Redox-conducting polymers based on metal-salen complexes for energy storage applications
-
Irina A. Chepurnaya
 ,
Mikhail P. Karushev
,
Elena V. Alekseeva
,
Mikhail P. Karushev
,
Elena V. Alekseeva


Abstract
Metal-salen polymers are electrochemically active metallopolymers functionalized with multiple redox centers, with a potential for high performance in various fields such as heterogeneous catalysis, chemical sensors, energy conversion, saving, and storage. In light of the growing world demand for the development of superior energy storage systems, the prospects of employing these polymers for advancing the performance of supercapacitors and lithium-ion batteries are particularly interesting. This article provides a general overview of the results of investigating key structure-property relationships of metal-salen polymers and using them to design polymer-modified electrodes with improved energy storage characteristics. The results of independent and collaborative studies conducted by the members of two research groups currently affiliated to the Saint–Petersburg State University and the Ioffe Institute, respectively, along with the related data from other studies are presented in this review.
Introduction
Electrical energy has become a crucial and indispensable part of our daily life. Ongoing efforts to develop green alternatives to fossil fuels come with a challenging task to design effective energy storage systems for harvested electricity. Existing energy storage technologies such as supercapacitors and rechargeable lithium ion batteries need to be improved to help us deal with an inevitable energy crisis. Developmental efforts in the design of new materials possessing versatile redox properties, good electronic conductivity, ability to participate in multi-electron oxidation-reduction reactions, combined with low cost, easy synthesis, and low toxicity are currently one of the primary strategies to obtain high-performance systems and devices for electrochemical energy storage. A novel generation of such multifunctional materials is represented by electrochemically active polymer networks comprised of transition metal complexes with “non-innocent” ligands arranged into various molecular and supramolecular architectures.
One class of “non-innocent” (redox active) ligands that has been extensively investigated are N2O2 bis-Schiff-base bis-phenolate ligands known as salen-type ligands. Traditionally, the term “salen” refers to a particular ligand prepared via condensation of two equivalents of salicylaldehyde and one equivalent of ethylenediamine but is currently used to describe ligands with various phenyl ring substituents and diamine backbones. Salen ligands can easily coordinate to a wide variety of transition metal ions in a number of oxidation states to form stable coordinatively unsaturated metal complexes [M(salen)] (Fig. 1).
![Fig. 1:
Metal-salen complex – a building block for poly-[M(salen)] networks. M – transition metal: Ni, Cu, Co, Pd, Pt, Mn, Fe; N–Y–N – imine “bridge“; R – substituents in the aromatic rings of the ligand; R1 = H, CH3, OCH3, NO2,
t
Bu; R2 = R3 = R4 = H in most known poly-[M(salen)] networks.](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_001.jpg)
Metal-salen complex – a building block for poly-[M(salen)] networks. M – transition metal: Ni, Cu, Co, Pd, Pt, Mn, Fe; N–Y–N – imine “bridge“; R – substituents in the aromatic rings of the ligand; R1 = H, CH3, OCH3, NO2, t Bu; R2 = R3 = R4 = H in most known poly-[M(salen)] networks.
The first successful attempts to produce electrochemically active polymers from metal-salen complexes were reported in 1989 [1], [2]. Over the next few decades, initial and mostly random fundamental research efforts in this area [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16] have evolved into an application-driven systematic investigation of the structure-property relationships of metal-salen polymers [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58] to enable next generation of advanced functional materials with easily tunable properties [59], [60], [61], [62], [63], [64], [65], [66], [67], [68], [69], [70], [71], [72], [73], [74], [75], [76], [77], [78], [79], [80], [81], [82], [83], [84], [85], [86], [87], [88], [89], [90], [91], [92], [93], [94], [95], [96], [97], [98], [99], [100], [101], [102], [103], [104], [105].
Complexes of various transition metal ions have been used as building blocks to assemble redox-conducting metal-salen polymers (Fig. 1). Nickel(II) [1], [2], [3], [4], [5], [6], [14], [15], [16], [17], [18], [19], [22], [23], [24], [25], [26], [27], [28], [32], [34], [35], [36], [37], [38], [39], [40], [41], [43], [44], [45], [46], [47], [48], [49], [50], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [62], [66], [67], [68], [69], [70], [71], [72], [77], [78], [79], [80], [81], [82], [83], [84], [85], [86], [87], [88], [89], [90], [91], [92], [93], [94], [95], [96], [97], [98], [99], [100], [101], [102], [103], [104], [105], [112], cobalt(II) [3], [4], [5], [8], [51], [63], [74], [75], [112], and copper(II) [1], [4], [5], [7], [11], [29], [30], [31], [32], [64], [72], [73], [84], [85], [87] polymers have been most extensively studied; polymeric manganese(III) [5], palladium(II) [20], [21], [22], [23], [24], [25], [26], [32], [33], [84], [85], [86], [87], platinum(II) [86], and iron(III) [65] complexes have also been reported. Metal centers have been combined with salen ligands decorated with various substituents in the phenolate moieties (R) and imine bridges (Y) (Fig. 1) for the tuning of the electronic properties of the ligand and steric hindrance around the metal, respectively. To achieve specific research goals, more easily polymerizable units have also been introduced into the molecule as ligand substituents R to ultimately produce co-polymers [9], [10], [11], [12], [13], [46], [76]. Strategies for modular synthesis of metal-salen monomers of most complex composition and molecular structure have been proposed and continue evolving to meet rising developmental challenges.
