Acetylenic antifolates as anticancer agents
-
Filiz Esra Önen Bayram
 ,
Hande Sipahi
,
Hande Sipahi

Abstract
Folates are crucial cofactors involved in the de novo generation of purine and deoxythymidine monophosphate, which are essential for DNA synthesis. Antifolates are structural analogues of folate derivatives that act as inhibitors of folate-dependent enzymes and constitute the oldest antimetabolite class of anticancer agents. This review focuses on antifolates with remarkable anticancer activities that include a terminal alkyne function in their molecular structure. The properties of CB3717, a tremendous inhibitor of thymidylate synthase, are described, and the development of raltitrexed and pralatrexate, a dihydrofolate reductase inhibitor approved by the U.S. Food and Drug Administration (FDA) as the first drug for the treatment of relapsed and refractory peripheral T cell lymphoma are presented.
Introduction
Folates, composed of a pterin ring coupled with p-aminobenzoate and glutamate moieties (Figure 1A), are cofactors of enzymes involved in DNA/RNA syntheses and methylation processes. The endogenous synthesis of these molecules is only possible in bacteria; thus they are provided to humans by food intake. Given the vital importance of folates in cellular mechanisms, health authorities have underlined their absolute necessity of absorption, issuing notifications for recommended daily intake ratios [1–3]. This family of compounds can exist in either an oxidized form or a reduced dihydrofolate (DHF) or tetrahydrofolate (THF) form (Figure 1B). THF and its derivatives include two chiral centers (C6 and Cα), and natural THF and its 5-substituted derivatives consist of the (6S, αS) diastereomers, and due to nomenclature rules the natural N10-subsituted reduced folates are designated as (6S, αS) diastereomers. (6S, αS) is the only active form of folates in cells and bears one-carbon groups with different oxidation states (Figure 1C). It can bear a methyl group on its N-5 position (most reduced form), methylene or methenyl moieties on positions 5 and 10 (intermediate), a formyl group on position 5 or 10 or a formimino group on position 5. These one-carbon groups are further transferred to specific substrates, as one-carbon transfer mechanisms are the main cellular processes in which folates are involved (Figure 2) [4–6].
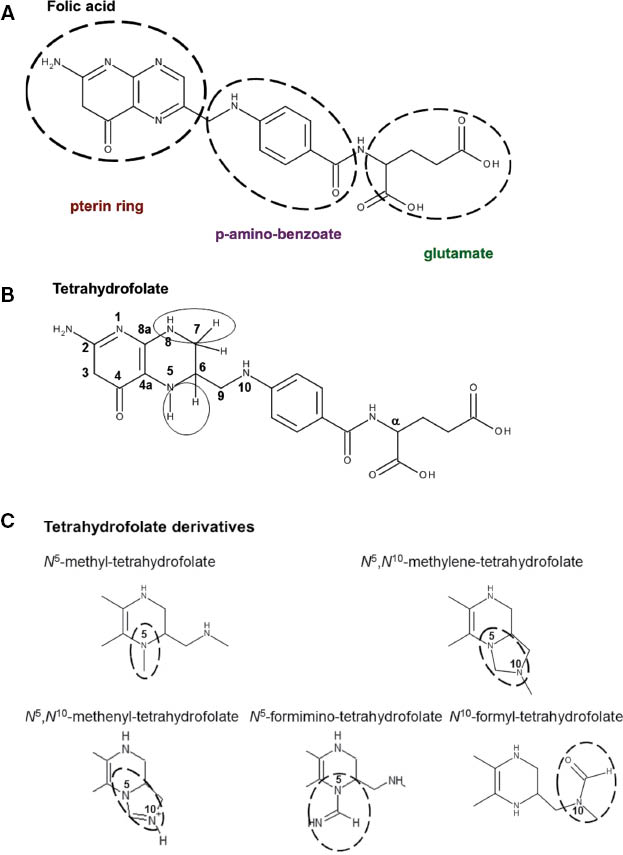
Chemical structures of folic acid (A), tetrahydrofolate (B) and its derivatives (C).
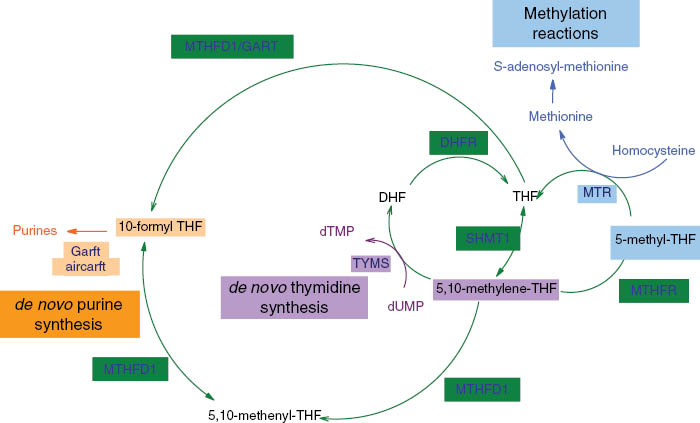
The folate-mediated one-carbon metabolic network. AICARFT, 5′-amino-4′-imidazolecarboxamide ribonucleotide transformylase; DHF, dihydrofolate; DHFR, dihydrofolate reductase enzyme; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; GARFT, β-glycinamide ribonucleotide transformylase; MTHFD1, C-1-tetrahydrofolate synthase; MTHFR, methylene tetrahydrofolate reductase; MTR, 5-methyltetrahydrofolate-homocysteine methyltransferase; SHMT1, serine hydroxymethyltransferase; THF, tetrahydrofolate; TYMS, thymidylate synthase.
