s, 2H, NH2), 7.76 (d, 2H, J5′.6′ = 8.6 Hz, Harom.-3′ + Harom.-5′), 7.46 (d, 2H, J2′,3′ = 8.6 Hz, Harom.-2′ + Harom.-6′), 3.88 (s, 3H, N3-CH3), 3.68 (s, 3H, N3-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.4 (C(2)pyrimid.), 164.8 (C(6)pyrimid.), 157.4 (C(4)pyrimid.), 148.4 (C(1′)arom.), 132.8 (C(3′)arom. + C(5′)arom.), 129.8 (C(2′)arom. + C(6′)arom.), 123.4 (C4′)arom.), 78.1 (C(5)pyrimid.), 37.6 (N3-CH3), 36.1 (N1-CH3) ppm. – Anal. for C12H12BrN5OS (354.23): calcd. C 40.69, H 3.41, N 19.77; found: C 46.48,H 3.38, N 19.53.
3.2.3 6-Amino-N1,N3-dimethyl-5-(4-nitrophenyl)diazenyl)-2-thioxo-pyrimidin-4-one (19)
From 4-nitroaniline 8 (390 mg). Yield: 806 mg (92 %), m. p. 228–230 °C (dec.), Rf = 0.63. – IR (KBr, ν, cm–1): 3297 (NH2, br.), 1626 (C=O), 1509 (C=C), 1414 (N=N), 1091 (C=S). – 1H NMR ([D6]DMSO): δ = 9.09 (s, 2H, NH2), 8.34 (d, 2H, J5′,6′ = 9.0 Hz, Harom.-3′ + Harom.-5′), 7.91 (d, 2H, J2′,3′ = 9.0 Hz, Harom.-2′ + Harom.-6′), 3.89 (s, 3H, N3-CH3), 3.69 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.7 (C(2)pyrimid.), 163.8 (C(6)pyrimid.), 157.3 (C(4)pyrimid.), 148.7 (C(1′)arom.), 148.4 (C(4′)arom.), 125.5 (C(3′)arom. + C(5′)arom.), 122.1 (C(2′)arom. + C(6′)arom.), 75.0 (C(5)pyrimid.), 37.7 (N3-CH3), 36.2 (N1-CH3) ppm. – Anal. for C12H12NO2N5OS (320.33): calcd. C 44.99, H 3.78, N 26.24; found: C 44.72, H 3.61, N 26.02.
3.2.4 6-Amino-N1,N3-dimethyl-2-thioxo-5-(p-tolyldiazenyl)-2-thioxo-pyrimidin-4-one (20)
From 4-methylaniline 9 (312 mg). Yield: 540 mg (70 %), m. p. 235–237 °C (dec.), Rf = 0.76. – IR (KBr, ν, cm–1): 3297, 3257 (NH2), 1630 (C=O), 1511 (C=C), 1418 (N=N), 1101 (C=S). – 1H NMR ([D6]DMSO): δ = 11.64, 8.64 (2xbr s., 2H, NH2), 7.61 (d, 2H, J5′,6′ = 8.0 Hz, Harom.-3′ + Harom.-5′), 7.29 (d, 2H, J2′,3′ = 8.0 Hz, Harom.-2′ + Harom.-6′), 3.88 (s, 3H, N3-CH3), 3.69 (s, 3H, N1-CH3), 2.36 (s, 3H, Ph-Me) ppm. – 13C NMR ([D6]DMSO): δ = 177.2 (C(2)pyrimid.), 163.9 (C(6)pyrimid.), 157.6 (C(4)pyrimid.), 148.2 (C(1′)arom.), 138.8 (C(4′)arom.), 130.6 (C(3′)arom. + C(5′)arom.), 128.1 (C(2′)arom. + C(6′)arom.), 76.8 (C(5)pyrimid.), 37.5 (N3-CH3), 36.1 (N1-CH3), 21.3 (Me) ppm. – Anal. for C13H15N5OS (289.10): calcd. C 53.96, H 5.23, N 24.20; found: C 53.72, H 35.16, N 24.03.
3.2.5 4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)benzoic acid (21)
From p-aminobenzoic acid 10 (402 mg). Yield: 800 mg (86 %), m. p. 247–249 °C, Rf = 0.42. – IR (KBr, ν, cm–1): 3381 (NH2, br.), 1629 (C=O), 1503 (C=C), 1416 (N=N), 1100 (C=S). – 1H NMR ([D6]DMSO): δ = 12.63 (s, 1H, CO2H), 11.80 (br s., 2H, NH2), 8.04 (d, 2H, J5′,6′ = 8.0 Hz, Harom.-3′ + Harom.-5′), 7.78 (d, 2H. J2′,3′ = 8.0 Hz, Harom.-2′ + Harom.-6′), 3.73 (s, 3H, N3-CH3), 3.69 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 179.1 (C(2)pyrimid.), 167.5 (CO2H), 159.5 (C(6)pyrimid.), 157.0 (C(4)pyrimid.), 153.0 (C(1′)arom.), 141.1 (C(4′)arom.), 131.0 (C(3′)arom. + C(5′)arom.), 121.4 (C(2′)arom. + C(6′)arom.), 75.0 (C(5)pyrimid.), 36.4 (N3-CH3), 35.7 (N1-CH3) ppm. – Anal. for C13H13N5O3S (319.34): calcd. C 48.89, H 4.10, N, 21.93; found: C 48.68, H 4.02, N 21.78.