Over the course of the study, metal-salen polymers have been discovered to possess a number of unique inherent properties such as electrocalatytic activity in various organic and inorganic reactions, sensing activity towards different analytes, electrochromism, which makes them promising candidates for a wide range of emerging applications in heterogeneous catalysis [59], [60], [61], [62], [63], [64], [65], chemical sensors [66], [67], [68], [69], [70], [71], [72], [73], [74], [75], and energy conversion and saving [76], [77], [78], [79], [80], [81], [82], [83], [84], [85], [86], [87]. All these properties of metal-salen polymers and related application areas have been described in detail in recently published reviews [106], [107]. This review will mainly focus on the functional properties of these materials relevant to energy storage applications. It will provide a general overview of the fundamental and applied studies in the area of metal-salen polymers conducted by the members of two research groups currently affiliated to the Saint-Petersburg State University and the Ioffe Institute, respectively, along with major related findings of other researchers worldwide.
It has to be emphasized that the overwhelming majority of investigated metal-salen complexes do not require to be linked to another polymerizable unit to efficiently form polymeric networks, which presents a distinctive advantage over other metal complexes. While some of the reported co-polymers of [M(salen)] and organic moieties such as, for example, 3,4-ethylene dioxythiophene (EDOT) may have some advantages over pure polymers, especially in terms of conductivity [12], [13], their substantially lower specific capacity due to a much higher molecular weight of a monomeric unit presents a serious disadvantage in energy storage applications. For this reason, such materials will not be addressed in detail in this paper.
In this review, we will show that metal-salen polymers have the ability to fulfill all main requirements to the multifunctional materials for energy storage applications: (a) easy synthesis; (b) easily tunable electrochemical activity to provide matching between redox potentials of the polymer and other system components; (c) high specific capacity; (d) high conductivity and charge transfer rates; (e) high stability. Examples of the successful use of these polymers in supercapacitors and lithium-ion batteries to improve their performance characteristics will be demonstrated. We will also show that the application of metal-salen polymers in energy storage devices requires going beyond studying inherent structure-dependent properties of the polymeric material. Research efforts should also include polymer-modified electrode design and investigation of its performance in simulated conditions (in laboratory scale prototype devices).
Synthesis and structure of metal-salen polymers
Despite over three decades of intensive investigation, the mechanism of polymerization and the structure of metal-salen polymers still remain a matter of controversy. Summarizing key findings of previous studies, it is fair to conclude that these polymers could be best described as three-dimensional networks composed of [M(salen)] monomers joined by different kinds of bonds. The first type is covalent carbon-carbon bonds formed via oxidative coupling of salen phenolates, primarily in the para-positions of aromatic rings (here and in the following discussion – with respect to the oxygen atom in the salen ligand core), as in conventional linear polymer chains (Fig. 2a). The second type is non-covalent (intermolecular) orthogonal interactions, the exact nature of which still remains poorly understood: d-π (metal-ligand) [6], [8], [16], [21], [22], [23], [24], [25], [26], [32], [34], [35], [40], [64], [71], [73] (Fig. 2b), π-π (ligand-ligand) [52], [53], and d-d (metal-metal) [47] (Fig. 2c) interactions have been proposed to account for the stabilization of different kinds of supramolecular networks, which are often referred to as stacks or molecular columns [40], [71]. The formation of linear chains can be preceded by monomer stacking [34], [35] or occur simultaneously with the organization of chains into secondary structures during polymerization [31]. In a mixed bonding structure, the ratio between covalent and non-covalent components seems to depend on the composition of a particular [M(salen)] monomer and the polymerization conditions. A large variety of metal-salen polymer networks has been demonstrated, ranging from stable supramolecular aggregates completely lacking carbon-carbon bonds [34], [35], [47] to chain polymers with seemingly hindered interchain interactions [44], [52]. In many cases, the relative extent of intermolecular interactions is directly proportional to the planarity and bulkiness of the imine bridge in the salen ligand [13], [52], [58].