Polyglutamated THF constitutes the major folate form of ingested folate. Once hydrolyzed in the gut lumen into monoglutamate by glutamate carboxypeptidase II [7], its transport into enterocytes is ensured mainly by proton-coupled folate transporter (PCFT) proteins. After internalization, the THF is converted into the 5-methylated form that circulates in peripheral blood vessels [8]. Another source of folate comes from the absorption of synthetic folic acid, which is provided by food fortification or as a nutritional supplement. Folic acid is also internalized in enterocytes either by active transport processes using PCFT proteins or simply by passive diffusion. To play its cofactor role, folic acid is then converted by the dihydrofolate reductase enzyme (DHFR) into THF and transformed, via several processes, to the plasma-circulating 5-methyl tetrahydrofolate monoglutamate. This latter molecule, resulting either from diet or synthetic source, is further internalized by RFC proteins or folate receptors in other somatic cells where it is polyglutamated by folylpolyglutamate synthetase (FPGS) to finally enter the one-carbon metabolic network [9–12].
Folate-mediated pathways are interdependent and are essential for many cellular biosyntheses. In DNA synthesis, for instance, the de novo generation of purine heterocycles is mediated by β-glycinamide ribonucleotide transformylase (GARFT) and 5′-amino-4′-imidazolecarboxamide ribonucleotide transformylase (AICARFT), two key enzymes for which 10-formyl THF acts as a cofactor. Moreover, thymidylate synthase (TYMS) requires 5,10-methylene THF as a cofactor when converting deoxyuridine monophosphate (dUMP) into deoxythymidine monophosphate (dTMP) for the de novo synthesis of deoxythymidine triphosphate, one of the four building blocks of DNA [4–6].
Antifolates are structural analogues of folate derivatives that tend to act as inhibitors of folate-dependent enzymes. These compounds constitute the oldest antimetabolite class of anticancer agents [13, 14]. In this review, the main emphasis will be placed on antifolates with a terminal alkyne function in their molecular structure showing remarkable anticancer activities.
Acetylenic antifolate as a TYMS inhibitor: CB3717, the forerunner of raltitrexed
Thymidylate synthase activation depends on the formation of a ternary complex composed of the protein, its substrate (dUMP), and 5,10-methylene THF, its cofactor. dTMP synthesis, which is essential for DNA synthesis and repair, is obtained via the reductive methylation of dUMP when 5,10-methylene THF is oxidized to 7,8-DHF. The inhibition of TYMS leads to an important decrease in the amount of available thymidine, thus resulting in severe cytotoxicity in dividing cells. Hence, TYMS is considered as a key target in anticancer therapy [15–23]. Its inhibition can be achieved by preventing the access of either its substrate or its cofactor to the active site via pyrimidine or folate analogues, respectively. For instance, the pyrimidine analogue 5-fluorouracil (5-FU), a chemotherapeutic agent widely used in anticancer therapy for about 50 years, is converted into fluorodeoxyuridine monophosphate, which forms, together with 5,10-methylene THF and TYMS, a relatively stable ternary complex [24, 25].
Substituted 2-amino-4-hydroxy series of quinazolines, which are 5,8-dideazofolic acid derivatives, were also demonstrated to be effective inhibitors of TYMS [26–28]. Considering this, and to develop novel potent TYMS inhibitors, Jones et al. [29] synthesized structures by introducing allyl and propargyl groups to the N-10 position of these folate analogues and investigated their anticancer properties. An N-propargylic compound called CB3717 (Figure 3) demonstrated a great capacity to inhibit TYMS (Ki≈3 nmol/L), competing with 5,10-methylene THF. Moreover, the authors noticed its remarkable antitumor activity both in vitro and in vivo on either murine or human systems [30, 31]. Further investigations actually indicated a polyglutamation of the intracellular CB3717, since the molecule was proven to show an affinity for FPGS [32]. This chemical modification was demonstrated to stabilize the active structure inside the cell as the polyglutamated metabolite cannot be carried back by RFC proteins [33–35], providing to the drug an extended intracellular half-life and thus a greater antimetabolic activity. Some additional studies also established that the glutamation degree was closely related to the inhibition ability of the TYMS protein, since the Ki values of these compounds were found to be considerably enhanced especially for those with four to five glutamate moieties (Ki≈40 pmol/L) [36, 37].
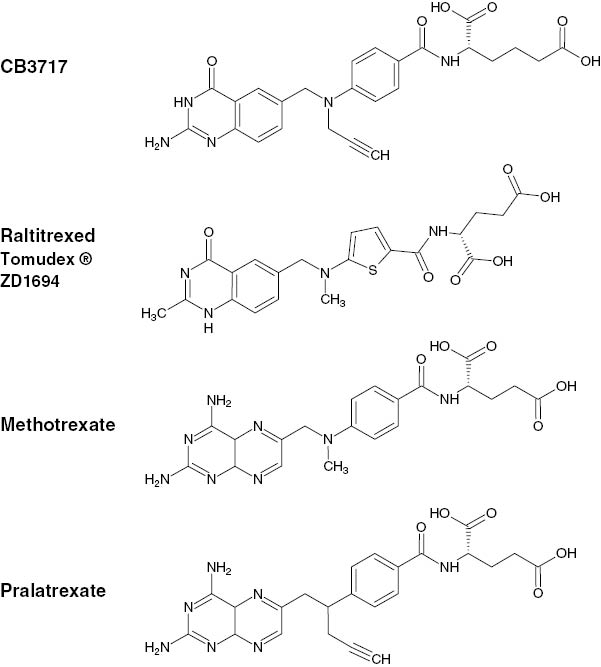
Chemical structures of THF and antifolate anticancer agents.