3.2.6 4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)benzensulfonamide (22)
From p-aminobenenezsulfonamide 11 (502 mg). Yield: 740 mg (81 %), m. p. 259–60 °C, Rf = 0.75. – IR (KBr, ν, cm–1): 3372, 3247 (NH2), 1630 (C=O), 1502 (C=C), 1406 (N=N), 1101 (C=S). – 1H NMR ([D6]DMSO): δ = 11.70, 8.92 (2 × s, 2H, NH2), 7.93 (d, 2H, J5′,6′ = 8.5 Hz, Harom.-3′ + Harom.-5′), 7.86 (d, 2H, J2′,3′ = 8.5 Hz, Harom.-2′ + Harom.-6′), 7.41 (s, 2H, SO2NH2), 3.89 (s, 3H, N3-CH3), 3.69 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.5 (C(2)pyrimid.), 157.4 (C(6)pyrimid.), 154.4 (C(4)pyrimid.), 148.5 (C(1′)arom.), 143.5 (C4′)arom.), 127.4 (C(2′)arom. + C(6′)arom.), 121.7 (C(3′)arom. + C(5′)arom.), 77.5 (C(5)pyrimid.), 37.6 (N3-CH3), 36.1 (N1-CH3) ppm. – Anal. for C12H14N6O3S2 (354.40): calcd. C 40.67, H 3.98, N 23.71; found: C 40.23, H 3.89, N 23.50.
3.2.7 4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)benzensulfonic acid (23)
From p-aminobenzenesulfonic acid 12 (400 mg). Yield: 830 mg (80 %), m. p. 273–275 °C, Rf = 0.2. – IR (KBr, ν, cm–1): 3442 (NH2, br.), 1641 (C=O), 1508 (C=C), 1414 (N=N), 1120 (C=S). – 1H NMR ([D6]DMSO): δ = 11.71, 8.86 (2 × s, 2H, NH2), 7.72 (d, 2H, J2′,3′ = 8.5 Hz, Harom.-2′ + Harom.-6′), 7.68 (d, 2H. J5′,6′ = 8.5 Hz, Harom.-3′ + Harom.-5′), 3.90 (s, 3H, N3-CH3), 3.70 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.3 (C(2)pyrimid.), 158.9 (C(6)pyrimid.), 157.5 (C(4)pyrimid.), 148.5 (C(4′)arom.), 127.1 (C(2′)arom. + C(6′)arom.), 121.0 (C(3′)arom. + C(5′)arom.), 77.3 (C(5)pyrimid.), 36.4 (N3-CH3), 35.7 (N1-CH3) ppm. – Anal. for C12H13N5O4S2 (355.39): calcd. C 40.27, H 3.55, N 19.49; found: C 40.27, H 3.55, N 19.49.
3.2.8 N-((4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)phenyl)sulfonyl) acetamide (24)
From N-((4-aminophenyl)sulfonyl)acetamide (13) (625 mg). Yield: 650 mg (56 %), m. p. 261–263 °C (dec.), Rf = 0.52. – IR (KBr, ν, cm–1): 3385, 3279 (NH2), 1631 (C=O), 1502 (C=C), 1408 (N=N), 1099 (C=S). – 1H NMR ([D6]DMSO): δ = 11.75, 9.02 (2 × br s, 2H, NH2), 8.00 (d, 2H, J2′,3′ = 7.8 Hz, Harom.-2′ + Harom.-6′), 7.88 (d, 2H, J5′,6′ = 7.8 Hz, Harom.-3′ + Harom.-5′), 3.90 (s, 3H, N3-CH3), 3.70 (s, 3H, N1-CH3), 1.96 (s, 3H, NHCOMe) ppm. – 13C NMR ([D6]DMSO): δ = 177.8 (C(2)pyrimid.), 169.3 (C=O), 157.3 (C(6)pyrimid.), 156.6 (C(4)pyrimid.), 148.8 (C(1′)arom.), 138.2 (C4′)arom.), 129.4 (C(2′)arom. + C(6′)arom.), 121.7 (C(3′)arom. + C(5′)arom.), 77.3 (C(5)pyrimid.), 37.6 (N3-CH3), 36.2 (N1-CH3), 23.8 (NHCOMe) ppm. – Anal. for C12H13N5O4S2 (396.44): calcd. C 42.41, H 4.07, N 21.2; found: C 42.20, H 3.95, N 20.92.