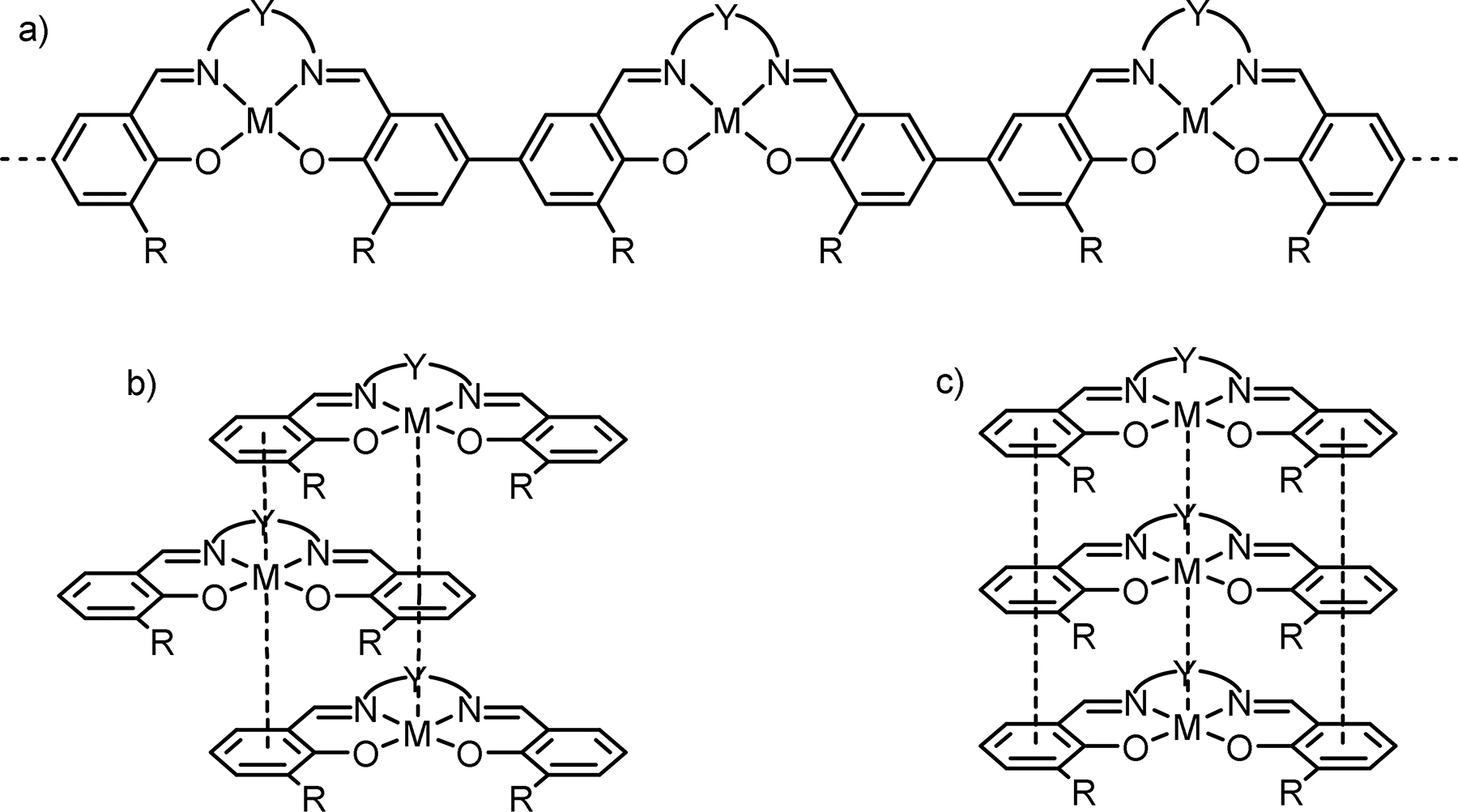
Proposed bonding in metal-salen polymers: covalent carbon-carbon bonds (a), non-covalent d-π (b), π-π and d-d (c) interactions.
The vast majority of metal-salen polymers exists and functions as an integral part of polymer-modified electrodes. It therefore comes as no surprise that the most widespread technique used to produce polymer films of different thicknesses on various substrates is electrochemical deposition. For the purposes of fundamental research, films of metal-salen complexes are usually electropolymerized onto standard working electrodes of different materials such as Pt, Au, glassy carbon, ITO, Pt-coated quartz crystal, etc. The working electrode is anodically polarized in a non-aqueous solution containing sufficient concentrations of a monomer and supporting electrolyte. The electropolymerization can be accomplished by different electrochemical methods such as potentiostatic or potentiodynamic polarization, as well as galvanostatic charging. The amount of the electrodeposited polymer (i.e., film thickness) can be easily controlled through the polymerization charge (for static methods) or the number of cycles (for a potentiodynamic method). The layer-by-layer polymerization of [M(salen)] complexes has also been demonstrated [27].
In most cases, the electrochemical deposition of metal-salen polymers involves the generation of carbon-carbon bonds between aromatic rings of the ligands (Fig. 2a). The presence of C–C linked aromatic fragments has been directly confirmed by subjecting the polymer films to strongly acidic conditions and identifying bis-salicylaldehyde among hydrolysis products [5], [6], [7], [101], [108]. It has been proposed by many research groups [1], [2], [4], [5], [7], [14], [15], [7], [14], [15], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [29], [30], [33], [41], [43], [45], [48], [49], [57] that the corresponding polymerization mechanism is solely ligand-based and does not involve the change in the oxidation state of the central metal ion. For nickel(II)-salen monomers, the first step has been shown to include an oxidative C–H bond cleavage, at least in the para-position of the aromatic ring, resulting in the formation of Ni(II)-phenoxyl radicals followed up by the polymerization via C–C coupling [29], [43], [45]. In some cases, however, further stepwise oxidation of the monomer to produce Ni(II)-bis-phenoxyl radicals and finally Ni(II)-phenoxonium cations can be observed before or along with the ligand coupling reaction [45]. This polymerization mechanism could be considered valid only for metal-salen complexes with d8 and d9 metal centers (Ni, Pd, Cu, etc.). The oxidation of cobalt(II) (d7) complexes of salen ligands to cobalt(III) species has been shown to occur at less positive potentials than the phenolate-to-phenoxyl oxidation [3], [4], [5], [8], [51], [63], [74], [75] so intermediates of [Co(salen)] polymerization could reasonably be expected to be different from those described above and are to be determined.
Metal-salen monomers should normally exhibit square planar geometry [24] and have no para-phenolate protecting groups for successful polymerization via radical C–C coupling. Various solvents (e.g., 1,2-dichloroethane (DCE), acetonitrile (AN), propylene carbonate (PC)) and supporting electrolytes containing different combinations of cations (e.g., Li+, Bu4N+, Et4N+, Et3MeN+) and anions (e.g., ClO4 −, BF4 −, PF6 −) can be used for the electrochemical synthesis of metal-salen polymers. However, only low and medium donor number solvents afford successful film formation. A reversible metal-centered oxidation of all [M(salen)] complexes takes place in solvents of high donor numbers (e.g., dimethylsulphoxide (DMSO), pyridine (py), N-Methyl-2-pyrrolidone (NMP)) [109], [110].
