The amide group and its preparation methods by acid-amine coupling reactions: an overview
-
Sandra Agudo-Álvarez
 ,
Sandra S. Díaz-Mínguez
,
Sandra S. Díaz-Mínguez


Abstract
The amide bond is one of the most important structural units in nature, as it is part of the backbone of peptides and natural proteins, as well as some essential amino acids, DNA, RNA, hormones, or vitamins found in the body. Furthermore, this bond is significant in the pharmaceutical industry due to its presence in the structure of numerous APIs contained in drugs. This paper reviews the most important methods collected in the bibliography for the preparation of this moiety.
Introduction
The amide bond is one of the most important structural units in nature, as it is part of the backbone of peptides and natural proteins, as well as some essential amino acids, DNA, RNA, hormones, or vitamins found in the body. Furthermore, this bond is significant in the pharmaceutical industry due to its presence in the structure of numerous active pharmaceutical ingredients (APIs) contained in drugs (Fig. 1) [1, 2].
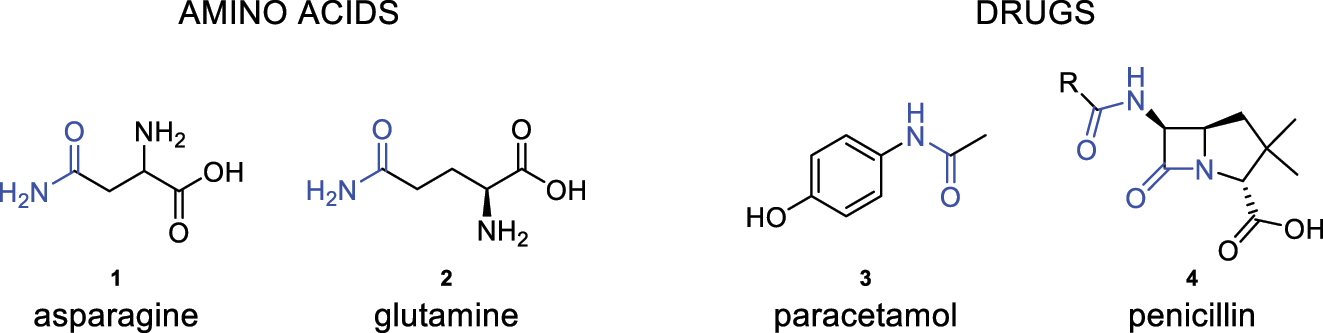
Amino acids and medications containing amide bonds in their structure.
The amide bond is easily formed, imparts structural rigidity, and is resistant to hydrolysis under mild acidic or basic conditions. The resonance forms of the amide bond, resulting from the delocalization of the non-bonding electron pair on the nitrogen atom with the carbonyl group, create a system with some planar character (Fig. 2). This delocalization partially restricts rotations around the nitrogen bond.

Resonance forms of the amide bond.
Most amides are solids at room temperature, except for formamide and some of its derivatives (N-methylformamide, N,N-dimethylformamide, N-ethylformamide, N,N-diethylformamide, etc.) and some acetamide derivatives (N,N-diethylacetamide, etc.) which are in a liquid state at room temperature. Primary and secondary amides have higher boiling points than other tertiary amides of the same molecular mass. This can be attributed to the intermolecular interactions through hydrogen bonding that occur between the molecules of these amides. On the other hand, amides are very weak bases compared to amines. This is explained by the delocalization of the non-bonding electron pair on the nitrogen with the carbonyl. Additionally, the nitrogen-supported proton does not dissociate easily, limiting its acidic character.
Amides are relatively polar due to the presence of the amino group and act as hydrogen bond acceptors. As a result, they can establish a network of hydrogen bonds with water and other protic solvents. The solubility of amides in water is high as a result of these interactions but decreases as the molecular mass of the amide increases.
The synthesis of amide bonds can be carried out by coupling amines and carboxylic acids. This process involves the prior activation of a carboxylic acid followed by the nucleophilic attack of the corresponding amine. Not all amidation methods are equally effective; some have limitations that have been overcome with the development of new reagents. Traditional methods use alkyl or aryl acid chlorides or mixed anhydrides that react with amines. Another strategy that employs milder conditions involves the use of activated carbodiimides, and recently, reagents with activated benzotriazole structures have been developed, which is the focus of this review.
Within this latter method for amide bond formation, carbodiimides (combined with tertiary amines, DMAP, or benzotriazole-like compounds, HOBt (5) [3]1 or HOAt (6)) can be used, as well as phosphonium salts (BOP (7), PyBOP (8) and others) and guanidinium and uronium salts (HBTU (9), TBTU (10), HATU (11) and others) (Fig. 3).
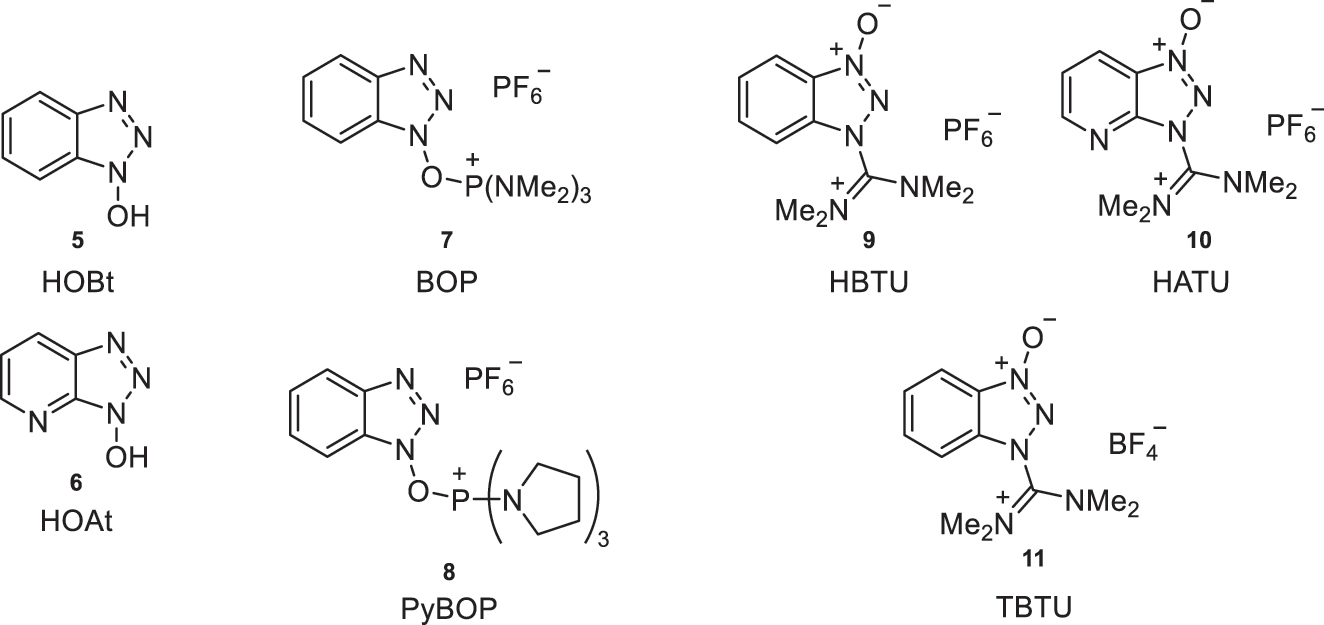
Examples of coupling agents for amide bond formation with benzotriazole-like structures.
Not all strategies work equally well for every acid-amine pair, and it is necessary to choose the most suitable method for each specific case. In the literature, various conditions are documented that provide high yields, selectivity, reproducibility, and low epimerization (if the substrates contain chiral centers) in the formation of these amide bonds [1].
Reagents for amidation
Next, we will discuss the main methods for amide bond formation as described in the literature, along with recent examples.
Acid chlorides
One of the oldest and simplest methods for amide bond formation involves the activation of a carboxylic acid, which is converted into its corresponding acid halide for subsequent reaction with an amine. The most common acid halides used are acid chlorides [1, 4, 5].
The preparation of these acid chlorides can be carried out on both small and industrial scales. In industrial production, selected acids are reacted with reagents like phosgene (12). For example, the reaction of nonanoic acid and phosgene (12) can yield nonanoyl chloride [6]. Another notable method on an industrial scale is the acidolysis of anhydrides using hydrochloric acid, such as the reaction of acetic anhydride with hydrogen chloride to produce acetyl chloride [7]. On the other hand, for the preparation of acid chlorides on a laboratory scale, the most commonly used reagents are thionyl chloride (13) and oxalyl chloride (14). Phosphorus chlorides such as phosphorus oxychloride (15), phosphorus trichloride (16), and phosphorus pentachloride (17) can also be employed (Fig. 4) [1, 2, 4].

Reagents used for the industrial-scale preparation of acid chlorides.
For the production of the desired acid chlorides using thionyl chloride (13) and oxalyl chloride (14) as reagents, it is common to use catalytic amounts of DMF [1], pyridine, or DMAP [4]. When DMF is used as a catalyst, the Vilsmeier-Haak intermediate (18) is formed (Fig. 5). This intermediate then reacts with the corresponding acid to yield its derived acid chloride [1, 4].
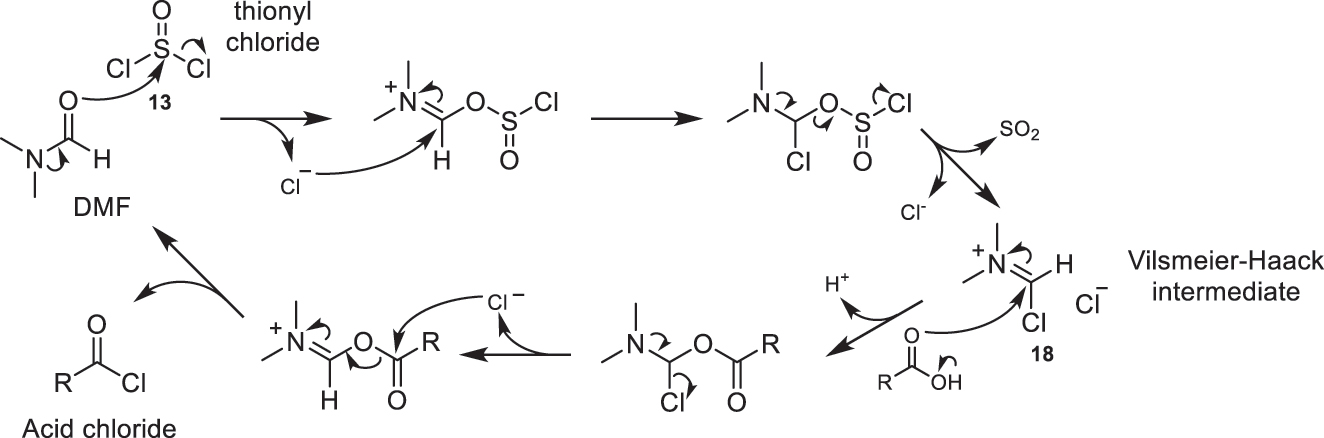
Proposed mechanism for the production of acid chlorides using thionyl chloride (13) as a reagent and DMF as a catalyst.
On the other hand, the use of thionyl chloride (13) is the most common in laboratories due to its cost-effectiveness and better yields compared to its counterparts. However, it has the drawback of forming dimethylcarbamoyl chloride (19), a carcinogen in animal models when DMF is used as a catalyst (Fig. 6) [1].