3.2.9 4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)-N-pyrimidin-2-yl-benzene sulfonamide (25)
From sulfadiazine (14) (731 mg). Yield: 750 mg (59 %), m. p. 158–160 °C (dec.), Rf = 0.57. – IR (KBr, ν, cm–1): 3385, 3279 (NH2), 1630 (C=O), 1514 (C=C), 1421 (N=N), 1099 (C=S). – 1H NMR ([D6]DMSO): δ = 11.65 (s, 2H, NH2), 9.07 (s, H, NH), 8.52 (d, 2H, J4″,5″ = 5.0 Hz, Hpyrimid.-4″ + Hpyrimid.-6″), 8.08 (d, 2H, J2′,3′ = 7.5 Hz, Harom.-2′ + Harom.-6′), 7.88 (d, 2H. J5′,6′ = 7.5 Hz, Harom.-3′ + Harom.-5′), 7.06 (t, 1H, J5″,6″ = 5.0 Hz, H-5″pyrimid.), 3.90 (s, 3H, N3-CH3), 3.69 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 176.4 (C(2)pyrimid), 168.2 (C(2″)pyrimid.), 157.1 (C(6)pyrimid.), 156.7 (C(4″)pyrimid. + C(6″)pyrimid.), 155.3 (C(4)pyrimid.), 149.9 (C(1′)arom.), 139.6 (C(4′)arom.), 131.3 (C(2′)arom. + (C6′)arom.), 123.8 (C(3′)arom. + C(5′)arom.), 100.0 (C(5″)pyrimid.), 82.7 (C(5)pyrimid.), 38.7 (N3-CH3), 36.2 (N1-CH3) ppm. – Anal. for C16H16N8O3S2 (432.48): calcd. C 44.43, H 3.73, N 25.91; found: C 44.17, H 3.69, N 25.63.
3.2.10 4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)-N-(4-methylpyrimidin-2-yl)benzensulfonamide (26)
From sulfamerazine (15) (772 mg). Yield: 520 mg (40 %), m. p. 207–209 °C (dec.), Rf = 0.76. – IR (KBr, ν, cm–1): 3392, 3282 (NH2), 1624 (C=O), 1512 (C=C), 1418 (N=N), 1100 (C=S). – 1H NMR ([D6]DMSO): δ = 12.78, 9.14 (2 × s, 2H, NH2), 8.36 (d, H, J5″,6″ = 5.5 Hz, Hpyrimid-6″), 8.18 (d, 2H, J2′,3′ = 7.5 Hz, Harom.-2′ + Harom.-6′), 7.83 (d, 2H, J5′,6′ = 7.5 Hz, Harom.-3′ + Harom.-5′), 7.05 (d, 1H, H-5″pyrimid.), 3.75 (s, 3H, N3-CH3), 3.72 (s, 3H, N1-CH3), 2.39 (s, 3H, C4″-Me) ppm. – 13C NMR ([D6]DMSO): δ = 179.0 (C(2)pyrimid.), 171.9 (C(4″)pyrimid.), 167.4 (C(2″)pyrimid.), 158.8 (C(6)pyrimid.), 155.2 (C(4)pyrimid.), 153.2 (C(6″)pyrimid.), 148.1 (C(1′)arom.), 140.6 (C(4′)arom.), 128.3 (C(2′)arom. + C(6′)arom.), 122.6 (C(3′)arom. + C(5′)arom.), 109.3 (C(5″)pyrimid.), 82.8 (C(5)pyrimid.), 38.6 (N3-CH3)pyrimid.), 35.8 (N1-CH3)pyrimid.), 23.0 (Mepyrimid.) ppm. – Anal. for C17H18N8O3S2 (446.51): calcd. C 45.73, H, 4.06, N 25.10; found: C 45.47, H 4.01, N 24.89.
3.2.11 4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)-N-(4,6-dimethyl-pyrimidin-2-yl)benzensulfonamide (27)
From sulfamethazine (16) (813 mg). Yield: 560 mg (42 %), m. p. 199–200 °C (dec.), Rf = 0.6. – IR (KBr, ν, cm–1): 3389, 3274 (NH2), 1622 (C=O), 1509 (C=C), 1422 (N=N), 1108 (C=S). – 1H NMR ([D6]DMSO): δ = 11.72 (br s., 2H, NH2), 8.96 (br s., 1H, NH), 8.08 (d, 2H, J2′,3′ = 8.0 Hz, Harom.-2′ + Harom.-6′), 7.83 (d, 2H. J5′,6′ = 8.0 Hz, Harom.-3′ + Harom.-5′), 6.67 (s, 1H, H-5″pyrimid.), 3.89 (s, 3H, N3-CH3), 3.69 (s, 3H, N1-CH3), 2.30 (2 × s, 6H, 2 × Mepyrimid.) ppm. – 13C NMR ([D6]DMSO): δ = 177.5 (C(2)pyrimid.), 169.6 (C(2″)pyrimid.), 167.4 (C(4″)pyrimid. + C(6″)pyrimid.), 164.9 (C(6)pyrimid.), 151.0 (C(4)pyrimid.), 143.5 (C(1′)arom.), 140.6 (C(4′)arom.), 129.0 (C(2′)arom. + C(6′)arom.), 121.1 (C(3′)arom. + C(5′)arom.), 113.1 (C(5″)pyrimid.), 72.2 (C(5)pyrimid.), 37.6 (N3-CH3)pyrimid.), 36.1 (N1-CH3)pyrimid.), 23.0 (2 × Mepyrimid.) ppm. – Anal. for C18H20N8O3S2 (460.53): calcd. C 46.94, H 4.38, N 24.33; found: C 46.73, H 4.27, N 24.07.