Based on these promising results, CB3717 was evaluated in clinical trials in patients with breast, ovarian and hepatocellular carcinoma. However, phase I [38–40] and phase II [41, 42] studies revealed a severe nephrotoxicity in patients with either weekly or 3-weekly administration schedules, probably due to the precipitation of the drug in renal tubules as it is poorly water soluble at physiological pH [43]. This considerable drawback marked the end of the clinical investigation of CB3717 but led to a subsequent collaboration between the Institute of Cancer Research and ICI Pharmaceuticals (now AstraZeneca) for the development of a library of water-soluble molecules with CB3717-like structures. The evaluation of this library revealed an N-10-methylthiophene analogue, namely, ZD1694 (Figure 3), that exhibited great inhibition properties toward TYMS [43–45]. This compound, better known as raltitrexed or Tomudex©, is now approved by many countries as an anticancer agent for the treatment of metastatic colorectal cancer, although its administration is often limited to specific cases only [46–50].
Acetylenic antifolate as a DHFR inhibitor: pralatrexate
DHFR that catalyzes the reduction of DHF to THF also constitutes a key target in anticancer therapy as its inhibition blocks the THF synthesis and thus leads to a depletion in purine and pyrimidine precursors, which are essential for DNA, RNA and hence protein synthesis. The DHFR inhibitor methotrexate (MTX, 4-amino-10-methylpteroyl-glutamic acid) was first demonstrated to exhibit an antineoplastic effect in 1948 [51] (Figure 3). This anticancer agent is now commonly used for the treatment of a wide range of cancers including leukemia, lymphoma, breast, lung, bladder carcinomas, head and neck cancer, and osteogenic carcinoma [52]. In the early 1980s, Sirotnak et al. [53, 54] developed a new family of folate analogues composed of a series of 10-deazaaminopterins (with a carbon atom instead of a nitrogen atom present at position 10) that were found to be more potent than MTX. Their exalted activity were attributed to an enhanced capacity of internalization coupled with a higher degree of glutamation, as these molecules were shown to have notable affinity for the RFC proteins [53] and the FPGS enzyme [55, 56].
Also, with the development of CB3717, the role of propargylic moiety for the generation of anticancer properties was explored by several research groups. While Jackman et al. [57, 58] were investigating the activity of the propargylic group of 2-amino-deficient folate analogues, Piper et al. [59] investigated the potency of the 10-N-propargylaminopterin, which was shown to be more active than MTX, although requiring a high-dose injection to be effective in vivo. Based on these results, DeGraw et al. [60] synthesized the 10-propargyl-10-deazaminopterin molecule or pralatrexate (PDX) (Figure 3) and evaluated its anticancer properties in in vitro and in vivo studies. Even if the structure was found to inhibit DHFR less efficiently than MTX (Ki=3-fold higher), it exhibited an outstanding cytotoxicity, being five times more potent than MTX in cell growth inhibition (IC50MTX=9.50 nmol/L, IC50PDX=2.0 nmol/L) and quite more effective on murine mammary models in vivo [60].
Given these promising results, PDX was then evaluated on human cancer cell lines in vitro and investigated on human tumor xenografted mice in vivo. Breast and non-small cell lung cancer (NSCLC) constituted the first human cell lines on which PDX demonstrated its efficacy, being up to 30-fold more cytotoxic than MTX. As the most sensitive cell line was detected to be the adenocarcinoma cell line, NSCLC tumor xenografts (LX-1 and A549 cell lines) were chosen to be studied in vivo. Whereas tumors of MTX-treated mice did not yield in any healing, PDX led to a complete regression of the tumors in 75% of the animals [56]. The superior antitumor activity of PDX on NSCLC tumor models was also further confirmed by Izbicka et al. [61].
The antitumor efficacy of PDX was also examined on human lymphoma, whether used as a single agent or combined with other cytotoxic agents such as the nucleoside analogue gemcitabine or the proteasome inhibitor bortezomib. The IC50 values obtained with all of the Hodgkin or non-Hodgkin lymphoma cell lines treated with PDX were ten-fold smaller than the values obtained with MTX-treated cells. In addition, no or very slight regression of tumors was observed in MTX-treated lymphoma xenografted mice (RL- and SKI-DLBCL), whereas PDX treatment led to complete regression in 30% of RL- and 56% of SKI-DLBCL-xenografted mice [62]. Toner et al. [63] have shown the synergic effect of PDX and gemcitabine especially in treatments occurring in a scheduled manner (gemcitabine administration 24 h after the PDX treatment) as they demonstrated the significantly superior activity of this combination when compared with the MTX→cytarabine combination on animals with SKI-DLBCL xenografts (3/5 complete remission for PDX→gemcitabine, whereas no remission for MTX→cytarabine). A similar synergetic efficacy was also noticed by Marchi et al. [64] when they tested PDX in combination with bortezomib in in vitro and in vivo models of T-cell lymphoid malignancies.