In our investigation of a series of electrochemically synthesized polymeric nickel(II) complexes of Salen, Salphen, and Saltmen ligands (R1 = H or CH3O – Fig. 1) [44], we have shown that most of them consist of short-chain oligomers formed by 2–5 monomer units. These oligomers are likely stacked to form a mechanically stable insoluble film on the electrode surface. The stacking is largely inhibited in complexes with imine bridges containing four methyl groups (Saltmen) so such complexes tend to form longer chains: poly-[Ni(CH3O-Saltmen)] could be considered a true covalently-linked polymer.
The composition of the monomer solution along with the polymerization potential has been shown to affect the morphology and subsequent electrochemical behavior of the forming polymer film [26], [40], thus representing a powerful tool for fine-tuning of the polymer properties at molecular level. For instance, we have demonstrated that larger ion transport rates could be achieved in polymer networks that were electrodeposited in the presence of bigger electrolyte ions and solvent molecules and then operated in solutions containing species of a smaller size [26]. Ultimately, the electrochemical polymerization conditions have to be chosen based on the specific peculiarities of metal-salen monomer chemistry and operating conditions of a polymer-modified electrode.
In selected cases, the electrochemical deposition of metal-salen polymers may involve the formation of polymeric materials with no or very few C–C linked chain fragments. For example, a stable electrochemically active electrode-modifying material has recently been prepared from [4,40-phenylazo-2,20-(ethylenediimino-k2N-methyl)diphenolate-k2 O]nickel ([Ni(Phazosalen)]) via potentiostatic deposition onto a working electrode in 0.1 M Bu4NBF4/DCE [47]. This monomer contains a para-protecting phenylazo-group, which prevents the conventional polymerization of [Ni(Phazosalen)] via oxidative coupling of the phenolates. The deposited material, [Ni(Phazosalen)] n , is therefore not a classical network of C–C linked chains but is rather a material consisting primarily of stacked molecules of the monomeric complex. The “stacked” structure is stabilized by intermolecular forces, including the dispersive interaction of the extended π-systems, the Coulombic attraction between metal ions and azo-groups as well as between positively charged [Ni(Phazosalen)]+ monocations and electrolyte anions (Fig. 3). The voltammetric response of this material in 0.1 M Et4NBF4/AN in the potential range 0–1.2 V (here and throughout the text, values of potentials are reported as V vs. an Ag|AgCl (saturated aqueous NaCl), unless indicated differently) closely resembles that of poly-[Ni(Salen)] under the same conditions.
![Fig. 3:
Proposed structure of [Ni(Phazosalen)]
n
.](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_003.jpg)
Proposed structure of [Ni(Phazosalen)] n .
Another example of the material stabilized only by intermolecular forces is polylayers of metal-salen molecules that have been shown to form on graphite and glassy carbon surfaces due to the adsorption from solution, even without the application of anodic potential to the electrode [34], [35]. These polylayers remained on the electrode surface after thorough washing and showed a globular structure that was quite similar to the structure of the polymer formed by a conventional oxidative electrochemical polymerization technique. It has been proposed that these polylayers are a conducting “stacked” material stabilized by the same intermolecular forces as those found in conventionally formed metal-salen polymers [35]. Interestingly, the electrochemical oxidation of these “stacked” structures yielded carbon-carbon linked polymers.
The oxidative C–H bond cleavage, an essential first step in the polymer chain fragment formation, can potentially be accomplished not only electrochemically but also chemically. Chemical oxidative polymerization is a widespread technique used to synthesize intrinsically conducting polymers such as polyaniline, polypyrrole etc. in the form of non-supported materials (e.g., bulk, nanostructures, free-standing films). Despite important advantages that this method offers in the field of materials for energy storage (ease of processing, mass production, etc.) [111], it has been very little explored to date to produce polymeric metal-salen-based materials. We have recently reported a suspension polymerization of [Ni(CH3-Salen)] in acetonitrile in the presence of NOBF4 as an oxidizing agent [101]. The obtained material featured nitro-groups in the end-fragments of short chains (the degree of polymerization was 3.2–4 as determined by acid hydrolysis) (Fig. 4). Its specific capacity turned out to be lower than for an electrochemically synthesized poly-[Ni(CH3-Salen)], presumably due to the presence of electrochemically inert fragments and/or issues of hindered charge transport in a disordered material.
![Fig. 4:
Chemical polymerization of [Ni(CH3-Salen)].](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_004.jpg)
Chemical polymerization of [Ni(CH3-Salen)].
In the following discussion of the functional properties of metal-salen polymers that are most relevant to their application in energy storage we will primarily focus on electrochemically synthesized films as the most common and widely investigated representatives of polymeric metal-salen-based materials.
Morphology and physical properties of poly-[M(salen)]
Three distinct types of morphology have been detected for metal-salen polymers by scanning electron microscopy (SEM): globules, “honeycomb”-like deposits, and dense continuous layers (Fig. 5). In our studies, the globular structure was displayed only by poly-[Ni(Salen)] and poly-[Ni(CH3OSalen)] [44] as well as by poly-[Ni(Salpn-1,2)] and poly-[Ni(KetoSalen)] [55]. Other polymers existed as films of different structure, which was affected by the nature of ligand substituents [28], [44], [51], [55], [102]. Interestingly, the morphology of nickel-salen polymer films was mostly influenced by the bulkiness of the imine bridge [44], [55] whereas the morphology of cobalt-salen polymers was more sensitive to the presence of CH3O-groups in ortho-positions of the phenolates [51]. Our findings are in good agreement with the results of other research groups [1], [6], [15], [49].