3.2.12 4-((6-Amino-N1-methyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)benzensulfonamide (28)
From 4-amino-benzenesulfonamide (11) (546 mg). Yield: 742 mg (68 %), m. p. 251–253 °C, Rf = 0.13. – IR (KBr, ν, cm–1): 3388, 3279 (NH2), 1627 (C=O), 1515 (C=C), 1420 (N=N), 1094 (C=S). – 1H NMR ([D6]DMSO): δ = 11.87, 8.93 (2 × s, 2H, NH2), 7.93 (d, 2H, J5′,6′ = 8.7 Hz, Harom.-3′ + Harom.-5′), 7.85 (d, 2H, J2′,3′ = 8.7 Hz, Harom.-2′ + Harom.-6′), 7.40 (s, 2H, SO2NH2), 3.81 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 176.8 (C(2)pyrimid.), 164.1 (C(6)pyrimid.), 157.4 (C(4)pyrimid.), 149.6 (C(1′)arom.), 143.3 (C(4′)pyrimid.), 127.4 (C(3′)arom. + C(5′)arom.), 121.6 (C(2′)arom. + C(6′)arom.), 78.8 (C(5)pyrimid.), 35.7 (N1-CH3)pyrimid.) ppm. – Anal. for C11H12N6O3S2 (340.38): calcd. C 38.81, H 3.55, N 24.69; found: C 38.60, H 3.46, N 24.45.
3.2.13 [Bis(4-amino-N3-methyl-6-oxo-2-thioxo-pyrimidin-1-yl)]Pt(II), [(AMeThPyrm)2Pt] (29)
To a stirred solution of 5 (116 mg, 0.74 mmol) in MeOH (2 mL) was added a solution of PtCl2 (99 mg, 0.34 mmol) in MeOH (3 mL) (2:1 molar ratio), and the mixture was heated under reflux for 2 h. The solid was separated on cooling, filtered, washed with MeOH and dried in a vacuum desiccator to give 138 (90 mg, 48 %), m. p. 270–271 °C (dec.). – IR (KBr, ν, cm–1): 3348, 3169 (NH2), 1650 (C=O), 1602 (C=C), 1134 (C=S). – 1H NMR ([D6]DMSO): δ = 6.90 (br s, 2H, NH2), 4.88 (s, 1H, H-5), 3.88 (s, 3H, N3-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 179.7 (C(2)pyrimid.), 164.4 (C(6)pyrimid.), 160.1 (C(4)pyrimid.), 79.6 (C(5)pyrimid.), 34.6 (N3-CH3) ppm. – Anal. for C10H12N6O2PtS2 (507.46): calcd. C 23.67, H 2.38, N 16.56; found: C 23.43, H 2.21, N 15.28.
3.3 General procedure for the preparation of metal pyrimidine complexes 30–33
To a solution of the aryl-azothiopyrimidine (0.37 mmol) in MeOH (3 mL) was added a solution of PtCl2 (99 mg, 0.37 mmol) in MeOH (3 mL) (1:1 molar ratio), and the mixture was heated under reflux for 2 h. After cooling, the solid was filtered and dried in a vacuum desiccator.
3.3.1 [(6-Amino-5-(4-chlorophenyl)diazenyl)-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one)]Pt(II)Cl2, [(AzPyrm-Cl)PtCl2] (30)
From 17 (115 mg). Yield: 120 mg (55 %), m. p. 245–247 °C (dec.). – IR (KBr, ν, cm–1): 3433, 3303 (NH2), 1656 (C=O), 1488 (C=C), 1437 (N=N), 1088 (C=S). – 1H NMR ([D6]DMSO): δ = 10.95, 9.26 (2 × s, 2H, NH2), 7.96 (d, 2H, J2′,3′ = 7.0 Hz, Harom.-2′ + Harom.-6′), 7.58 (d, 2H. J5′,6′ = 7.0 Hz, Harom.- 3′ + Harom.-5′), 3.92 (s, 3H, N3-CH3), 3.68 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.6 (C(2)pyrimid.), 156.7 (C(6)pyrimid.), 154.8 (C(4)pyrimid.), 149.8 (C(1′)arom.), 133.1 (C4′)arom.), 129.9 (C(2′)arom. + C(6′)arom.), 122.3 (C(3′)arom. + C(5′)arom.), 38.6 (N3-CH3), 35.8 (N1-CH3), 77.8 (C(5)pyrimid.) ppm. – Anal. for C12 H12Cl3N5OPtS (575.76): calcd. C 25.03, H 2.10, N 12.16; found: C 24.82, H 2.01, N 11.89.
3.3.2 [(4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)benzoic acid)]Pt(II)Cl2, [(AzPyrm-CO2H)PtCl2] (31)
From 21 (118 mg). Yield: 103 mg (46 %), m. p. 251–253 °C. – IR (KBr, ν, cm–1): 3452 (NH2, br.), 1675 (C=O), 1503 (C=C), 1421 (N=N), 1104 (C=S). – 1H NMR ([D6]DMSO): δ = 12.36 (s, 1H, CO2H), 11.14, 9.38 (2 × s, 2H, NH2), 8.04–8.00 (m, 4H, Harom.-2′ + Harom.-6′ + Harom.-3′ + Harom.-5′), 3.93 (s, 3H, N3-CH3), 3.69 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 178.2 (C(2)pyrimid.), 167.4 (C2OH), 157.4 (C(6)pyrimid.), 154.6 (C(4)pyrimid.), 154.2 (C(1′)arom.), 131.1 (C(2′)arom. + C(6′)arom.), 128.1 ((C4′)arom.), 121.3 (C(3′)arom. + C(5′)arom.), 82.0 (C(5)pyrimid.), 38.0 (N3-CH3), 36.9 (N1-CH3) ppm. – Anal. for C13H15Cl2N5O4PtS2 (603.34): calcd. C 25.88, H 2.51, N 11.61; found: C 25.88, H 2.51, N 11.78.