To understand the molecular basis underlying the enhancement of the cytotoxic effect with the propargylic molecule, studies evaluating the expression levels of the genes coding for proteins involved in one-carbon metabolisms were conducted. As the RFC1 that codes for the RFC protein was found to be more expressed in PDX-sensitive cell lines (diffuse large B cell and HT cell line), it was suggested that a better internalization of the drug could lead to the enhancement of its antiproliferative activity [63]. A similar investigation was further carried out on a wide panel of cancer cells including colon, breast, melanoma, NSCLC, ovarian, prostate and head and neck cell lines by Serova et al. [65]. The authors analyzed the expression levels of genes coding for DHFR, FPGS, RFC, TYMS, GARFT, SLC25A32 (mitochondrial folate transporter/carrier) and ATP-binding cassette transporters (ABCB1) and did not notice any correlation between PDX sensitivity and the expression levels of TYMS, GARFT, LC25A32 or ABCB1, nor of RFC1, and thus they could not support the findings previously established. However, they determined a significant increase in the levels of FPGS genes, indicating again the important role of glutamation in acquiring cytotoxicity. A moderate but not statistically significant increase in the expression of DHFR was also reported. To better characterize the molecular mechanisms implied in PDX-sensitive cell lines, the group also generated PDX- and MTX-resistant cells from cells detected to be the most sensitive to the corresponding drugs. The examination of gene expression levels in DU-PDX and HEP-PDX cell lines, both being PDX resistant, revealed a considerable decrease in the expression of RFC1, indicating a decisive role of the folate transport protein for PDX to be active, as previously suggested [53, 63]. Furthermore, an increase in the ABCB1 gene was also observed. Modification in ABCB1 expression levels did not show any correlation with PDX sensitivity though, as the inhibition of the ABC proteins did not restore sensitivity to the drug. Interestingly, even though MTX-resistant cells exhibited an extensive increase in DHFR levels as expected since DHFR over-regulation constitutes the main mechanism for the generation of MTX resistance [66], the increase observed for PDX-resistant cells was not statistically significant, suggesting different molecular mechanisms of action for these two antifolates. This hypothesis is actually in agreement with Zain and O’Connor’s [67] findings, which indicated that modifications in gene expression levels in MTX-treated cells were mainly occurring in genes involved in folate metabolism, whereas the gene expression of PDX-treated cells was essentially disrupted for genes implied in pathways regulating immunomodulation and transcription factors.
Based on the encouraging results obtained during the first preclinical studies, the clinical evaluation of PDX began with NSCLC patients. The phase I studies revealed mucositis as the drug’s dose-limiting toxicity. Its antitumor activity was confirmed as two of 33 patients with stage IV NSCLC responded favorably to the treatment and the conditions of five of 33 patients were stabilized [68]. The phase II study of 38 NSCLC patients provided satisfactory results, with 10% of objective responses and 31% of disease stabilization. Stomatitis and mucositis constituted the main toxicities associated with the treatment [69]. When examining the efficacy of the drug in combination with probenecid on patients presenting solid tumors, Fury et al. [70] mentioned the possibility of supplementation with vitamin B12 and folate in order to prevent mucositis, hence allowing a dose escalation during treatments. Azzoli et al. [71] evaluated PDX in combination with taxanes, incorporating to their protocol the co-administration of vitamin B12and folic acid. This supplementation allowed the patients to tolerate safely higher doses of the drug [71], confirming the predictions of Fury et al. [70]. From then on, investigators always incorporated vitamin B12 and folic acid supplementation into their protocols.
PDX was further evaluated on other carcinomas. Although the drug did not show any activity on patients with malignant pleural mesothelioma [72], it demonstrated successful activity on lymphoma, with achievement of complete regression in all of the patients with T-cell lymphoma in a study carried out on 20 patients, 16 of whom presenting B-cell and four presenting T-cell lymphomas [73]. The authors tested two different doses for the treatment of T-cell lymphoma: the first treatment at the recommended dose (135 mg/m2 every other week) resulted in the development of severe mucositis, whereas the administration of 30 mg/m2 of PDX weekly for 6 weeks was well tolerated by three other patients. These findings led to a new phase I clinical study that redefined the maximum-tolerated dose of PDX, which decreased the ratio of patients developing stomatitis from almost 100%–17% [74]. Based on these results, a multicenter phase II study, carried out with patients with relapsed or refractory peripheral T-cell lymphoma, demonstrated the drug’s outstanding efficacy, as on 109 evaluable patients 29% experienced objective responses and 38% achieved complete remission. These data led to the approval of PDX by the U.S. FDA in 2009 for the treatment of peripheral T-cell lymphoma on patients who have relapsed or have not responded to other chemotherapy drugs [75–77]. PDX is the first drug approved for this disease [78].
To conclude, this review focused on antifolates with a terminal alkyne function. Antifolates constitute the first class of anticancer antimetabolites, and the development of folate antagonists bearing a propargyl group led to the generation of the first clinically evaluated folate-based TYMS inhibitor, CB3717. Yet, the clinical studies of this highly potent molecule revealed a serious nephrotoxicity of the structure due to its poor solubility. CB3717 structure optimization studies contributed to the development of two marketed anticancer agents: raltitrexed, a TYMS inhibitor used for the treatment of colorectal cancer, and pralatrexate, a propargylic DHFR inhibitor, which is the first drug approved for relapsed or refractory peripheral T-cell lymphoma. These results highlight the potential of the propargyl group for the development of novel potent anticancer molecules.