![Fig. 5:
SEM images of poly-[M(salen)] films: poly-[Ni(Salen)] (a) [44], poly-[Со(CH3OSalEn)] (b), poly-[Co(SaltmEn)] (c) [51].](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_005.jpg)
The investigation of a poly-[Pd(Salen)] film by fast ellipsometry showed that the morphology of a metal-salen polymer could vary across the film thickness: the film could be relatively close packed near the electrode surface, but less close-packed at the film/electrolyte interface [20]. This observation has been partially confirmed by SEM results showing a more compact film closer to the electrode surface [6], [15]. The metal-salen polymer film could also be prone to substantial swelling on anodic cycling [20], [57]. The experimental density values for a series of nickel(II) polymers of Salen, Salphen, and Saltmen ligands (R1 = H or CH3O – Fig. 1) determined ex situ in a neutral (non-charged) state, by a flotation method, were in the range 1.8–2.4 g cm−3 [44]. They are higher than the density of crystalline [Ni(Salen)] (1.325 g cm−3), which could be an indication of very close packing of polymer chains in the material. If swelled, positively charged polymers could reasonably be expected to have lower densities but they have yet to be determined by in situ methods.
Our recent studies of poly-[Ni(CH3-Salen)]-based electrodes for lithium-ion batteries [104] showed that metal-salen polymers could exhibit very good and stable mechanical properties. The Young’s modulus (elastic modulus) of poly-[Ni(CH3-Salen)] was found to be ∼1.7 GPa in the dry state, and in the range 1.74−2.83 GPa in the wet state. The similarity of the Young’s modulus values for a dry and a wet polymer is a promising feature for its application as a conductive binder (see Section 8). To provide a reference point, PVdF binder experienced significant decrease in Young’s modulus value (approximately 1 order of magnitude smaller) when its dry sample was immersed in an electrolyte solution. The stable mechanical property at wetted state is desirable for binders since it offers high resistance to elastic and plastic deformation of electrodes that occurs due to repeated volume expansion of active materials. Our results are in good agreement with another recent study of a poly-[meso-Ni(SaldMe)] film [57], which demonstrated that changes in the Young’s modulus during charging-discharging of the polymer were within 10% of the initial value, thus confirming high mechanical stability of the polymer structure in redox processes.
Last but not least, metal-salen polymers demonstrate excellent thermal stability (decomposition begins above 300 °C) [112] and shelf life.
Electrochemical activity and charge transport in metal-salen polymers
Composition and structural parameters of metal-salen polymers govern their electrochemistry. Poly-[M(salen)] films demonstrate electrochemical activity in a wide range of potentials: the polymer oxidation usually occurs in the 0–1.5 V potential range whereas the reduction processes take place in the range 0–(−2.0) V. The exact potentials of redox transformations in the polymer depend on the metal center, ligand substituents, and electrolyte composition (Fig. 6). Such broad electrochemical activity window is highly advantageous for energy storage applications because it provides extensive opportunities for matching the redox potentials of the polymer and other active electrode materials. Metal-salen polymers exhibit electrochemical activity in electrolytes that are employed in commercial supercapacitors (tetraalkylammonium salts/acetonitrile) and lithium-ion batteries (lithium salts/organic carbonates), which is one of the advantages determining their potential use as electrode materials in these energy storage devices. It has to be noted that the studies of the reduction processes in metal-salen polymers have been quite limited and primarily driven by the applications of these polymers in catalysis [3], [6], [26], [59], [60]. Most capacitive energy storage applications take advantage of the redox processes in poly-[M(salen)] films at anodic potentials (0–1.5 V) so further discussion will focus on the peculiarities of the reversible (or quasi-reversible) oxidation reactions, which will also be referred to as oxidation-reduction (in this notation, reduction means conversion of the positively charged polymer to the initial, non-charged state).
![Fig. 6:
Cyclic voltammograms of poly-[M(salen)] films in 0.1 M Et4NBF4/AN: nickel-salen polymers on the surface of the ITO electrode, scan rate 0.004 V s−1 (a) (adapted from [52]); cobalt-salen polymers on the surface of the glassy carbon electrode, scan rate 0.05 V s−1 (b) (adapted from [51]).](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_006.jpg)
The biggest challenges of understanding the redox transformations in metal-salen polymers arise from the existence of two distinctly different electrochemically active entities in the system: metal centers on one hand, and a conjugated organic backbone, on the other hand. Metal-centered oxidation-reduction is usually encountered in pure redox polymers and described in terms of the electron hopping mechanism. The charge transport in conjugated organic polymers is normally described using a polaron-bipolaron model. The potential co-existence of both metal- and ligand-based redox processes in immediately adjacent or overlapping potential ranges presents a major challenge for modeling oxidation-reduction processes in metal-salen polymers. For a long time, the question of whether these polymeric networks should be considered redox polymers [3], [16], [21], [22], [23], [24], [25], [26] or intrinsically conducting ones [1], [2], [4], [5], [7], [14], [15], [17], [18], [19], [29], [30] has been the reason for substantial disagreement among different research groups.