Proposed mechanism for the formation of dimethylcarbamoyl chloride (19) using thionyl chloride (13) as a reagent and DMF as a catalyst.
The most commonly used solvents for reagent 13 are aprotic solvents like THF, n-heptane, toluene, MeCN, and DME, although this same reagent can sometimes be used as a solvent [1].
From an experimental standpoint, oxalyl chloride (14) offers advantages over thionyl chloride (13). Among these, its lower boiling point (bp (COCl)2: 62 °C, bp SOCl2: 75 °C) can be highlighted, allowing for more rapid removal of excess reagent through evaporation. On the other hand, when used with DMF as a catalyst, it does not form dimethylcarbamoyl chloride. However, it generates CO as a byproduct in the formation of the Vilsmeier-Haack intermediate (18), which is highly toxic (Fig. 7).

Proposed mechanism for the production of acid chlorides using oxalyl chloride (14) and DMF as a catalyst.
Similar to thionyl chloride (13), the most common solvents used with oxalyl chloride (14) are aprotic solvents such as toluene, THF, EtOAc, i PrOAc, or MeCN [1].
As mentioned earlier, the formation of the amide bond occurs through the reaction of an amine and the desired acid chloride. This reaction can be carried out under anhydrous conditions using organic bases like Et3N, i Pr2NEt, pyridine [1], NMM [3], or DIEA [4]. Despite their sensitivity to water, acid chlorides can react with amines in the presence of aqueous bases like NaOH, NaHCO3, K2CO3, or K3PO4 under Schotten-Baumann conditions [1, 4, 5].
Below are some examples of this methodology used for amide bond formation with thionyl chloride (13) and oxalyl chloride (14) (Fig. 8) [8, 9].

Amidation reaction using acid chlorides.
Anhydrides
An alternative method for amide bond formation involves the use of anhydrides [1]. These compounds are derived from carboxylic acids [10] and react with a wide range of nucleophiles, including amines [4].
Symmetrical anhydrides
The preparation of symmetrical anhydrides can be carried out by heating the selected acid to dehydrate it using a Dean-Stark system or through dehydrating agents to remove water. Additionally, the acid can be reacted with DCC under mild conditions to yield the anhydride [4].
The primary limitation of these anhydrides is their molecular economy and waste generation, as only half of the equivalents of the acid used effectively couples with the amine. Moreover, if the acid used is excessively expensive, these anhydrides have economic limitations [4, 10].
Mixed anhydrides
As a solution to the drawbacks of using symmetrical anhydrides, mixed anhydrides can be employed where the second carboxylic residue is cheap and easy to couple with the acid. The most common reagents used for obtaining mixed anhydrides are derivatives of pivaloyl chloride (24), isobutyl chloroformate (25), and EEDQ (26), as well as derivatives of acetic anhydride (27) and tert-butoxycarbonyl anhydride (28) (Fig. 9) [1, 4, 10].

Reagents used for the preparation of mixed anhydrides.
The advantage of these anhydrides is due to the proper regioselectivity they exhibit in nucleophilic addition to the more reactive position a, which is more electrophilic than position b. To further favor this selectivity, sterically hindered reagents are often used in the formation of the anhydride to improve the regioselectivity of the nucleophilic attack by the amine [1]. Anhydrides derived from pivaloyl chloride (24) and isobutyl chloroformate (25) are noteworthy, as their high selectivity is believed to be a result of the steric hindrance they impose on position b (Fig. 10) [1, 4].