References
1. Bailey LB, Gregory JF. Folate metabolism and requirements. J Nutr 1999;129:779–82.10.1093/jn/129.4.779Suche in Google Scholar
2. Institute of Medicine, Food and Nutrition Board, National Academy of Sciences. Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, pantothenic acid, biotin, and choline. National Academy Press, 1998.Suche in Google Scholar
3. Iyer R, Tomar SK. Folate: a functional food constituent. J Food Sci 2009;74:R114–22.10.1111/j.1750-3841.2009.01359.xSuche in Google Scholar
4. Litwack G. Folic acid and folates. New York, NY: Academic Press, 2008.Suche in Google Scholar
5. Bailey LB. Folate in health and disease. Gainesville, FL, USA: CRC Press, 2009.10.1201/9781420071252Suche in Google Scholar
6. Milstien S, Kapatos G, Levine RA. Chemistry and biology of pteridines and folates: Proceedings of the 12th International Symposium on Pteridines and Folates, National Institutes of Health, Bethesda, Maryland, June 17–22, 2001. Springer, 2002:649–53.10.1007/978-1-4615-0945-5Suche in Google Scholar
7. Halsted CH. The intestinal absorption of dietary folates in health and disease. J Am Coll Nutr 1989;8:650–8.10.1080/07315724.1989.10720340Suche in Google Scholar
8. Laanpere M, Altmäe S, Stavreus-Evers A, Nilsson TK, Yngve A, Salumets A. Folate-mediated one-carbon metabolism and its effect on female fertility and pregnancy viability. Nutr Rev 2010;68:99–113.10.1111/j.1753-4887.2009.00266.xSuche in Google Scholar
9. Zhao R, Matherly LH, Goldman ID. Membrane transporters and folate homeostasis: intestinal absorption and transport into systemic compartments and tissues. Exp Rev Mol Med 2009;11:e4.10.1017/S1462399409000969Suche in Google Scholar
10. Yuasa H, Inoue K, Hayashi Y. Molecular and functional characteristics of proton-coupled folate transporter. J Pharm Sci 2009;98:1608–16.10.1002/jps.21515Suche in Google Scholar
11. Matherly LH, Hou Z, Deng Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metast Rev 2007;26:111–28.10.1007/s10555-007-9046-2Suche in Google Scholar
12. Matherly LH, Goldman DI. Membrane transport of folates. Vitam Horm 2003;66:403–56.10.1016/S0083-6729(03)01012-4Suche in Google Scholar
13. Jackman AL. Antifolate drugs in cancer therapy. Totowa, NJ: Humana Press, 1999.10.1007/978-1-59259-725-3Suche in Google Scholar
14. McGuire JJ. Anticancer antifolates: current status and future directions. Curr Pharm Design 2003;9:2593–613.10.2174/1381612033453712Suche in Google Scholar PubMed
15. Hagner N, Joerger M. Cancer chemotherapy: targeting folic acid synthesis. Cancer Man Res 2010;2:293–301.10.2147/CMAR.S10043Suche in Google Scholar
16. Jarmula A. Antifolate inhibitors of thymidylate synthase as anticancer drugs. Mini-Rev Med Chem 2010;10:1211–22.10.2174/13895575110091211Suche in Google Scholar PubMed
17. Xia W, Low PS. Folate-targeted therapies for cancer. J Med Chem 2010;53:6811–24.10.1021/jm100509vSuche in Google Scholar PubMed
18. Bertino JR. Cancer research: from folate antagonism to molecular targets. Best Pract Res Cl En 2009;22:577–82.10.1016/j.beha.2009.09.004Suche in Google Scholar PubMed
19. Gangjee A, Jain HD. Antifolates – past, present and future. Curr Med Chem Anti-Cancer Agents 2004;4:405–10.10.2174/1568011043352803Suche in Google Scholar PubMed
20. Jackman AL, Theti DS, Gibbs DD. Antifolates targeted specifically to the folate receptor. Adv Drug Deliver Rev 2004;56:1111–25.10.1016/j.addr.2004.01.003Suche in Google Scholar PubMed
21. Touroutoglou N, Pazdur R. Thymidylate synthase inhibitors. Clin Cancer Res 1996;2:227–43.Suche in Google Scholar
22. Jackman AL, Calvert AH. Folate-based thymidylate synthase inhibitors as anticancer drugs. Ann Oncol 1995;6:871–81.10.1093/oxfordjournals.annonc.a059353Suche in Google Scholar PubMed
23. Garg D, Henrich S, Salo-Ahen OMH, Myllykallio H, Costi MP, Wade RC. Novel approaches for targeting thymidylate synthase to overcome the resistance and toxicity of anticancer drugs. J Med Chem 2010;53:6539–49.10.1021/jm901869wSuche in Google Scholar PubMed
24. Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, et al. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957;179:663–6.10.1038/179663a0Suche in Google Scholar PubMed
25. Santi DV, McHenry CS, Sommer H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 1974;13:471–81.10.1021/bi00700a012Suche in Google Scholar
26. Bird OD, Vaitkus JW, Clarke J. 2-Amino-4-hydroxyquinazolines as inhibitors of thymidylate synthetase. Mol Pharmacol 1970;6:573–5.10.1016/S0026-895X(25)15262-0Suche in Google Scholar