Structural units of metal-salen polymers that undergo oxidation upon application of an appropriate potential are [M(salen)] fragments. It comes as no surprise that certain similarities between redox conversions of the monomers and corresponding polymers, especially regarding the locus of oxidation, type of charged species and charge delocalization, have been detected. Each [M(salen)] complex is a multi-redox system wherein a metal center and two ligand phenolates are potential subjects to redox reactions. Metal-salen monomers containing substituents in ortho- and para-positions of the phenolate moieties form relatively stable species upon oxidation, which have been systematically investigated by several research groups [113], [114], [115], [116], [117]. The locus of one-electron oxidation of such complexes is dictated by the relative ordering of the metal and ligand frontier molecular orbitals, which is affected by the central metal ion, peripheral substituents on the ligand, solvent and temperature: the formation of metal(III)-salen complexes or metal(II)-salen •+ phenoxyl radical cation structures has been observed. In the latter case, the radical can remain localized on one of the aromatic rings or delocalize if the metal ion can effectively mediate the electronic coupling between two phenolates [113], [114], [115]. Two-electron oxidation of metal(II)-salen complexes leads to the formation of metal(II)-(salen•+•+) structures, in which each phenolate moiety is oxidized to a phenoxyl radical cation (for example, in Ni(II)-salen complexes [116]), or metal(III)-(salen •+) species (for example, in Co(II)-salen complexes [117]).
It is currently acknowledged by most researchers that the oxidation of Ni(II) and Cu(II)-salen polymers is primarily ligand-centered [41], [43], [48], [49], [52], [53], [54] whereas the oxidation of Co(II)-salen polymers is indicative of both Co(II)/Co(III) reaction and ligand oxidation, which seems to be very similar to the ligand-centered redox process in nickel-salen analogs [51]. To date, comparatively little research work on polymeric cobalt complexes of salen ligands has been carried out so many aspects of their electrochemical behavior still remain unknown. In contrast, nickel- and copper-salen polymers have been objects of many studies. These research efforts made it possible to elucidate various aspects of charge injection and transport in these polymeric systems, which we will discuss below.
The injection of charge into salen polymers (p-doping during oxidation) is immediately coupled with charge transport, which proceeds through the polymer backbone (intrinsic conductivity) and is accompanied by the ingress of charge compensating anions into the polymer network to maintain electroneutrality [17], [18], [19], [20], [23], [28], [32], [57], [58]. The charge transport rate in the polymer film is controlled by one of the two processes, more often by the counterion transport [23]. Once the redox processes were identified as ligand-centered and similar to those observed in organic conducting polymers, the polaron-bipolaron model of conduction was mainly used to describe charge transfer processes in metal-salen polymers [14], [15], [18], [19], [30].
Many nickel-salen polymers previously investigated by our research groups demonstrate multiple partially (and sometimes, almost completely) overlapping waves on cyclic voltammograms (see Fig. 6 as an example) and unusual electrochemical impedance spectra in electrolyte solutions at positive potentials: Nyquist plots of polymer films are often characterized by the absence of high frequency semicircles (at least, below 100 kHz) and occurrence of Warburg and pseudo-capacitive impedance constituents at the same frequencies [37], [44]. Such highly complex nature of polymer oxidation can only be explained by the presence of several (at least two) different overlapping redox transitions in the polymer. It has been suggested in [36] that oxidized nickel-salen polymers feature three different charge carriers: mobile ones (counterions) and two types of immobile ones. Three different models of immobile charge carriers in the polymer have been proposed. The simplest model describes a polymer film with two types of immobile charge carriers that cannot convert into each other (a so called “mechanical mixture of immobile charge carriers”). The second model corresponds to the polymer with polaron and bipolaron charge carriers: a delocalized cation radical (polaron) is formed in the beginning of oxidation and converted into a dication (bipolaron) at higher anodic potentials. The third model refers to the polymer film possessing both metal- and ligand-centered charge carriers (i.e., both redox and polaron conductivity). Theoretical parameters of cyclic voltammetry (CV) curves [36] and electrochemical impedance spectra [37] have been calculated according to the proposed models. Solving the thermodynamic equations revealed that splitting of voltammetric peaks could be observed in all three cases. A fairly good correlation between experimental and calculated current-voltage curves has been observed in the framework of the model describing charge transfer along conjugated polymer chains due to sequential generation of polarons and bipolarons. The aforementioned model was further advanced by including the concept of polaron and bipolaron fragments containing different number of repeat units [42]. Yet, the charge transfer mechanism had to be confirmed by elucidating the structure of charge carriers in metal-salen polymers by in situ analytical techniques.
We have recently undertaken the first step towards elucidating the charge carriers and their delocalization pathways in poly-[M(salen)] films: the formation of different charged species in nickel-salen polymers upon their electrochemical oxidation (p-doping) was followed by using a combined in situ ESR/UV-vis-NIR spectroelectrochemical technique [52], [53]. Based on the obtained results, it has been proposed that the oxidation of nickel-salen polymers (Fig. 7a) is a ligand-based process that proceeds in two sequential steps. Step 1 involves one electron per a monomer unit oxidation and first leads to the generation of ESR active biphenoxyl radical cations that either remain largely localized (in the polymers consisting of monomeric units with more electron rich ligands) (Fig. 7b) or become delocalized through the central nickel ion (intrachain charge delocalization) to produce an oligomeric radical cation (polaron) in the case of less electron rich ligands (Fig. 7c). At high concentration of such radical cations, ESR silent intrachain radical pairs with antiferromagnetically coupled spins are formed (Fig. 7d–f). At Step 2, further oxidation of each monomer unit results in the formation of diamagnetic dications (Fig. 7g). Interestingly enough, some of the investigated nickel-salen polymers show two types of ESR signal. An anisotropic signal is observed at the beginning of charge injection into the polymer, an isotropic one – at a higher doping level. We attributed both signals to the phenoxyl radical structure and proposed that the anisotropic ESR signal could originate from the radicals generated in sterically regular polymer domains such as, for example, stacked structures formed by adjacent chain fragments and likely stabilized through π-π interactions. Such intermolecular π-π stacking between adjacent chains provides a path for interchain charge delocalization in nickel-salen polymers (Fig. 8) and is substantially hindered in polymeric complexes of salen ligands containing bulky non-planar imine bridge substituents such as for example four methyl groups in a Saltmen ligand.