Proposed mechanism and regioselectivity of amidation using isobutyl chloroformate (25).
In the literature, there are several examples of the use of in situ formed mixed anhydrides for amidation reactions, as shown in the figures. These anhydrides are formed using reagents 24 and 25 along with Et3N as a base (Fig. 11) [10, 11].
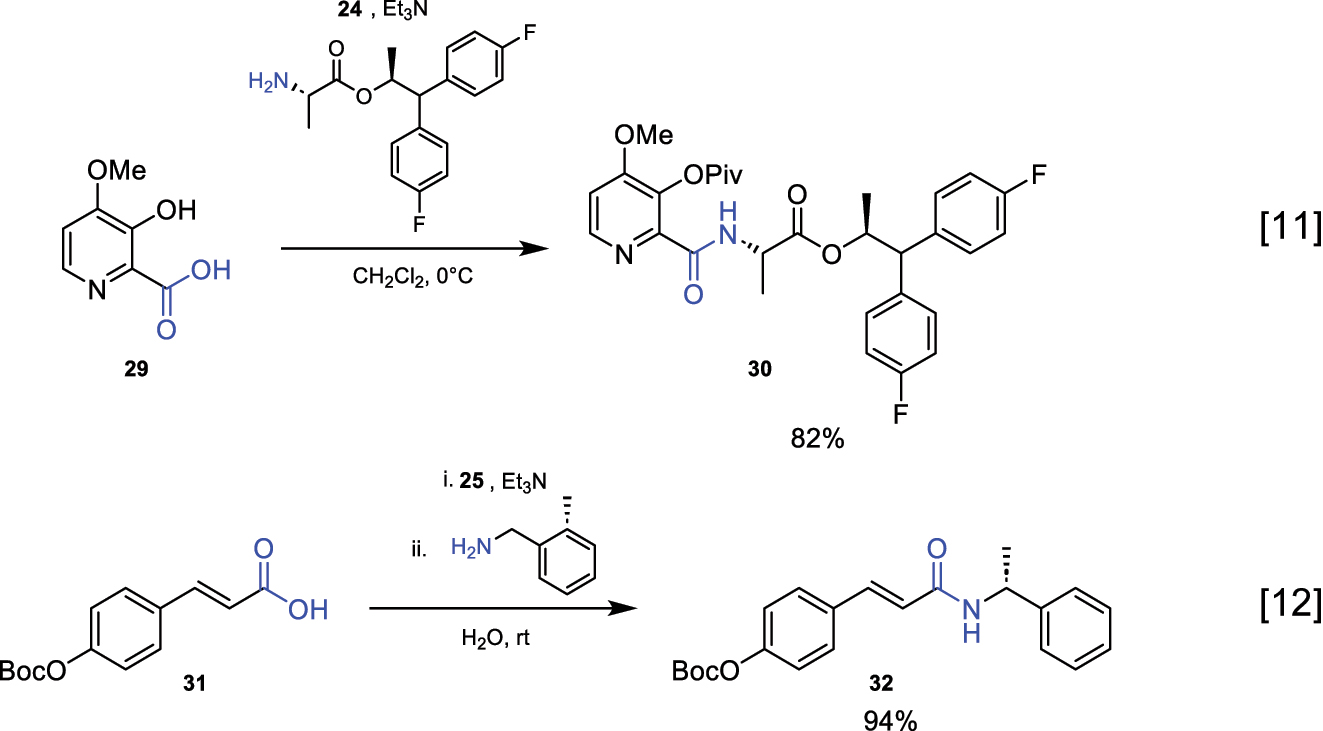
Reaction for amide bond formation via mixed anhydrides.
In the literature, there are examples where the use of chloroformates for the generation of mixed anhydrides results in a two-step synthetic process for amide formation: first, the acid is activated by forming the corresponding anhydride, and then a subsequent step where the nucleophilic attack of the amine occurs. However, when mixed anhydrides are used with EEDQ (26) as the reagent in the presence of the corresponding amine, the coupling is direct, as the anhydride reacts with the amine as soon as it forms [10]. This reagent is cost-effective, commercially available, and any byproducts can be easily removed through aqueous extraction (Fig. 12) [1], [12], [13], [14.
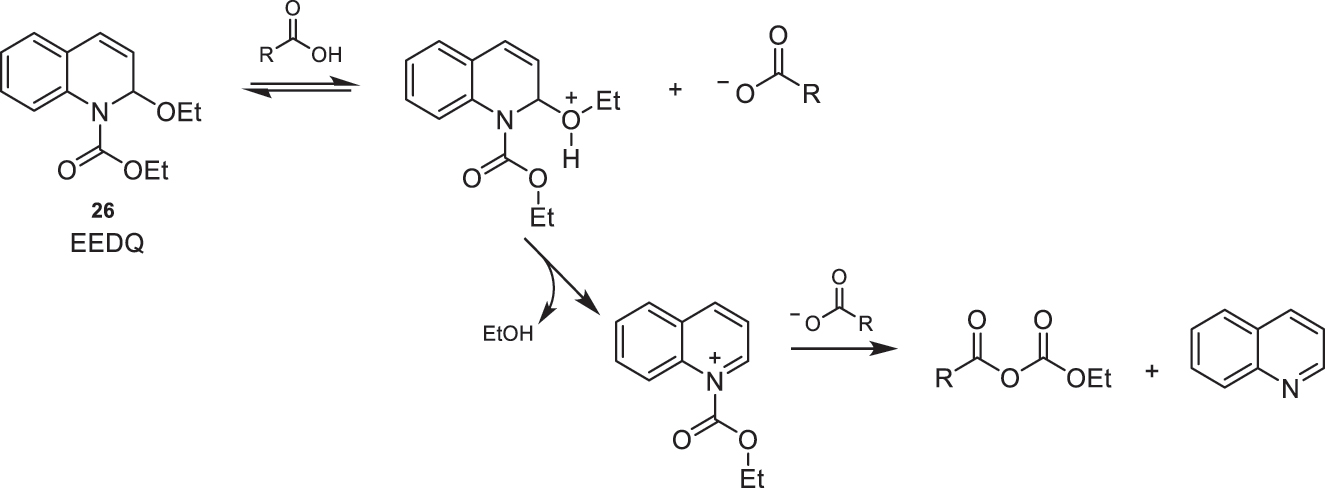
Proposed mechanism for the formation of mixed anhydrides using EEDQ (26) as the reagent.
An example of the use of the reagent EEDQ (26) is described for the formation of the amide bond, as shown in Fig. 13 [15].
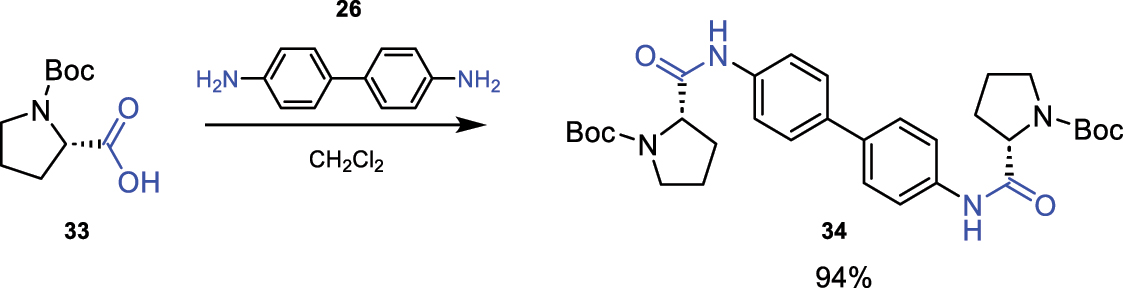
Amidation reaction using EEDQ (26).
1,1′-Carbonyldiimidazole
1,1′-Carbonyldiimidazole (35) (Fig. 14) is an economical reagent commercially available on a kilogram scale and is a crystalline solid at room temperature. Its use does not pose significant safety issues under typical reaction conditions. Furthermore, this reagent can also be used in the absence of a solvent in amidation reactions. This approach significantly reduces reaction times compared to using dry solvents, and it is considered a protocol that falls within Green Chemistry, as it eliminates the need for organic solvents [16].
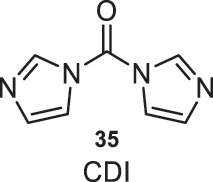
Structure of the CDI (35) reagent.
In amidation reactions using CDI (35) as the activating agent, a mixed anhydride is formed through the reaction of the corresponding acid with reagent 35. This anhydride is highly reactive and disproportionates in the presence of imidazole to yield the corresponding imidazolide, imidazole, and CO2 (Fig. 15). The formation of the amide derived from imidazole occurs through the nucleophilic attack of the departing imidazole on the more electrophilic carbon (position a) of the mixed anhydride, which originates from the carboxylic acid used. If the attack were to occur on the other carbon (position b), the starting materials would be regenerated. Finally, to obtain the desired amide, the reaction of the generated imidazolide with the corresponding amine takes place, as shown in Fig. 15. As mentioned earlier, imidazole is produced as a byproduct, which can be easily removed by treatment with acidic aqueous solutions. This methodology has the advantage of not requiring additional bases, reducing the economic cost and environmental impact of the process [1].
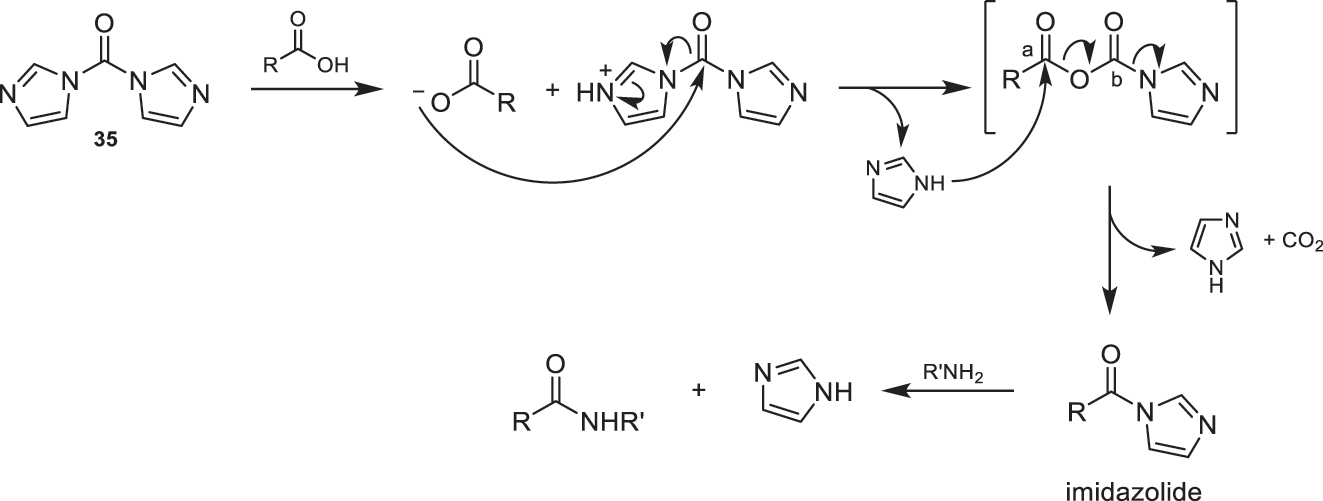
Proposed mechanism for amidation reaction using CDI (35).
In the case that the desired product contains a tertiary amide, and due to the challenges that can arise when using secondary amines due to steric hindrance, an elegant method has been developed for the preparation of such amides. In this approach, the amidation reaction can use methyl iodide in combination with CDI (35). When reacting with the selected acid (Fig. 16), they form an imidazolyl urea, which subsequently gives rise to an amiduro anion. This significantly enhances the nucleophilic character of the starting secondary amine, which, upon reaction with the imidazolide generated in situ in the reaction medium, yields the desired tertiary amide [4].
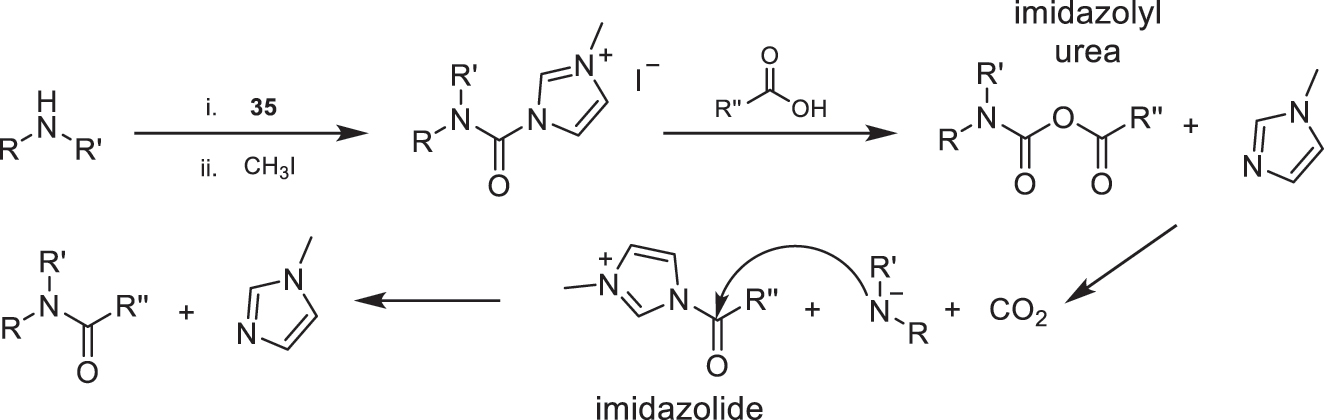
Proposed mechanism for the formation of tertiary amides.
In Fig. 17, an example of amide bond formation using CDI (35) as the reagent is illustrated [17].
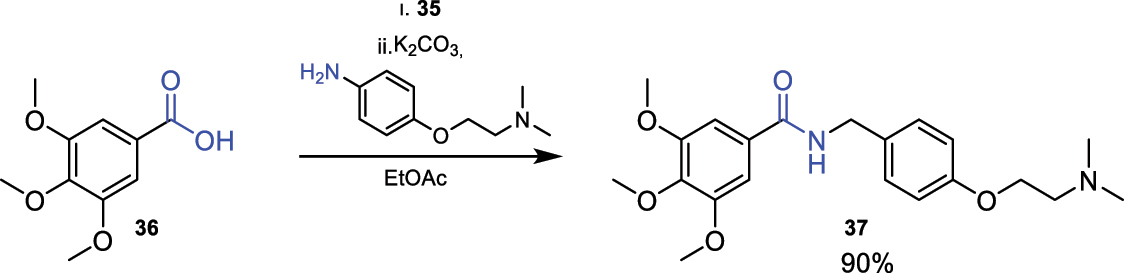
Reaction for amide bond formation using CDI (35).
Phosphorus and boron-derived reagents
As an alternative for amide bond formation, reagents derived from phosphorus and boron are used (Fig. 18). In particular, propanophosphoric anhydride (38) and boric acid (39) are noteworthy. The latter has significantly gained popularity as a reagent for amidation reactions in recent years due to its cost-effectiveness and high solubility in water, making it easy to remove through extraction methods.
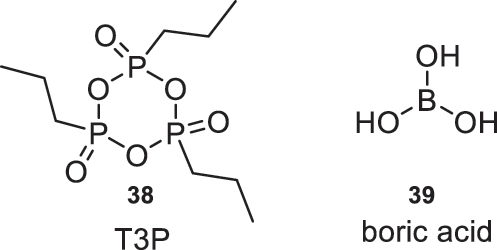
Phosphorus and boron-derived reagents used for amide bond formation.
The proposed mechanism described in the literature for amidation using these reagents is shown in Fig. 19. The presence of a base forms the carboxylate anion of the corresponding acid. This species, through a nucleophilic attack on the desired reagent and favored by the oxophilic character of phosphorus and boron, generates an activated phosphate or borate. This activated species will subsequently undergo a nucleophilic attack by the corresponding amine for the formation of the desired amide.