27. Carlin SC, Rosenberg RN, VandeVenter L, Friedkin M. Quinazoline antifolates as inhibitors of growth, dihydrofolate reductase, and thymidylate synthetase of mouse neuroblastoma cells in culture. Mol Pharmacol 1974;10:194–203.10.1016/S0026-895X(25)13843-1Suche in Google Scholar
28. Jones TR. 5-Substituted quinazoline antifolates. Eur J Cancer 1980;16:707–11.10.1016/0014-2964(80)90213-3Suche in Google Scholar
29. Jones TR, Calvert AH, Jackman AL, Brown SJ, Jones M, Harrap KR. A potent antitumour quinazoline inhibitor of thymidylate synthetase: synthesis, biological properties and therapeutic results in mice. Eur J Cancer 1981;17:11–9.10.1016/0014-2964(81)90206-1Suche in Google Scholar
30. Jackman AL, Taylor GA, O’Connor BM, Bishop JA, Moran RG, Calvert AH. Activity of the thymidylate synthase inhibitor 2-desamino-N10-propargyl-5,8-dideazafolic acid and related compounds in murine (L1210) and human (W1L2) systems in vitro and in L1210 in vivo. Cancer Res 1990;50:5212–8.Suche in Google Scholar
31. Jackson RC, Jackman AL, Calvert AH. Biochemical effects of a quinazoline inhibitor of thymidylate synthetase, N-(4-(N-((2-amino-4-hydroxy-6-quinazolinyl)methyl)prop- 2-ynylamino)benzoyl)-l-glutamic acid (CB3717), on human lymphoblastoid cells. Biochem Pharmacol 1983;32:3783–90.10.1016/0006-2952(83)90150-8Suche in Google Scholar
32. Moran RG, Colman PD, Rosowsky A, Forsch RA, Chan KK. Structural features of 4-amino antifolates required for substrate activity with mammalian folylpolyglutamate synthetase. Mol Pharmacol 1985;27:156–66.10.1016/S0026-895X(25)11444-2Suche in Google Scholar
33. Westerhof GR, Schornagel JH, Kathmann I, Jackman AL, Rosowsky A, Forsch RA, et al. Carrier- and receptor-mediated transport of folate antagonists targeting folate-dependent enzymes: correlates of molecular-structure and biological activity. Mol Pharmacol 1995;48:459–71.10.1016/S0026-895X(25)10494-XSuche in Google Scholar
34. Jackman AL, Gibson W, Brown M, Kimbell R, Boyle FT. The role of the reduced-folate carrier and metabolism to intracellular polyglutamates for the activity of ICI D1694. Adv Exp Med Biol 1993;339:265–76.10.1007/978-1-4615-2488-5_26Suche in Google Scholar
35. Manteuffel-Cymborowska M, Kaminska B, Grzelakowska-Sztabert B. Dose dependent retention and polyglutamation of N10-propargyl-5,8-dideazafolic acid (CB 3717) in the tumour-bearing mouse. Anticancer Res 1988;8:791–6.Suche in Google Scholar
36. Pawelczak K, Jones TR, Kempny M, Jackman AL, Newell DR, Krzyzanowski L, et al. Quinazoline antifolates inhibiting thymidylate synthase: synthesis of four oligo(L-gamma-glutamyl) conjugates of N10-propargyl-5,8-dideazafolic acid and their enzyme inhibition. J Med Chem1989;32:160–5.10.1021/jm00121a029Suche in Google Scholar
37. Sikora E, Jackman AL, Newell DR, Calvert AH. Formation and retention and biological activity of N10-propargyl-5,8-dideazafolic acid (CB3717) polyglutamates in L1210 cells in vitro. Biochem Pharmacol 1988;37:4047–54.10.1016/0006-2952(88)90094-9Suche in Google Scholar
38. Sessa C, Zucchetti M, Ginier M, Willems Y, D’Incalci M, Cavalli F. Phase I study of the antifolate N10-propargyl-5,8-dideazafolic acid, CB 3717. Eur J Cancer Clin Oncol 1988;24:769–75.10.1016/0277-5379(88)90313-6Suche in Google Scholar
39. Vest S, Bork E, Hansen HH. A phase I evaluation of N10-propargyl-5,8-dideazafolic acid. Eur J Cancer Clin Oncol 1988;24:201–4.10.1016/0277-5379(88)90253-2Suche in Google Scholar
40. Calvert AH, Alison DL, Harland SJ, Robinson BA, Jackman AL, Jones TR, et al. A phase I evaluation of the quinazoline antifolate thymidylate synthase inhibitor, N10-propargyl-5,8-dideazafolic acid, CB3717. J Clin Oncol 1986;4:1245–52.10.1200/JCO.1986.4.8.1245Suche in Google Scholar PubMed
41. Bassendine MF, Curtin NJ, Loose H, Harris AL, James OF. Induction of remission in hepatocellular carcinoma with a new thymidylate synthase inhibitor, CB3717. A phase II study. J Hepatol1987;4:349–56.10.1016/S0168-8278(87)80545-7Suche in Google Scholar
42. Cantwell BM, Macaulay V, Harris AL, Kaye SB, Smith IE, Milsted RA, et al. Phase II study of the antifolate N10-propargyl-5,8-dideazafolic acid (CB 3717) in advanced breast cancer. Eur J Cancer Clin Oncol 1988;24:733–6.10.1016/0277-5379(88)90307-0Suche in Google Scholar
43. Jodrell DI, Newell DR, Morgan SE, Clinton S, Bensted JP, Hughes LR, et al. The renal effects of N10-propargyl-5, 8-dideazafolic acid (CB3717) and a non-nephrotoxic analogue ICI D1694, in mice. Br J Cancer 1991;64:833–8.10.1038/bjc.1991.409Suche in Google Scholar
44. Jackman AL, Harrap KR, Boyle FT. TomudexTM (ZD1694): from concept to care, a programme in rational drug discovery. Invest New Drugs 1996;14:305–16.10.1007/BF00194534Suche in Google Scholar