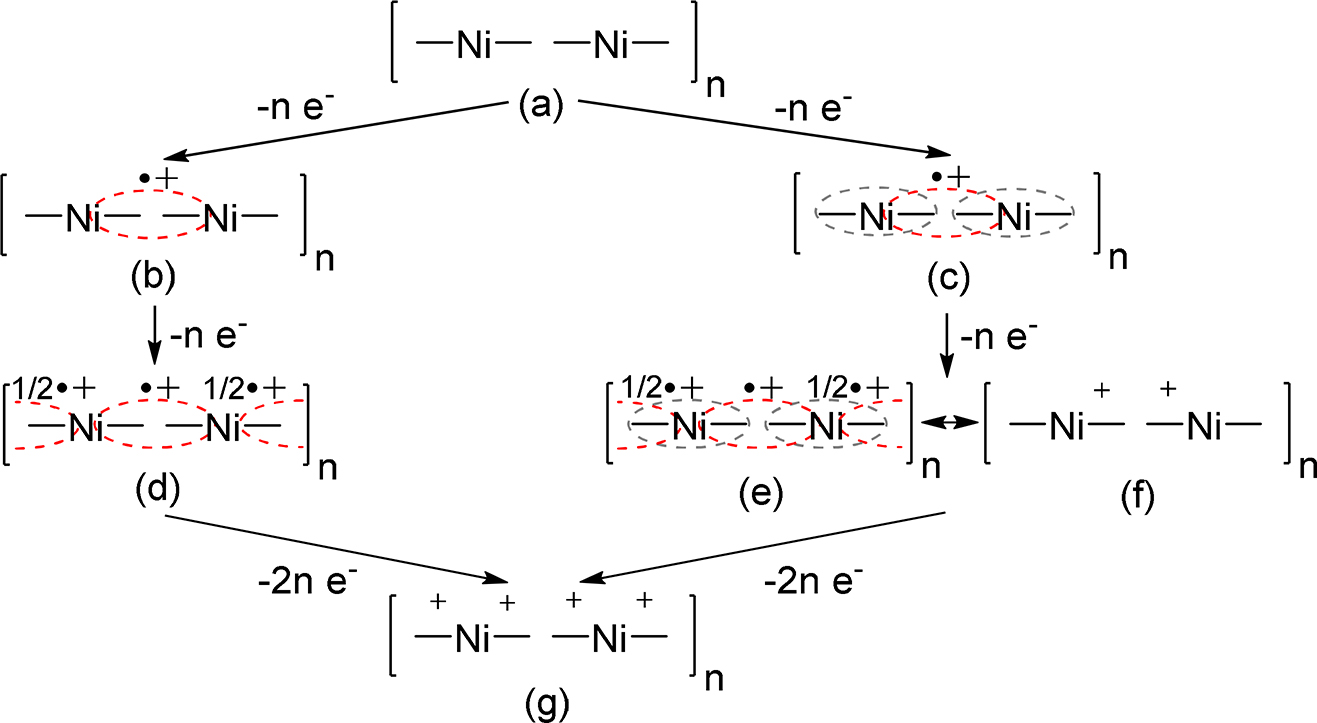
Proposed structure of charged species in chains of nickel-salen polymers: undoped polymer (a); radical cation (b) and (c); polymer doped with one positive charge per one monomer unit (d–f); polymer doped with two positive charges per one monomer unit (g). Red dashed line represents a biphenoxyl radical cation. Grey dashed line represents an intrachain radical cation delocalization through the nickel ion.
![Fig. 8:
Proposed structure of charged species in π-stacks of poly-[Ni(salen)] films: dimer radical cation (a); π-dimer of radical cations (b). Red dashed line represents a biphenoxyl radical cation. Grey dashed line represents an interchain radical cation delocalization.](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_008.jpg)
Proposed structure of charged species in π-stacks of poly-[Ni(salen)] films: dimer radical cation (a); π-dimer of radical cations (b). Red dashed line represents a biphenoxyl radical cation. Grey dashed line represents an interchain radical cation delocalization.
Our results support the earlier finding of those researches who suggested describing nickel- and copper-salen polymers as conducting rather than redox (discrete) systems and proposed the existence of a ···(−Ph−O−M−O−Ph-) n ··· conduction path which included metal ions as redox inactive bridges between two phenolates [14], [43]. At the same time, they do not contradict another possible pathway for charge delocalization, which has been recently proposed as an additional one to the conduction through the metal center in nickel-salen polymers. According to the authors of [41], [49], the propagation of charge via imine bridges (a ···(−Ph–HC=N–Y–N=CH–Ph-) n ··· path) is also feasible, especially in the polymers with Salphen ligands. In this case, the mechanism of energy storage is related to the reversible conversion of the azomethine nitrogen group (−N=CH−), so poly-[M(salen)] networks can be best described as both poly-phenylene-type and poly-imine-type polymers.
Despite convincing experimental evidence for the non-involvement of metal ions in the electrochemical transformations in nickel-salen polymers, the question on the exact role of nickel centers is still debatable. Based on the combined results of spectroelectrochemical studies and DFT calculations for a series of polymeric nickel(II) complexes of salen ligands containing different substituents in the phenolates and imine bridges, we proposed that two different oxidized forms of the polymers could be observed, depending on the coordination ability of the electrolyte [38]. The first oxidized form of the polymer formed in poorly coordinating solvents is purely ligand-centered and characterized by positive charges delocalized across aromatic moieties. The second oxidized form of the polymer could potentially be observed in strongly coordinating solvents and involves the formation of six-coordinated Ni(III). The direct involvement of nickel ions in the anodic processes along with ligand fragments has resently been proposed to account for redox transformations in poly-[Ni(CH3-Salcn)] films [54].