Proposed mechanism for amidation reactions using reagents derived from phosphorus and boron.
Among the phosphorus-derived reagents, T3P (38) stands out for its low toxicity, stability, and ease of handling. Moreover, the byproducts of the amidation reaction are water-soluble and can be easily extracted. The drawback of this reagent is its moderate cost and environmental impact, as it generates phosphonate-derived residues that cause water eutrophication in rivers, lakes, aquifers, seas, and oceans [1].
On the other hand, among the boron-derived analogs, boric acid (39) is noteworthy as a reagent for amide bond formation because it is cost-effective. Furthermore, once the reaction is complete, this reagent is water-soluble and can be extracted through an aqueous workup [1].
Below are some examples described in the literature where these reagents are used for amide bond formation (Fig. 20) [18, 19].
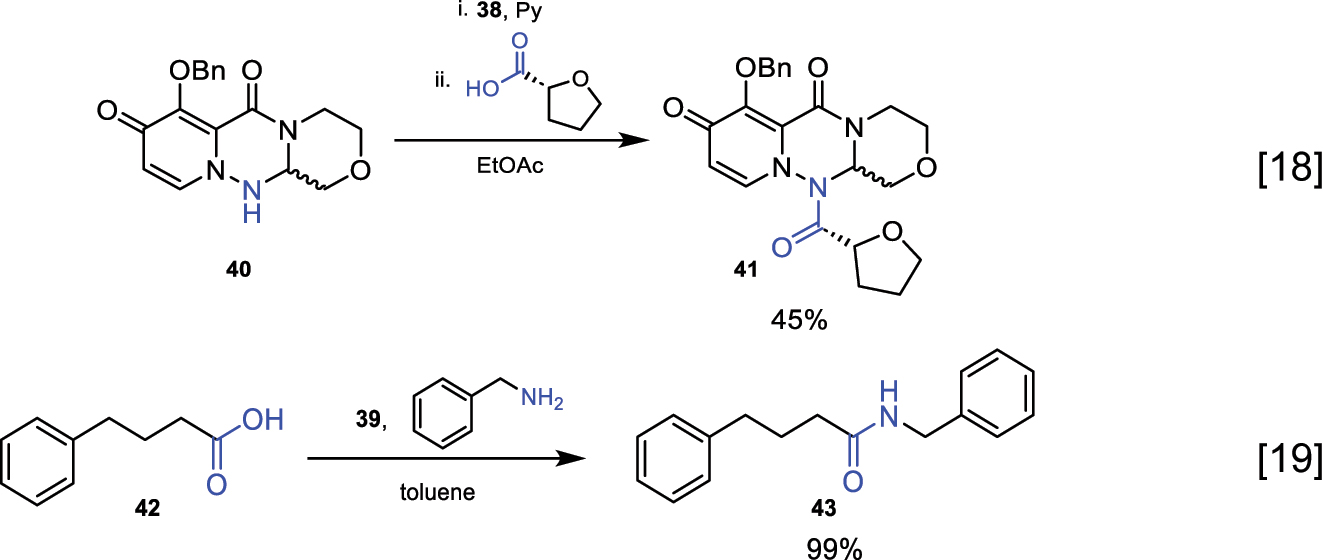
Reaction for amide bond formation using phosphorus and boron-derived reagents.
Benzotriazole-type reagents
As in previous cases, the most commonly used strategy for amide bond formation is based on the reaction between acids and amines through the activation of the –OH group in the acid functionality. In this case, carbodiimide-type reagents, reagents based on phosphorus salts, and guanidinium and uronium salt-derived reagents are used.
Carbodiimides
A very common method for amide bond formation is the use of carbodiimides. These compounds have in their structure two nitrogen atoms with basic character that can promote the reaction between an acid and an amine, leading to the formation of the amide bond. For this, it is necessary to form a more reactive species in situ, an O-acylisourea, which leads to the formation of the desired product in the presence of the required amine (Fig. 21).
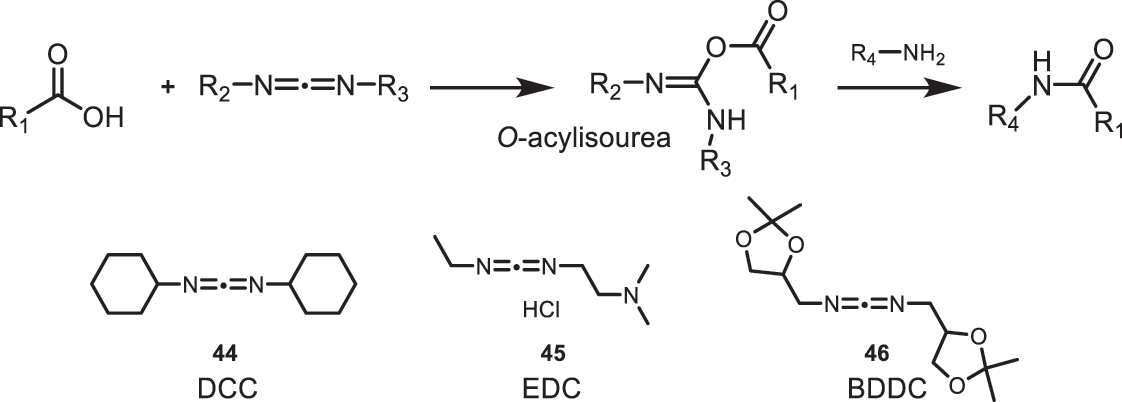
Mechanism for the formation of amide bonds through O-acylisourea.
There are numerous examples of carbodiimides used in organic synthesis with different substitutions, among which DCC (44) can be highlighted. In reactions using DCC (44) as a coupling agent, DCU is formed as a byproduct, which is difficult to remove by column chromatography. A widely used alternative is EDC (45), which has a significant advantage over DCC (44) because both EDC (45) and its urea derivative have high solubility in aqueous solutions and can be easily eliminated in the workup through aqueous washing. In coupling reactions with amino acids using BDDC (46), moderate yields are obtained, and the formed byproducts can be easily removed through an acidic wash.
The use of N-hydroxylamine derivatives (HOXt) in combination with carbodiimides leads to the formation of esters that, while less reactive than O-acylisoureas [20], allow for good yields, increasing the efficiency of the process by avoiding the possible formation of N-acylureas. This advantage is due to the ability of N-hydroxylamines to protonate O-acylisoureas, thus preventing the intramolecular reaction that would lead to the formation of N-acylureas, shifting the reaction towards the formation of activated esters (Fig. 22). The presence of tertiary amines promotes the formation of activated esters [21, 22]. As an alternative to the use of N-hydroxylamines, this type of reaction can be carried out using a catalytic amount of a tertiary amine, DMAP, which favors the progress of the reaction through the formation of activated amides (Fig. 22) [23].

Synthetic routes for carbodiimide-mediated amidation reactions.
Some of the most commonly used N-hydroxylamines, in combination with carbodiimides, as coupling agents in the formation of amide bonds from their precursor acids, are N-hydroxylamines HOSu (NHS) (50), HOBt (5), and HOAt (6), with the latter two having a benzotriazole-like structure. In terms of performance, activated esters derived from HOAt (6) show better yields than those derived from HOBt (5) [24]. This could be because the nitrogen at position 7 of benzotriazole, due to stereochemical effects, allows for the formation of a hydrogen bond with the amine, which facilitates its proper orientation for the subsequent nucleophilic attack leading to amide bond formation (Fig. 23) [4, 10].
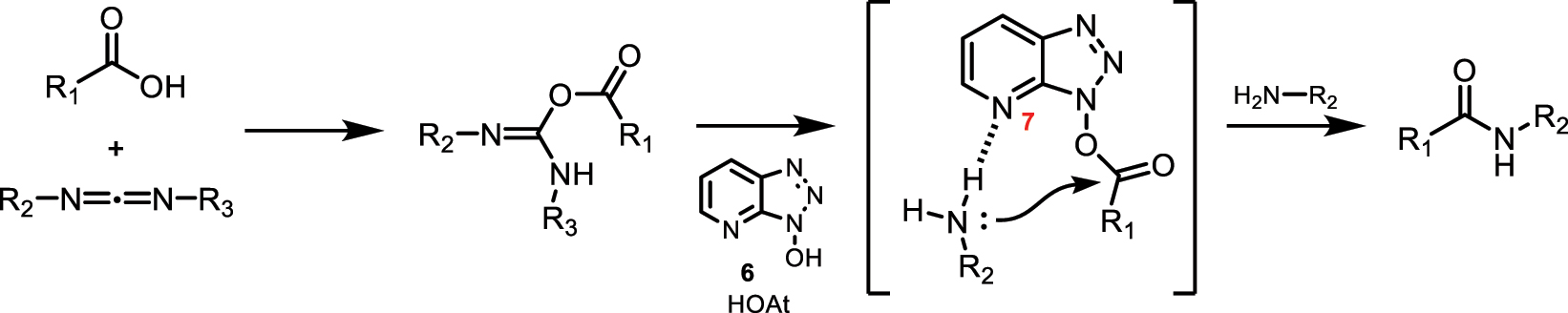
Intermolecular interaction via hydrogen bonding when using HOAt (6) as a coupling agent.
The benzotriazoles 6-HOAt (47), 5-HOAt (48), and 4-HOAt (49) do not allow for the formation of this intermolecular hydrogen bond, resulting in a loss of process efficiency.
Some examples in which this methodology is used are as follows (Fig. 24) [23, 25, 26].

Carbodiimide-mediated amidation reaction.
Phosphonium salts
Alternatively, for amide bond formation, reagents incorporating phosphonium salts in their structure were developed, which prevent secondary reactions and epimerization processes. In this regard, a phosphorus-derived reagent, BOP (7), was optimized, incorporating an N-hydroxybenzotriazolyl subunit into its structure. Despite yielding fast couplings and containing the mentioned functionality to prevent the epimerization of chiral centers in the starting materials, BOP (7) has had limited use in the industry due to the generation of HMPA as a byproduct, a highly carcinogenic compound [1, 4, 5].
As an alternative to avoid the formation of this toxic byproduct, PyBOP (8) was developed. However, this reagent comes with a high cost, preventing its use on a large scale (Fig. 25) [1, 4, 5].

Phosphonium salt-derived reagents.
As an example of the preparation of such reagents, Fig. 26 shows the synthesis of the BOP reagent (7). For its preparation, tris(dimethylamino)phosphine (61) is reacted with carbon tetrachloride in the presence of HOBt (5), using THF as a solvent, followed by exchanging the chloride anion with the hexafluorophosphate anion [5].