3.3.3 [4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)benzenesulfonamide]Pt(II)Cl2, [(AzPyrm-SO2NH2)PtCl2] (32)
From 22 (118 mg). Yield: 110 mg (45 %), m. p. 249–250 °C (dec.). – IR (KBr, ν, cm–1): 3362, 3268 (NH2), 1653 (C=O), 1484 (C=C), 1450 (N=N), 1100 (C=S). – 1H NMR ([D6]DMSO): δ = 11.70, 8.92 (2 × s, 2H, NH2), 8.11 (d, 2H, J5′,6′ = 8.0 Hz, Harom.-3′ + Harom.-5′), 7.93 (d, 2H. J2′,3′ = 8.0 Hz, Harom.-2′ + Harom.-6′), 7.45 (s, 2H, SO2NH2), 3.94 (s, 3H, N3-CH3), 3.60 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.4 (C(2)pyrimid.), 158.5 (C(6)pyrimid.), 153.2 (C(4)pyrimid.), 149.6 (C(1′)arom.), 149.3 (C4′)arom.), 127.5 (C(2′)arom. + (C6′)arom.), 120.4 (C(3′)arom. + (C5′)arom.), 82.6 (C(5)pyrimid.), 38.4 (N3-CH3), 35.8 (N1-CH3) ppm. – Anal. for C12H16Cl2N6O4PtS2 (638.41): calcd. C 22.58, H 2.58, N 13.16; found: C 22.36, H 2.39, N 13.31.
3.3.4 [4-((6-Amino-N1,N3-dimethyl-4-oxo-2-thioxo-pyrimidin-5-yl)diazenyl)benzenesulfonic acid]Pt(II)Cl2, [(AzPyrm-SO3H)PtCl2] (33)
From 23 (131 mg). Yield: 140 mg (58 %), m. p. 281–283 °C. – IR (KBr, ν, cm–1): 3432, 3067 (NH2), 1657 (C=O), 1488 (C=C), 1443 (N=N), 1120 (C=S). – 1H NMR ([D6]DMSO): δ = 11.23 (br s., 2H, NH2), 7.91 (d, 2H, J3′,5′ = 8.3 Hz, Harom.-3′ + Harom.-5′), 7.71 (d, 2H, J2′,3′ = 8.3 Hz, Harom.-2′ + Harom.-6′), 3.94 (s, 3H, N3-CH3), 3.68 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.5 (C(2)pyrimid.), 157.1 (C(6)pyrimid.), 153.4 (C(4)pyrimid.), 148.6 (C(1′)arom.), 143.8 (C4′)arom.), 127.2 (C(2′)arom. + (C6′)arom.), 122.0 (C(3′)arom. + (C5′)arom.), 84.1 (C(5)pyrimid.), 36.4 (N3-CH3), 35.7 (N1-CH3) ppm. – Anal. for C12H15Cl2N5O5PtS2 (639.40): calcd. C 22.54, H 2.36, N 10.95; found: C 22.37, H 2.48, N 10.80.
3.3.5 [Bis(4-amino-N3-methyl-6-oxo-2-thioxo-pyrimidin-1-yl)]Ru(III)Cl·H2O, [(AMeThPyrm)2RuCl·H2O] (34)
To a stirred solution of 5 (116 mg, 0.74 mmol) in MeOH (3 mL) was added a solution of RuCl3·3H2O (97 mg, 0.37 mmol) in MeOH (3 mL) (2:1 molar ratio), and the mixture was heated under reflux for 2 h. After cooling, the solid complex was filtered, washed with MeOH and dried in a vacuum desiccator to give 34 (150 mg, 43 %), m. p. >295 °C (dec.). – IR (KBr, ν, cm–1): 3351, 3183 (NH2), 1646 (coord. C=O), 1600 (non-coord. C=O), 1509 (C=C), 1124 (coord. C=S), 1100 (non-coord. C=S). – 1H NMR ([D6]DMSO): δ = 7.42 (br s, 2H, NH2), 7.30 (br s, 2H, NH2), 5.72 (s, H, H-5), 3.68 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 182.8 (C(2)pyrimid.), 162.5 (C(6)pyrimid.), 156.6 (C(4)pyrimid.), 82.3 (C(5)pyrimid.), 34.9 (N1-CH3) ppm. – Anal. for C10H14ClN6O3RuS2 (466.91): calcd. C 25.72, H 3.02, N 18.00; found: C 25.41, H 3.14, N 17.38.