45. Jackman AL, Taylor GA, Gibson W, Kimbell R, Brown M, Calvert AH, et al. ICI D1694, a quinazoline antifolate thymidylate synthase inhibitor that is a potent inhibitor of LL210 tumor cell growth in vitro and in vivo: a new agent for clinical study. Cancer Res1991;51:5579–86.Suche in Google Scholar
46. Wilson KS, Malfair Taylor SC. Raltitrexed: optimism and reality. Exp Opin Drug Met 2009;5:1447–54.10.1517/17425250903307455Suche in Google Scholar
47. Jackman AL, Farrugia DC, Gibson W, Kimbell R, Harrap KR, Stephens TC, et al. ZD1694 (Tomudex): a new thymidylate synthase inhibitor with activity in colorectal cancer. Eur J Cancer 1995;31A:1277–82.10.1016/0959-8049(95)00166-GSuche in Google Scholar
48. Clarke SJ, Beale PJ, Rivory LP. Clinical and preclinical pharmacokinetics of raltitrexed. Clin Pharmacokinet 2000;39:429–43.10.2165/00003088-200039060-00004Suche in Google Scholar
49. Van Cutsem E, Cunningham D, Maroun J, Cervantes A, Glimelius B. Raltitrexed: current clinical status and future directions. Ann Oncol 2002;13:513–22.10.1093/annonc/mdf054Suche in Google Scholar
50. Cunningham D, Zalcberg J, Maroun J, James R, Clarke S, Maughan TS, et al. Efficacy, tolerability and management of raltitrexed (Tomudex) monotherapy in patients with advanced colorectal cancer. a review of phase II/III trials. Eur J Cancer 2002;38:478–6.10.1016/S0959-8049(01)00413-0Suche in Google Scholar
51. Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. New Engl J Med 1948;238:787–93.10.1056/NEJM194806032382301Suche in Google Scholar PubMed
52. Cronstein BN, Bertino JR. Methotrexate. Birkhäuser, 2000.10.1007/978-3-0348-8452-5Suche in Google Scholar
53. Sirotnak FM, DeGraw JI, Moccio DM, Samuels LL, Goutas LJ. New folate analogs of the 10-deaza-aminopterin series. Basis for structural design and biochemical and pharmacologic properties. Cancer Chemother Pharmacol 1984;12:18–25.10.1007/BF00255903Suche in Google Scholar PubMed
54. Sirotnak FM, DeGraw JI, Schmid FA, Goutas LJ, Moccio DM. New folate analogs of the 10-deaza-aminopterin series. Further evidence for markedly increased antitumor efficacy compared with methotrexate in ascitic and solid murine tumor models. Cancer Chemother Pharmacol 1984;12:26–30.10.1007/BF00255904Suche in Google Scholar
55. Rumberger BG, Barrueco JR, Sirotnak FM. Differing specificities for 4-aminofolate analogues of folylpolyglutamyl synthetase from tumors and proliferative intestinal epithelium of the mouse with significance for selective antitumor action. Cancer Res 1990;50:4639–43.Suche in Google Scholar
56. Sirotnak FM, DeGraw JI, Colwell WT, Piper JR. A new analogue of 10-deazaaminopterin with markedly enhanced curative effects against human tumor xenografts in mice. Cancer Chemother Pharmacol 1998;42:313–8.10.1007/s002800050823Suche in Google Scholar PubMed
57. Jones TR, Thornton TJ, Flinn A, Jackman AL, Newell DR, Calvert AH. Quinazoline antifolates inhibiting thymidylate synthase: 2-desamino derivatives with enhanced solubility and potency. J Med Chem 1989;32:847–52.10.1021/jm00124a018Suche in Google Scholar PubMed
58. Hughes LR, Jackman AL, Oldfield J, Smith RC, Burrows KD, Marsham PR, et al. Quinazoline antifolate thymidylate synthase inhibitors: alkyl, substituted alkyl, and aryl substituents in the C2 position. J Med Chem 1990;33:3060–7.10.1021/jm00173a024Suche in Google Scholar PubMed
59. Piper JR, McCaleb GS, Montgomery JA, Kisliuk RL, Gaumont Y, Sirotnak FM. 10-Propargylaminopterin and alkyl homologues of methotrexate as inhibitors of folate metabolism. J Med Chem 1982;25:877–80.10.1021/jm00349a024Suche in Google Scholar PubMed
60. DeGraw JI, Colwell WT, Piper JR, Sirotnak FM. Synthesis and antitumor activity of 10-propargyl-10-deazaaminopterin. J Med Chem 1993;36:2228–31.10.1021/jm00067a020Suche in Google Scholar PubMed
61. Izbicka E, Diaz A, Streeper R, Wick M, Campos D, Steffen R, et al. Distinct mechanistic activity profile of pralatrexate in comparison to other antifolates in in vitro and in vivo models of human cancers. Cancer Chemother Pharmacol 2009;64:993–9.10.1007/s00280-009-0954-4Suche in Google Scholar PubMed PubMed Central
62. Wang ES, O’Connor O, She Y, Zelenetz AD, Sirotnak FM, Moore MA. Activity of a novel anti-folate (PDX, 10-propargyl 10-deazaaminopterin) against human lymphoma is superior to methotrexate and correlates with tumor RFC-1 gene expression. Leuk Lymphoma 2003;44:1027–35.10.1080/1042819031000077124Suche in Google Scholar PubMed
63. Toner LE, Vrhovac R, Smith EA, Gardner J, Heaney M, Gonen M, et al. The schedule-dependent effects of the novel antifolate pralatrexate and gemcitabine are superior to methotrexate and cytarabine in models of human non-Hodgkin’s lymphoma. Clin Cancer Res 2006;12:924–32.10.1158/1078-0432.CCR-05-0331Suche in Google Scholar PubMed