Preparation of BOP (7).
The mechanistic proposal described in the literature for amidation using reagents with oxyphosphonium salt-like structure is depicted in Fig. 27, using BOP (7) as an example of a coupling agent. In the presence of a base, the carboxylate anion generated from the corresponding acid undergoes a nucleophilic attack on the phosphonium cation, favored by the oxophilic nature of phosphorus. This species leads to the activation of the O-benzotriazole ester and the formation of HMPA, the previously mentioned toxic product. Finally, the nucleophilic attack of the corresponding amine on this activated ester takes place to obtain the desired amide [1, 4, 5].
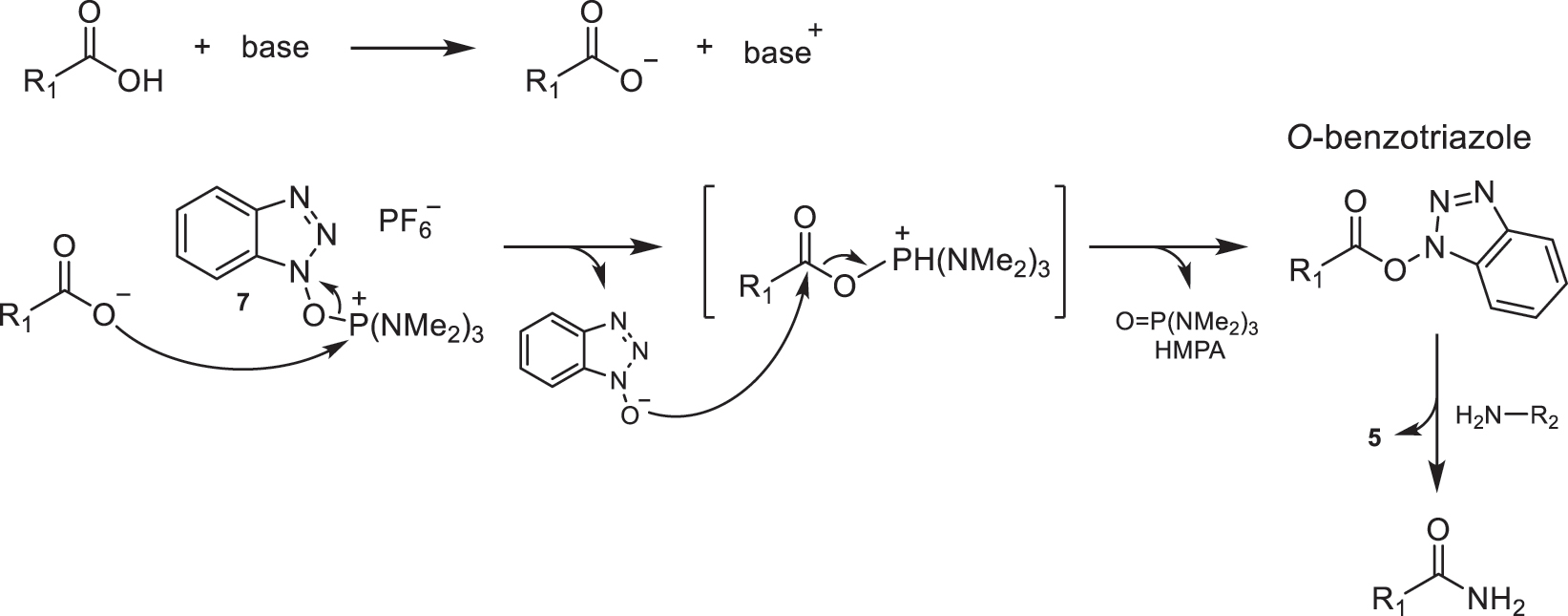
Proposed mechanism for amide bond formation using BOP (7) as a coupling agent.
It has been observed that the use of these types of reagents for the preparation of peptides containing N-methyl-α-amino acids in their structure leads to slow and low-yield couplings, with epimerization occurring in some cases. This may be because the activated benzotriazolyl ester is bulky and highly effective in reactions with primary amines. However, it does not react as readily with secondary amines, leading to the degradation of reaction intermediates. Therefore, more efficient derivatives such as PyBroP (63) for peptide couplings [4] and PyCloP (62) have been developed. These derivatives are characterized by being less bulky than their predecessors and therefore have less steric hindrance (Fig. 28), which promotes the amidation reaction [5].

Amidation of N-methyl-α-amino acids.
Additionally, phosphonium salt derivatives of HOAt (6) have been developed, such as AOP (64) and PyAOP (65), which are more efficient as coupling reagents due to the potential formation of a hydrogen bond in the reaction intermediate, as mentioned in previous sections (Fig. 29) [5].
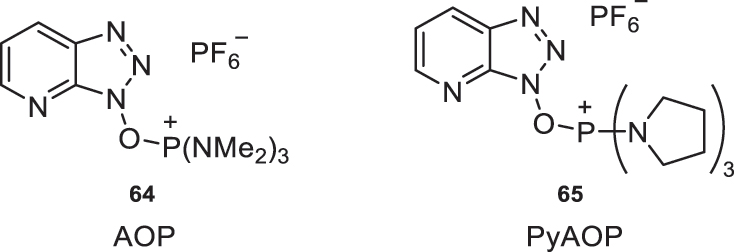
Phosphonium salt derivatives of HOAt (6).
Figure 30 displays some of the reagents described in the literature, which inhibit racemization in the coupling of peptides containing N-methyl-α-amino acids. For example, PyNOP (66), PyFOP (67), PyFNBOP (68), PyCloK (69), PyPOP (70), PyTOP (71), PyDOP (72), and PyDAOP (73) [5].
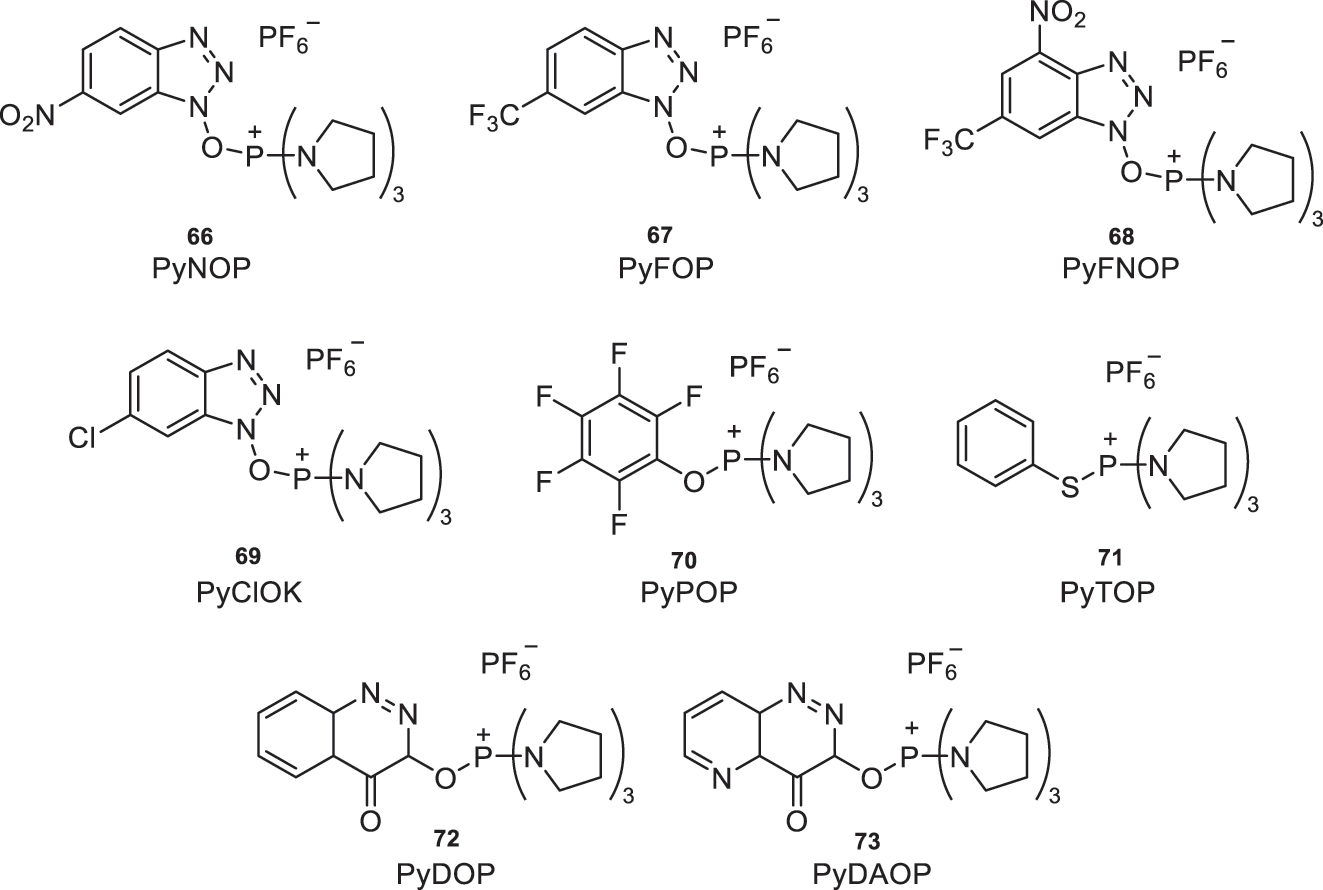
Reagents used in the amidation reaction of N-methyl-α-amino acids.
Below, some examples using reagents 7 and 8 in amidation reactions are presented (Fig. 31) [27, 28].
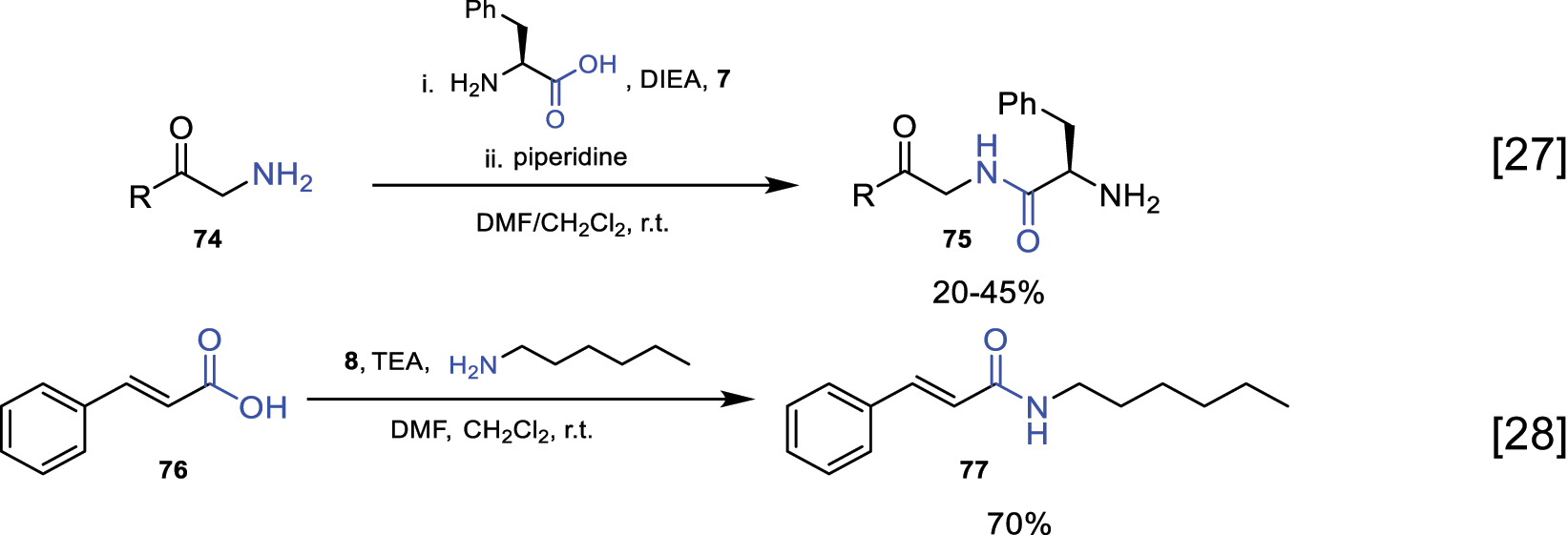
Amidation reaction using phosphonium salt-derived reagents.
Guanidinium and uronium salts
In a manner similar to phosphorus-derived benzotriazolic reagents, other reagents incorporating guanidinium and uronium cations have been developed for use as coupling agents in amide bond formation. As an alternative to the BOP reagent (7), HBTU (9) has been developed. Since then, numerous guanidinium and uronium salts with a benzotriazole-like structure have been reported for use as coupling agents in amidation reactions [1].
In Fig. 32, examples of these coupling agents for amide bond formation are shown.