3.3.6 [6-Amino-5-((4-chlorophenyl)diazenyl)-N1,N3-dimethyl-2-thioxo-pyrimidin-4-one)]Ru(III)Cl3·H2O, [(AzPyrm-Cl)RuCl3·H2O] (35)
This complex was prepared by following the procedure for the preparation of 34, from the azo-pyrimidine 17 (109 mg, 0.37 mmol) and RuCl3·3H2O (97 mg, 0.37 mmol) (1:1 molar ratio). Yield: 107 mg (54 %), m. p. 241–244 °C (dec.). IR (KBr, ν, cm–1): 3418 br. (NH2, br.), 1649 (C=O), 1489 (C=C), 1437 (N=N), 1088 (C=S). – 1H NMR ([D6]DMSO): δ = 8.28 (br s, 2H, NH2), 7.99 (d, 2H, J5′,6′ = 7.6 Hz, Harom.-3′ + Harom.-5′), 7.56 (d, 2H. J2′,3′ = 7.6 Hz, Harom.-2′ + Harom.-6′), 3.94 (s, 3H, N3-CH3), 3.56 (s, 3H, N1-CH3) ppm. – 13C NMR ([D6]DMSO): δ = 177.8 (C(2)pyrimid.), 163.2 (C(6)pyrimid.), 158.2 (C(4)pyrimid.), 150.0 (C(1′)arom.), 133.1 (C4′)arom.), 129.9 (C(2′)arom. + (C6′)arom.), 122.3 (C(3′)arom. + (C5′)arom.), 80.4 (C(5)pyrimid.), 36.5 (N3-CH3), 35.8 (N1-CH3) ppm. – Anal. for C12H14Cl4N5O2RuS (535.22): calcd. C 26.93, H 2.64, N 13.09; found: C 26.61, H 2.54, N 13.21.
Acknowledgments
We thank Prof. C. Pannecouque of Rega Institute for Medical Research, Katholieke Universiteit, Leuven, Belgium, for the anti-HIV screening. Mr. U. Haunz and Miss A. Friemel of the Chemistry Department, University of Konstanz, Germany, are highly acknowledged for the NMR experiments.
References
[1] K. S. Jain, T. S. Chitre, P. B. Miniyar, M. K Kathiravan, V. S. Bendre, V. S. Veer, S. R. Shahane, C. Shishoo, J.Curr. Sci.2006, 90, 793 (review).Search in Google Scholar
[2] Y. Yamazi, M. Takahashi, Y. Todome, Proc. Soc. Exp. Biol. Med.1970, 133, 674.Search in Google Scholar
[3] M. N. Prichard, D. C. Quenelle, C. B. Hartline, E. A. Harden, G. Jefferson, S. L. Frederick, S. L. Daily, R. J. Whitley, K. N. Tiwari, J. A. Maddry, J. A. Secrist, J. R. Kern, Antimicrob. Agents Chemother.2009, 53, 5251.Search in Google Scholar
[4] M. M. Ghorab, S. G. Abdel-Hamide, Indian J. Heterocycl. Chem.1995, 5, 115.Search in Google Scholar
[5] D. H. Patel, B. D. Mistry, K. R. Desai, Indian J. Heterocyclic Chem.2003, 13, 179.Search in Google Scholar
[6] S. H. Bantawal, M. Manjathuru, K. S. Mari, K. M. Padiyath, Bioorg. Med. Chem.2006, 4, 2040.Search in Google Scholar
[7] P. Sharma, N. Rane, V. K. Gurram, Bioorg. Med. Chem. Lett.2004, 14, 4185.Search in Google Scholar
[8] A. B. Adnan, T. Z. Hesham, A. F. Sherif, M. B. Azza, Eur. J. Med. Chem.2003, 38, 37.Search in Google Scholar
[9] M. T. Cocco, C. Congiu, V. Lilliu, Bioorg. Med. Chem.2006, 14, 366.Search in Google Scholar
[10] C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven, J. Scheiner, Nature1957, 179, 663.10.1038/179663a0Search in Google Scholar
[11] J. F. Beattie, G. A. Breault, R. P. A. Ellston, S. Green, P. J. Jewsbury, C. J. Midgley, R. Naven, C. A. Minshull, R. A. Pauptit, J. A. Tucker, J. E. Pease, Bioorg.Med. Chem. Lett.2003, 13, 2955.Search in Google Scholar
[12] H. Kimura, T. Katoh, T. Kajimoto, M. Node, M. Hisaki, Y. Sugimoto, T. Majima, Y. Ueheda, T. Yamori, Anticancer Res.2006, 26, 91.Search in Google Scholar
[13] I. M. Lagoja, Chem. Biodiver.2005, 2, 1.Search in Google Scholar
[14] R. Dudhea, P. K. Sharmab, P. Vermae, A. J. Chaudhary, Adv. Sci. Res.2011, 2, 10.Search in Google Scholar
[15] R. K. Russell, J. B. Press, R. A. Rampulla, J. J. McNally, R. Falotico, J. A. Keiser, D. A. Bright, A. Tobia, J. Med. Chem.1988, 31, 1786.Search in Google Scholar
[16] H. W. Leea, B. Y. Kim, J. B. Ahn, S. K. Kang, J. Hwa-Lee, J. S. Shin, S. K. Ahn, S. J. Lee, S. S. Yoon, Eur. J. Med. Chem.2005, 40, 862.Search in Google Scholar
[17] H. Tanaka, H. Takashima, M. Ubasawa, K. Sekiya, I. Nitta, M. Baba, S. Shigeta, R. T. Walker, E. De Clercq, T. Miyasaka, J. Med. Chem.1992, 35, 337.Search in Google Scholar
[18] J. Balzarini, M. Baba, E. De Clercq, E. Antimicrob. Agents Chemother.1995, 39, 998.Search in Google Scholar