64. Marchi E, Paoluzzi L, Scotto L, Seshan VE, Zain JM, Zinzani PL, et al. Pralatrexate is synergistic with the proteasome inhibitor bortezomib in in vitro and in vivo models of T-cell lymphoid malignancies. Clin Cancer Res 2010;16:3648–58.10.1158/1078-0432.CCR-10-0671Suche in Google Scholar PubMed
65. Serova M, Bieche I, Sablin MP, Pronk GJ, Vidaud M, Cvitkovic E, et al. Single agent and combination studies of pralatrexate and molecular correlates of sensitivity. Br J Cancer 2011;104:272–80.10.1038/sj.bjc.6606063Suche in Google Scholar PubMed PubMed Central
66. Assaraf YG. Molecular basis of antifolate resistance. Cancer Metast Rev 2007;26:153–81.10.1007/s10555-007-9049-zSuche in Google Scholar PubMed
67. Zain J, O’Connor O. Pralatrexate: basic understanding and clinical development. Exp Opin Pharmacother 2010;11:1705–14.10.1517/14656566.2010.489552Suche in Google Scholar PubMed
68. Krug LM, Ng KK, Kris MG, Miller VA, Tong W, Heelan RT, et al. Phase I and pharmacokinetic study of 10-propargyl-10-deazaaminopterin, a new antifolate. Clin Cancer Res 2000;6:3493–8.Suche in Google Scholar
69. Krug LM, Azzoli CG, Kris MG, Miller VA, Khokhar NZ, Tong W, et al. 10-Propargyl-10-deazaaminopterin: an antifolate with activity in patients with previously treated non-small cell lung cancer. Clin Cancer Res 2003;9:2072–8.Suche in Google Scholar
70. Fury MG, Krug LM, Azzoli CG, Sharma S, Kemeny N, Wu N, et al. A phase I clinical pharmacologic study of pralatrexate in combination with probenecid in adults with advanced solid tumors. Cancer Chemother Pharmacol 2006;57:671–7.10.1007/s00280-005-0080-xSuche in Google Scholar PubMed
71. Azzoli CG, Krug LM, Gomez J, Miller VA, Kris MG, Ginsberg MS, et al. A phase 1 study of pralatrexate in combination with paclitaxel or docetaxel in patients with advanced solid tumors. Clin Cancer Res 2007;13:2692–8.10.1158/1078-0432.CCR-06-1754Suche in Google Scholar PubMed
72. Krug LM, Heelan RT, Kris MG, Venkatraman E, Sirotnak FM. Phase II trial of pralatrexate (10-propargyl-10-deazaaminopterin, PDX) in patients with unresectable malignant pleural mesothelioma. J Thorac Oncol 2007;2:317–20.10.1097/01.JTO.0000263715.84567.5fSuche in Google Scholar PubMed
73. O’Connor OA, Hamlin PA, Portlock C, Moskowitz CH, Noy A, Straus DJ, et al. Pralatrexate, a novel class of antifol with high affinity for the reduced folate carrier-type 1, produces marked complete and durable remissions in a diversity of chemotherapy refractory cases of T-cell lymphoma. Br J Haematol 2007;139:425–8.10.1111/j.1365-2141.2007.06658.xSuche in Google Scholar PubMed
74. O’Connor OA, Horwitz S, Hamlin P, Portlock C, Moskowitz CH, Sarasohn D, et al. Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol 2009;27:4357–64.10.1200/JCO.2008.20.8470Suche in Google Scholar PubMed PubMed Central
75. O’Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L, Coiffier B, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 2011;29:1182–9.10.1200/JCO.2010.29.9024Suche in Google Scholar PubMed PubMed Central
76. Malik SM, Liu K, Qiang X, Sridhara R, Tang S, McGuinn WD, Jr., et al. Folotyn (pralatrexate injection) for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res 2010;16:4921–7.10.1158/1078-0432.CCR-10-1214Suche in Google Scholar PubMed
77. Marchi E, Mangone M, Zullo K, O’Connor OA. Pralatrexate pharmacology and clinical development. Clin Cancer Res 2013;19:6657–61.10.1158/1078-0432.CCR-12-2251Suche in Google Scholar PubMed
78. Foss FM. Evaluation of the pharmacokinetics, preclinical and clinical efficacy of pralatrexate for the treatment of T-cell lymphoma. Exp Opin Drug Met 2011;7:1141–52.10.1517/17425255.2011.595404Suche in Google Scholar PubMed
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Review
- Acetylenic antifolates as anticancer agents
- Original articles
- Neopterin and 7,8-dihydroneopterin are generated within atherosclerotic plaques
- Late-stage systemic immune effectors in Plasmodium berghei ANKA infection: biopterin and oxidative stress
- Conference abstracts
- 34th International Winter Workshop / Clinical, Chemical and Biochemical Aspects of Pteridines and Related Topics
Artikel in diesem Heft
- Frontmatter
- Review
- Acetylenic antifolates as anticancer agents
- Original articles
- Neopterin and 7,8-dihydroneopterin are generated within atherosclerotic plaques
- Late-stage systemic immune effectors in Plasmodium berghei ANKA infection: biopterin and oxidative stress
- Conference abstracts
- 34th International Winter Workshop / Clinical, Chemical and Biochemical Aspects of Pteridines and Related Topics