Reagents for amide bond formation derived from guanidinium and uronium salts.
For the preparation of these benzotriazole reagents, the synthesis of HBTU (9) is shown as an example in Fig. 33. To achieve this, tetramethylurea (78) is reacted with (COCl2)2 (14) in toluene to obtain the corresponding chlorotetramethylguanidinium salt, followed by an anion exchange with potassium hexafluorophosphate. Next, this species undergoes nucleophilic attack by HOBt (5) using Et3N as a base, resulting in the formation of HBTU (9) [5, 29].
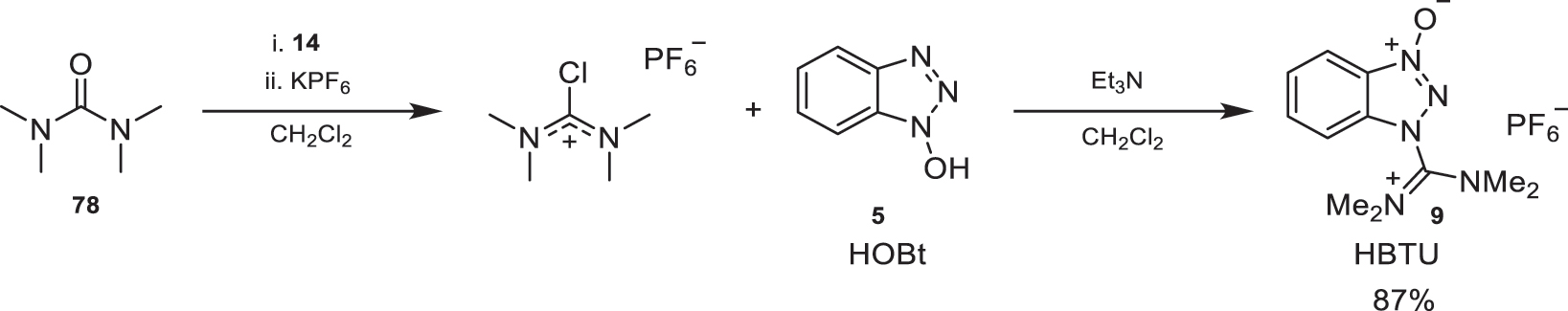
Preparation conditions of HBTU (9).
Among the main differences within these reagents, we can highlight: (a) the cation incorporated into their structure, some contain the guanidinium cation, while others contain the uronium cation (Fig. 34) and (b) the substitution of the aromatic ring; some have a nitrogen atom in the ring, while others do not, which imparts differences in reactivity. These structures have been determined both by NMR in solution and by X-ray crystallography in the solid state, both for HBTU (9) and HATU (10) [30].

Difference between uronium and guanidinium cations.
It has been demonstrated that uronium salts are more efficient as coupling agents for amide bond formation than their guanidinium analogs. This is because the activation of the acid through uronium salts (which involves breaking the C–O bond) is faster than through guanidinium salt species (which involves breaking the C–N bond). For example, during a study of peptide cyclization, this reactivity difference was observed. When using N-HBTU as the reagent, more than 91 % of the peptide had not reacted after 15 s. However, when using O-HBTU, more than half of the peptide had disappeared [30, 31].
The mechanism of amide bond formation using these coupling agents is equivalent to that described for phosphonium salts (Fig. 27). In Fig. 35, the proposed mechanism for the amidation reaction using HBTU (9) as the reagent is shown. In this mechanism, the acid, in the presence of a tertiary base, forms the carboxylate anion, which undergoes a nucleophilic attack on the electrophilic center of the guanidinium moiety. This leads to the activation of a benzotriazolyl ester, which subsequently undergoes nucleophilic attack by the corresponding amine, resulting in the desired amide. As byproducts of this reaction, HOBt (5) and tetramethylurea are obtained [1].
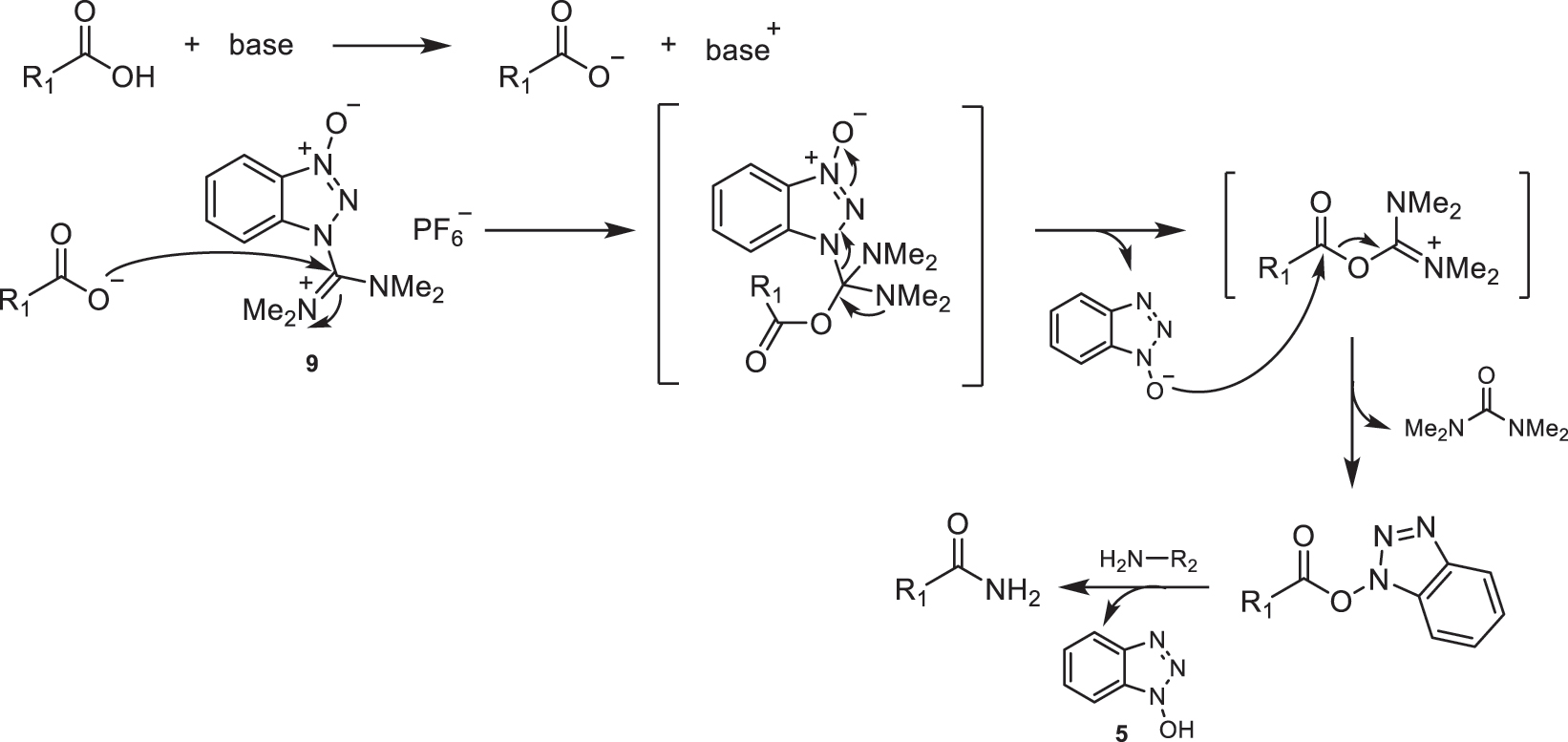
Proposed mechanism for amidation using HBTU (9).
The use of tertiary bases such as Et3N, i Pr2EtN, or collidine is necessary because these compounds act as bases and not as nucleophiles. In this way, these bases deprotonate the carboxylic acid, and their steric hindrance shields the nitrogen atom, enhancing their basicity.
Furthermore, in amidation reactions using this methodology, in addition to the standard reaction conditions where a tertiary amine is used along with the coupling agent (HBTU (9), HATU (10), TBTU (11), TPTU (79), etc.), they can also be used in combination with HOBt (5) to promote the formation of the activated benzotriazolyl ester and thereby increase the yield of the desired reaction [4].
The reagent HBTU (9) is not commonly used on a large scale for amide bond formation due to its high economic cost and lower yields. Conversely, its structural analog, HATU (10), is used because it provides faster and more efficient couplings with less epimerization, as well as milder reaction conditions. This might be attributed to the potential formation of a hydrogen bond in the reaction intermediate, as mentioned earlier in previous sections. Additionally, it has been found that the counterion of both reagents has no significant effect on the course of the reaction, with no substantial changes in its yield [1, 4].
The main drawback of these reagents is their high molecular weight, which, along with their high cost, increases the cost of the amidation process. On the other hand, N,N,N′,N′-tetramethylurea is formed as a byproduct, which is a cytotoxic compound and challenging to purify. However, their use is justified when, for example, the carboxylic acid contains an α-epimerizable chiral center and mild reaction conditions are required [1].
Other derivatives also commercially available and developed more recently are: TDTU (81), HDTU (82), TDATU (83), HDATU (84), TSTU (85) (Fig. 36) [5].