[19] T. Miyasaka, H. Tanaka, M. Baba, H. Hayakawa, R. T. Walker, J. Balzarini, E. De Clercq, J. Med. Chem.1989, 32, 2507.Search in Google Scholar
[20] L. C. Fillios, C. Naito, S. Andrews, A. M. Roach, Circ. Res.1960, 8, 71.Search in Google Scholar
[21] A. Kreutzberger, K. Burgwitz, Arch. Pharm.1981, 314, 394.Search in Google Scholar
[22] A. Monge, V. Martinez-Merino, C. Sanmartin, F. J. Fernandez, M. C. Ochoa, C. Bellver, P. Artigas, Arzneimittelforsch.1990, 40, 1230.Search in Google Scholar
[23] O. Fhid, M. Pawłowski, B. Filipek, R. Horodyńska, D. Maciak, Pol. J. Pharmacol.2002, 54, 245.Search in Google Scholar
[24] K. W. Witzel, J. M. Wickman, S. G. Augustin, J. G. Strom, Clin. Ther.2000, 22, 1254.Search in Google Scholar
[25] P. A. J. Janssen, P. J. Lewi, E. Arnold, F. Daeyaert, M. de Jonge, J. Heeres, L. Koymans, M. Vinkers, J. Guillemont, E. Pasquier, M. Kukla, D. Ludovici, K. Andries, M.-P. de Béthune, R. Pauwels, K. Das, A. D. Clark Jr., Y. V. Frenkel, S. H. Hughes, B. Medaer, F. De Knaep, H. Bohets, E. De Clerck, A. Lampo, P. Williams, P. Stoffels, J. Med. Chem.2005, 48, 1901.Search in Google Scholar
[26] D. W. Ludovici, B. L. de Corte, M. J. Kukla, H. Ye, C. Y. Ho, M. A. Lichtenstein, R. W. Kavash, K. Andries, M. P. de Bethune, H. Azijn, R. Pauwels, P. J. Lewi, J. Heeres, L. M. Koymans, M. R. de Jonge, K. J. V. Aken, F. F. Daeyaert, K. Das, E. Arnold, P. A. Janssen, Bioorg. Med. Chem.2001, 11, 2235.Search in Google Scholar
[27] K. Das, A. D. Clark, P. J. Lewi, J. Heeres, M. R. de Jonge, L. M. H. Koymans, H. M. Vinkers, F. Daeyaert, D. W. Ludovici, M. J. Kukla, B. De Corte, R. W. Kavash, C. Y. Ho, H. Ye, M. A. Lichtenstein, K. Andries, R. Pauwels, M.-P. d Béthune, P. L. Boyer, Clark, S. H. Hughes, P. A. J. Janssen, E. Arnold, E. J. Med. Chem.2004, 47, 2550.Search in Google Scholar
[28] J. J. Reddick, S. Saha, J.-M. Lee, J. S. Melinck, J. Perkins, T. P. Begley, Bioorg. Med. Chem. Lett.2001, 11, 2245.Search in Google Scholar
[29] N. Saha, D. Muherjee, Inorg. Chim. Acta1987, 137, 161.Search in Google Scholar
[30] N. A. Al-Masoudi, B. Salih, A. N. Abdul-Kareem, Heteroatom Chem.2011, 22, 44.Search in Google Scholar
[31] T. Joshi, V. Pierroz, C. Mari, L. Gemperle, S. Ferrari, G. Gasser, Angew. Chem. Int. Ed.2014, 53, 2960.Search in Google Scholar
[32] N. A. Al-Masoudi, N. N. A. Jafar, S. J. Baqir, C. Pannecouque, P. Leyssen, J. Neyts, Antivir.Chem. Chemother.2012, 23, 103.Search in Google Scholar
[33] N. A. Al-Masoudi, A. G. Kassim, N. A. Abdul-Reda, Nucleos. Nucleot. Nucleic Acids2014, 33, 141.10.1080/15257770.2014.880475Search in Google Scholar
[34] N. A. Al-Masoudi, Y. A. Marich, N. J. Al-Salihi, B. Saeed, Z. Naturforsch.2014, 69B, 913.Search in Google Scholar
[35] A. L. Davis, J. Keeler, E. D. Laue, D. Moskau, J. Magn. Reson.1992, 98, 207.Search in Google Scholar
[36] W. Willker, D. Leibfritz, R. Kerssebaum, W. Bermel, Magn. Reson. Chem.1993, 31, 287.Search in Google Scholar
[37] M. S. Masouda, A. I. Amany, A. K. Ekram, E. Adel, Spectrochim. Acta2007, 67A, 662.Search in Google Scholar
[38] G. Parthasarathi, K. M. Tapas, R. S. Asit, Trans. Metal Chem.1984, 9, 46.Search in Google Scholar
[39] R. Wysokiński, J. Kuduk-Jaworska, D. Michalska, Mol. Struct. THEOCHEM2006, 758, 169.10.1016/j.theochem.2005.10.032Search in Google Scholar
[40] G. L. McLeod, J. Res. Nat. Bur. Stand. A.1962, 66, 65.Search in Google Scholar
[41] T. S. Lobana, P. Kaur, G. Hunda, R. J. Butcher, A. Castineiras. Z. Anorg. Allg. Chem.2008, 634, 747.Search in Google Scholar
[42] R. M. Silverstein, F. X. Webster, D. Kiemle in Spectromteric Identification of Organic Compounds, 7th ed. (Ed.: D. Brennan), Wiley, New York, 2005.Search in Google Scholar
[43] M. S. Masoud, A. A. Soayed, A. E. Ali, O. K. Sharsherh, J. Coord. Chem.2003, 56, 725.Search in Google Scholar
[44] M. S. Masoud, A. A. Hasanein, A. K. Ghonaim, E. A. Khalil, A. A. Mahmoud, Z. Phys. Chim.1999, 209, 223.Search in Google Scholar