Reagents derived from guanidinium and uronium salts.
These coupling agents follow the reactivity trend already mentioned above, with uronium salts being more efficient than their guanidinium analogues.
Some examples described in the bibliography that use this methodology are described below (Fig. 37) 32], [33], [34], [35], [36.
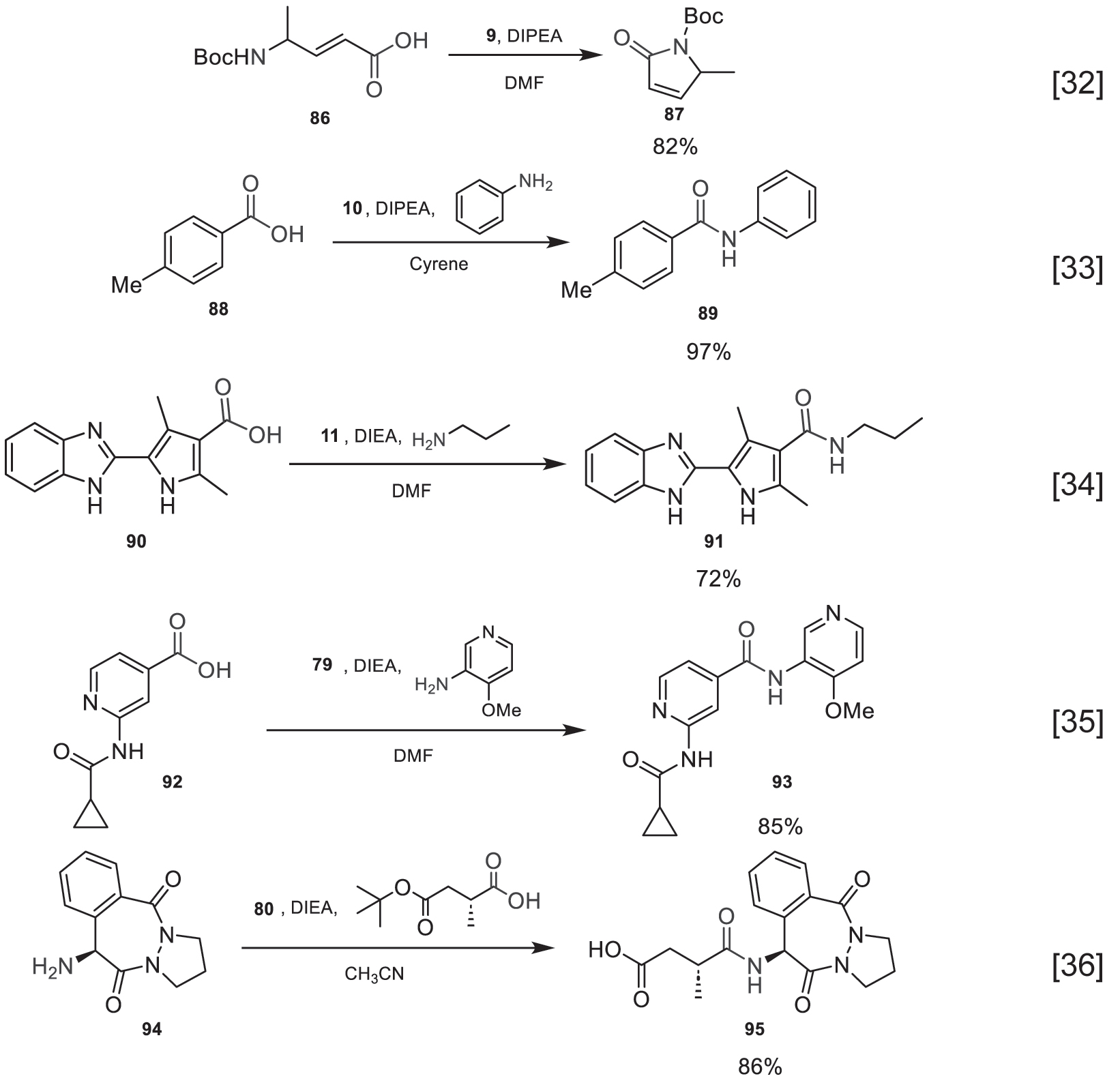
Amide bond formation reaction using reagents derived from guanidinium and uronium salts.
Conclusions
The amide bond has great biological importance. Therefore, this review aims to show a general review of the main methods of formation of these amide bonds that are collected in the bibliography. In this review and within the different methodologies described, the importance of using benzotriazole reagents is highlighted since they allow the obtaining of amides with good yields and using mild reaction conditions, in addition to significantly reducing epimerization in the case of that the starting compounds contain chiral centers in their structure. However, there are other methods that stand out for their good yields and for using affordable reagents, so their use cannot be ruled out. In each case, an exhaustive analysis of the requirements of the starting products to be used and the particular considerations of each case must be carried out to use the most convenient method.
Abbreviations
- AOP
-
(7-azabenzo-triazol-1-yloxy)tris-(dimethylamino)phosphonium hexafluorophosphate
- BDDC
-
bis{[4-(2,2-dimethyl-1,3-dioxolyl)]methyl}carbodiimide
- BOP
-
(benzotriazol-1-yloxy)-tris-(dimethylamino)phosphonium hexafluorophosphate
- DCC
-
dicyclohexylcarbodiimide
- DCU
-
dicyclohexylurea
- DIEA
-
N,N-diisopropylethylamine
- DMAP
-
N,N-dimethylaminopyridine
- DME
-
1,2-dimethoxyethane
- DMF
-
N,N-dimethylformamide
- EDC
-
N-ethyl-Nʹ-(3-dimethylaminopropyl)carbodiimide
- HATU
-
N-[(dimethylamino)-1H-1,2,3-triazolo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide
- HBTU
-
N-[(1H-benzotriazol-1-yl) (dimethylamino)-methylene]-N-methylmethanaminium hexafluorophosphate N-oxide
- HDATU
-
O-(3,4-dihydro-4-oxo-5-azabenzo-1,2,3-triazin-3-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HDTU
-
2-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HOAt
-
1-hydroxy-7-azabenzotriazole
- HOBt
-
1-hydroxybenzotriazole
- NMM
-
N-methylmorpholine
- PyAOP
-
(7-azabenzotriazol-1-yloxy)tris-(pyrrolidino)phosphonium hexafluorophosphate
- PyBOP
-
(benzotriazol-1-yloxy)-tris(pyrrolidine)phosphonium hexafluorophosphate
- PyBrOP
-
bromotri(pyrrolidino)phosphonium hexafluorophosphate
- PyCloK
-
(6-chloro-benzotriazol-1-yloxy)tris-(pyrrolidino)phosphonium hexafluorophosphate
- PyCloP
-
chlorotri(pyrrolidino)phosphonium hexafluoro-phosphate
- PyDAOP
-
[(3,4-dihydro-4-oxo-5-azabenzo-1,2,3-triazin)-3-yl]tris-(pyrrolidino)phosphonium hexafluorophosphate
- PyDOP
-
[(3,4-dihydro-4-oxo-1,2,3-benzotriazin)-3-(pyrrolidino) phosphonium hexafluorophosphate
- PyFNBOP
-
[4-nitro-6-(trifluoromethyl-1-yl)oxy]tris-(pyrrolidino)phosphonium hexafluorophosphate
- PyFOP
-
[(6-trifluoromethyl-1-yl)oxy]tris-(pyrrolidino)phosphonium hexafluorophosphate
- PyNOP
-
[(6-nitrobenzotriazol-1-yl)oxy]tris-(pyrrolidino)phosphonium hexafluorophosphate
- PyPOP
-
N,N,N′,N′-bis(tetramethylene)-O-pentafluoro phenyluronium hexafluorophosphate
- PyTOP
-
(pyridyl-2-thio)tris(pyrrolidino)phosphonium hexafluorophosphate
- T3P
-
n-propamephosphonic anhydride
- TBTU
-
N-[(1H-benzotriazol-1-yl)(dimethylamino)methylene]-N-methylmethanaminium tetrafluoroborate N-oxide
- TDU
-
2-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate
- TDATU
-
O-(3,4-dihydro-4-oxo-5-azabenzo-1,2,3-triazin-3-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate
- THF
-
tetrahydrofuran
- TOTU
-
O-[(cyano(ethoxycarbonyl)methylene)amino]-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- TPTU
-
2-(2-oxo-1(2H)-pyridyl)-1,1,3,3-tetramethyluronium tetrafluoroborate
- TSTU
-
2-succinimido-1,1,3,3-tetramethyluronium tetrafluoroborate
References
[1] J.R. Dunetz, J. Magano, G.A. Weisenburger. Org. Proc. Res. Dev. 20, 140 (2016), https://doi.org/10.1021/op500305s.Search in Google Scholar
[2] L. Zhu, L. Deng, Y. Xie, L. Liu, X. Ma, R. Liu. Results Chem. 5, 100882 (2023), https://doi.org/10.1016/j.rechem.2023.100882.Search in Google Scholar
[3] Guidelines for Authors of the American Chemical Society, A.M. Coghill, L.R. Garson. The ACS Stile Guide: Effective Communication of Scientific Information, pp. 169–202, Oxford University Press, New York (2006).Search in Google Scholar
[4] A. El-Faham, F. Albericio. Chem. Rev. 111, 6557 (2011), https://doi.org/10.1021/cr100048w.Search in Google Scholar PubMed
[5] C.A.N.G. Montalbetti, V. Falque. Tetrahedron 61, 10827 (2005), https://doi.org/10.1016/j.tet.2005.08.031.Search in Google Scholar
[6] R. Worms, H.J. Niederkirchen, T. Ludwigshafen, W. Mannheim, A. Mainz. J. Mannheim. US Patent 6,770,783 B1, Filed 31 August 2000, Issued 03 August 2004.Search in Google Scholar
[7] U.-R. Samel, W. Kohler, A.O. Gamer, U. Keuser, S.-T. Yang, Y. Jin, M. Lin, Z. Wang, J.H. Teles. Propionic acid and derivatives. In Ullmann’s Encyclopedia of Industrial Chemistry, p. 1, Wiley-VCH, Weinheim, Germany (2018).10.1002/14356007.a22_223.pub4Search in Google Scholar
[8] J.T. Bristow. P.C.T. WO 2023/051311 A1, Filed 06 April 2023, Issued 20 September 2022.Search in Google Scholar
[9] E.M. Beccalli, A. Bernasconi, E. Borsini, G. Broggini, M. Rigamonti, G. Zecchi. J. Org. Chem. 75, 6923 (2010), https://doi.org/10.1021/jo101501u.Search in Google Scholar PubMed
[10] M.M. Joullié, K.M. Lasse. Arkivoc 8, 189 (2010), https://doi.org/10.3998/ark.5550190.0011.816.Search in Google Scholar
[11] N.R. Babij, N. Choy, M.A. Cismesia, D.J. Coulin, N.M. Hough, P.L. Johnson, J. Klosin, X. Li, Y. Lu, E.O. McCusker, K.G. Meyer, J.M. Renga, R.B. Rogers, K.E. Stockman, N.J. Webb, G.T. Whiteker, Y. Zhu. Green Chem. 22, 6047 (2022), https://doi.org/10.1039/d0gc02063j.Search in Google Scholar
[12] M. DellaGreca, L. Longobardo. ChemistrySelect 5, 4588 (2020), https://doi.org/10.1002/slct.202000176.Search in Google Scholar
[13] J. Chen, S.P. Corbin, N.J. Holman. Org. Process Res. Dev. 9, 185 (2005), https://doi.org/10.1021/op049777t.Search in Google Scholar
[14] A.H. Davulcu, D.D. McLeod, J. Li, K. Katipally, A. Littke, W. Doubleday, Z. Xu, C. W. McConlogue, C. J. Lai, M. Gleeson, M. Schwinden, R. L. ParsonsJr.. J. Org. Chem. 74, 4068 (2009), https://doi.org/10.1021/jo9003508.Search in Google Scholar PubMed
[15] S.E.A. Karim, Y.H. Youssef, M. Abdel-Halim, E. Frakolaki, N. Vassilaki, G. Zoidis, N.S. Ahmed, A.H. Abadi. Bioorg. Chem. 102, 104089 (2020).10.1016/j.bioorg.2020.104089Search in Google Scholar PubMed
[16] S.K. Verma, R. Ghorpade, A. Pratap, M.P. Kaushik. Tetrahedron Lett. 53, 2373 (2012), https://doi.org/10.1016/j.tetlet.2012.01.125.Search in Google Scholar
[17] E.R. Monteith, P. Mampuys, L. Summerton, J.H. Clark, B.U.W. Maes, C.R. McElroy. Green Chem. 22, 123 (2020), https://doi.org/10.1039/c9gc01537j.Search in Google Scholar
[18] D.L. Hughes. Org. Proc. Res. Dev. 23, 1298 (2019), https://doi.org/10.1021/acs.oprd.9b00144.Search in Google Scholar
[19] R. Pal. Arkivoc 1, 346 (2018), https://doi.org/10.24820/ark.5550190.p010.462.Search in Google Scholar
[20] J.M. Humphrey, A.R. Chamberlin. Chem. Rev. 97, 2243 (1997), https://doi.org/10.1021/cr950005s.Search in Google Scholar PubMed
[21] L.A. Carpino, A. El-Faham. Tetrahedron 55, 6813 (1999), https://doi.org/10.1016/s0040-4020(99)00344-0.Search in Google Scholar
[22] A.K. Ghosh, D. Shahabi. Tetrahedron Lett. 63, 152719 (2021), https://doi.org/10.1016/j.tetlet.2020.152719.Search in Google Scholar PubMed PubMed Central
[23] A.P. Silvestri, J.S. Oakdale. Chem. Commun. 56, 13417 (2020), https://doi.org/10.1039/d0cc05706a.Search in Google Scholar PubMed
[24] R. Subirós-Funosas, R. Prohens, R. Barbas, A. El-Faham, F. Albericio. Chem. Eur. J. 15, 9394 (2009), https://doi.org/10.1002/chem.200900614.Search in Google Scholar PubMed
[25] R.A. Kumar, D.V. Jawale, E. Oheix, V. Geertsen, E. Gravel, E. Doris. Adv. Synth. Catal. 362, 4425 (2020), https://doi.org/10.1002/adsc.202000795.Search in Google Scholar
[26] M.D. Morin, Y. Wang, B.T. Jones, Y. Mifune, L. Su, H. Shi, R.M.Y. Moresco, H. Zhang, B. Beutler, D.L. Boger. J. Am. Chem. Soc. 140, 14440 (2018), https://doi.org/10.1021/jacs.8b09223.Search in Google Scholar PubMed PubMed Central
[27] E. Rémond, J.A. Fehrentz, L. Liénat, S. Clément, J.L. Banères, F. Cavelier. Chem. Eur. J. 28, 47 (2022), https://doi.org/10.1002/chem.202201526.Search in Google Scholar PubMed
[28] C. Varela, A. Cabral, I. Barbosa, S. Costa, E. Soilva, F. Roleira, F. J. Chem. Educ. 97, 2366 (2020), https://doi.org/10.1021/acs.jchemed.8b00604.Search in Google Scholar
[29] V. Dourtoglou, B. Gross, V. Lambropoulou. C. Zioudrou. Synthesis. 7, 572 (1984), https://doi.org/10.1055/s-1984-30895.Search in Google Scholar
[30] L.A. Carpino, H. Imazumi, A. El-Faham, F.J. Ferrer, C. Zhang, Y. Lee, B.M. Foxman, P. Henklein, C. Hanay, C. Mügge, Holger Wenschuh, J. Klose, M. Beyermann, M. Bienert. Angew. Chem., Int. Ed. 41, 442 (2002).10.1002/1521-3773(20020201)41:3<441::AID-ANIE441>3.0.CO;2-NSearch in Google Scholar
[31] J. Klose, M. Bienert, C. Mollenkopf, D. Wehle, C. Zhang, L. A. Carpino, P. Henklein. Chem. Commun. 1847, 1847 (1999), https://doi.org/10.1039/a905021c.Search in Google Scholar
[32] M. Singh, S.A. Nalawade, K. Punneth, K. Veeresh, S. Pahan, S. Dey, H.N. Gopi. Eur. J. Org Chem. 26, e2023006 (2023), https://doi.org/10.1002/ejoc.202300682.Search in Google Scholar
[33] K.L. Wilson, J. Murray, C. Jamieson, A.J.B. Watson. Org. Biomol. Chem. 16, 2851 (2018), https://doi.org/10.1039/c8ob00653a.Search in Google Scholar
[34] N.K. Rasal, R.B. Sonawane, S.V. Jagtap. Bioorg. Chem. 97, 103660 (2020), https://doi.org/10.1016/j.bioorg.2020.103660.Search in Google Scholar PubMed
[35] J. Prabhakaran, K.K. Solingapuram, A. Sattiraju, A. Mintz, J.J. Mann, JD Kumar. J.S. Dileep. Bioorg. Med. Chem. Lett. 29, 778 (2019), https://doi.org/10.1016/j.bmcl.2019.01.033.Search in Google Scholar PubMed PubMed Central
[36] J. Velcicky, U. Bodendorf, P. Rigollier, R. Epple, D.R. Beisner, D. Guerini, P. Smith, B. Liu, R. Feifel, P. Wipfli, R. Aichholz, P. Cottet, I. Dix, T. Widmer, B. Wen, T. Brandl. J. Med. Chem. 61, 865 (2018), https://doi.org/10.1021/acs.jmedchem.7b01371.Search in Google Scholar PubMed
© 2024 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Frontmatter
- In this issue
- Special topic papers
- Ozone-initiated degradation of 1,2-dichlorobenzene over ceria-supported manganese, nickel, vanadium and iron catalysts
- The effect of chemical modification using citraconic anhydride on the stability of α-amylase from Aspergillus fumigatus
- Decarbonizing our environment via the promotion of biomass methanation in developing nations: a waste management tool
- Maximising the consistency of the presentation of the molecular level with its quantum mechanical description: challenges and opportunities
- The amide group and its preparation methods by acid-amine coupling reactions: an overview
- Surface tension measurement of FAP-based ionic liquid pendant drops in a high vacuum/gas cell
- Investigating the bioactive compounds from Capsicum annum as a probable alternative therapy for prostate cancer treatment: a structure-based drug design approach
- Crucial role of the internalisation of the distinction between dependent and independent variables for clearer chemistry understanding
- 3-(4-methoxyphenyl) acrylic acid halts redox imbalance and modulate purinergic enzyme activity in iron-induced testicular injury
- Computational study on reactivity, aromaticity, and absorption spectra of chrysene: effect of BN doping and substituents
- Health benefit of lipid composition of orange (Citrus sinensis) fruit pulp at different maturation stages
Articles in the same Issue
- Frontmatter
- In this issue
- Special topic papers
- Ozone-initiated degradation of 1,2-dichlorobenzene over ceria-supported manganese, nickel, vanadium and iron catalysts
- The effect of chemical modification using citraconic anhydride on the stability of α-amylase from Aspergillus fumigatus
- Decarbonizing our environment via the promotion of biomass methanation in developing nations: a waste management tool
- Maximising the consistency of the presentation of the molecular level with its quantum mechanical description: challenges and opportunities
- The amide group and its preparation methods by acid-amine coupling reactions: an overview
- Surface tension measurement of FAP-based ionic liquid pendant drops in a high vacuum/gas cell
- Investigating the bioactive compounds from Capsicum annum as a probable alternative therapy for prostate cancer treatment: a structure-based drug design approach
- Crucial role of the internalisation of the distinction between dependent and independent variables for clearer chemistry understanding
- 3-(4-methoxyphenyl) acrylic acid halts redox imbalance and modulate purinergic enzyme activity in iron-induced testicular injury
- Computational study on reactivity, aromaticity, and absorption spectra of chrysene: effect of BN doping and substituents
- Health benefit of lipid composition of orange (Citrus sinensis) fruit pulp at different maturation stages