[45] M. J. Clarke in Metal Complexes as Anticancer Agents, (Ed.: H. Sigel), CRC Press, Boca Raton, 1980, p. 231.Search in Google Scholar
[46] E. M. Antonarakis, A. Emadi, Cancer Chemother. Pharmacol.2010, 66, 1.Search in Google Scholar
[47] R. Pauwels, J. Balzarini, M. Baba, R. Snoeck, D. Schols, P. Herdewijn, J. Desmyter, E. De Clercq, J. Virol. Methods1988, 20, 309.10.1016/0166-0934(88)90134-6Search in Google Scholar
[48] K. D. Hargrave, J. R. Proudfoot, K. G. Grozinger, E. Cullen, S. R. Kapadia, U. R. Patel, V. U. Fuchs, S. C. Mauldin, J. Vitous, M. L. Behnke, J. M. Klunder, K. Pal, J. W. Skiles, D. W. McNeil, J. M. Rose, G. C. Chow, M. T. Skoog, J. C. Wu, G. Schmidt, W. W. Engel, W. G. Eberlein, T. D. Saboe, S. J. Campbell, A. S. Rosenthal, J. Adams, J. Med. Chem.1991, 34, 2231.Search in Google Scholar
[49] H. Mitsuya, K. J. Weinhold, P. A. Furman, M. H. St. Clair, S. N. Lehrmann, R. Gallo, D. Bolognesi, D. W. Barry, S. Broder, Proc. Natl. Acad. Sci. USA1985, 82, 7096.10.1073/pnas.82.20.7096Search in Google Scholar PubMed PubMed Central
[50] P. Zhan, X. Liu, Z. Li, Z. Fang, C. Pannecouque, E. De Clercq, Chem. Biodivers.2010, 7, 1717.Search in Google Scholar
[51] S. Seeliger, B. L. de Groot, J. Comput.-Aided Mol. Des.2010, 24, 417.Search in Google Scholar
[52] F. H. Dowling, E. Dumoff-Stanley, M. H. Lepper, L. K. Sweet, J. Am. Med. Assoc.1944, 12, 103.Search in Google Scholar
©2015 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- Review
- Cerium intermetallics with ZrNiAl-type structure – a review
- Original Communications
- In vitro cytotoxicity of hydrazones, pyrazoles, pyrazolo-pyrimidines, and pyrazolo-pyridine synthesized from 6-substituted 3-formylchromones
- A supramolecular 3-D organic-inorganic hybrid structure based on {PMo12} layers arranged in an alternating mode
- Ionic binuclear ferrocenyl compounds containing 1,1,3,3-tetracyanopropenide anions – synthesis, structural characterization and catalytic effects on thermal decomposition of main components of solid propellants
- Zur Chemie der 1,3,5-Triaza-2-phosphorinan- 4,6-dione. Teil XIV. Darstellung von weiteren P-alkyl- und P-arylsubstituierten 1,3,5-Trimethyl-1,3,5-triaza-2-phosphorinan-4,6-dionen
- Synthesis, biological activity and modeling study of some thiopyrimidine derivatives and their platinum(II) and ruthenium(III) metal complexes
- Synthesis of functionalized benzene using Diels–Alder reaction of activated acetylenes with synthesized phosphoryl-2-oxo-2H-pyran
- Synthesis, structure, and optical nonlinearity of soluble ternary cadmium copper tellurolate complex [Cd(μ-TeTol)4{Cu(PPh3)2}2] (Tol = 4-tolyl)
Articles in the same Issue
- Frontmatter
- In this Issue
- Review
- Cerium intermetallics with ZrNiAl-type structure – a review
- Original Communications
- In vitro cytotoxicity of hydrazones, pyrazoles, pyrazolo-pyrimidines, and pyrazolo-pyridine synthesized from 6-substituted 3-formylchromones
- A supramolecular 3-D organic-inorganic hybrid structure based on {PMo12} layers arranged in an alternating mode
- Ionic binuclear ferrocenyl compounds containing 1,1,3,3-tetracyanopropenide anions – synthesis, structural characterization and catalytic effects on thermal decomposition of main components of solid propellants
- Zur Chemie der 1,3,5-Triaza-2-phosphorinan- 4,6-dione. Teil XIV. Darstellung von weiteren P-alkyl- und P-arylsubstituierten 1,3,5-Trimethyl-1,3,5-triaza-2-phosphorinan-4,6-dionen
- Synthesis, biological activity and modeling study of some thiopyrimidine derivatives and their platinum(II) and ruthenium(III) metal complexes
- Synthesis of functionalized benzene using Diels–Alder reaction of activated acetylenes with synthesized phosphoryl-2-oxo-2H-pyran
- Synthesis, structure, and optical nonlinearity of soluble ternary cadmium copper tellurolate complex [Cd(μ-TeTol)4{Cu(PPh3)2}2] (Tol = 4-tolyl)