Performance parameters of poly-[M(salen)] relative to energy storage applications
Discovered peculiarities of charge injection and transport in metal-salen polymers and their dependence on the substituents in the monomeric fragments are reflected in the experimentally determined values of the amount of charge stored per unit weight, charge transport diffusion coefficient, and conductivity (performance characteristics that are most relevant to the polymer application in energy storage). These values for different metal-salen polymers have been obtained from the results of CV, chronoamperometry, galvanostatic charge-discharge (GCD), and electrochemical impedance spectroscopy (EIS) studies by us and other research groups [5], [7], [15], [19], [23], [25], [26], [28], [30], [37], [43], [44], [49], [51], [52], [54], [55], [56], [100], [102].
Two parameters are used in the capacitive energy storage to define the amount of charge that can be stored in the electrochemcially active material: specific capacitance (CF, F g−1 – in material characterization for supercapacitor appications) and specific capacity (CmAh, mAh g−1 – in material characterization for battery appications). Since the capacitance of metal-salen polymers is potential dependent, average values of specific capacitance can be determined for a particular range of potentials. Alternatively, the polymer capacitance at each selected potential can be calculated. Average capacitance values will be disclosed in the discussion that follows, unless otherwise specified.
The amount of charge stored in the polymer depends on the number of electrons transferred per each monomer unit in a redox process. We have recently examined a series of nickel- and cobalt-salen polymers by the combination of CV and microgravimetry [51], [52], [53] to unambiguously confirm that their metal-salen monomer units could each be oxidized by more than one electron. We also demonstrated direct correlation between the doping level achieved in the oxidation process and the overall extent of charge delocalization across the polymer film: multi-electron oxidation could be observed only in polymers with hindered intra- and interchain charge delocalization [52], [53]. Although this conclusion was made based on the studies of nickel-salen polymers, it seems to be applicable to the explanation of multi-redox transformations in cobalt-salen polymers, in particular those related to the ligand-centered oxidation-reduction. We have shown that poly-[Ni(salen)] films could be doped to the level corresponding to the transfer of two electrons per a monomer unit, whereas the doping level of poly-[Co(salen)] polymers could correspond to the transfer of up to three electrons per a monomer, as illustrated in Fig. 9 (in the latter case, the extra electron must be a result of Co(II)/Co(III) conversion).
![Fig. 9:
Charge storage characteristics of nickel- and cobalt-salen polymers (values of Ne−, CF [F g−1], and CmAh [mAh g−1] are adapted from [51], [52]).](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_009.jpg)
The values of specific capacitance and specific capacity disclosed in Fig. 9 correspond to the redox switching of “ideal” polymers (highly ordered thin films on perfectly flat electrode surfaces) under “ideal” conditions (ultra-pure electrolytes, oxygen- and moisture-free atmosphere). The values of these parameters for “real” polymers largely depend on the polymer film thickness, morphology, and operating conditions; some of them are listed in Table 1. The observed discrepancies could also be related to the uncertainties in the determination of the polymer mass on different electrode surfaces.
Properties of metal-salen polymers relevant to energy storage.
| Polymer | Specific capacitance CF, F g−1 and/or specific capacity CmAh, mAh g−1 (determination method) | Effective charge diffusion coefficient DCT, cm2 s−1, or cDct 1/2, mol cm−2 s−1/2 (determination method) | Conductivity (determination method) |
|---|---|---|---|
| poly-[Ni(Salen)] | Max – 210 F g−1 (EIS) Average – 90 F g−1 (CV) [44] 10 mAh g−1 (GCD at 1C) [102] |
Max – 1.1 × 10−8 cm2 s−1 (EIS) [44] 3 × 10−10 cm2 s−1 (CV) [4] 2.5 × 10−10 cm2 s−1 (CV) [23] |
∼10−5–10−6 S cm−1 (in dry state by a four probe method) [4] 0.35 × 10−3 S (in-situ on IDA electrode) [105] |
| poly-[Ni(CH3O-Salen)] | Max – 220 F g−1 (EIS) Average – 140 F g−1 (CV) [44] 28 mAh g−1 (GCD at 1C) [102] |
4.5 × 10−7 cm2 s−1 (CV) [4] 4.9 × 10−8 cm2 s−1 (EIS) [44] 5.5 × 10−10 cm2 s−1 (CV) [23] |
∼10−3 S cm−1 (in dry state by a four probe method) [4] ∼1 × 10−3 S (in-situ on IDA electrode) [105] |
| poly-[Ni(CH3Salen)] | 50 mAh g−1 (GCD at 1C) [102] | ‒ | 2.6 10−3 S (in-situ on IDA electrode) [105] |
| poly-[Ni(Saltmen)] | Max – 190 F g−1 (EIS) Average – 70 F g−1 (CV) [44] 230 F g−1 (CV) [55] |
3.5 × 10−9–4.6 × 10−7 A s−1/2 (chronoamperometry) [15] 3 × 10−9 cm2 s−1 (CVA) [26] Max – 3.6 × 10−7 cm2 s−1 (EIS) [44] |
‒ |
| Poly-[Ni(CH3O-Saltmen)] | Max – 130 F g−1 (EIS) Average – 100 F g−1 (CV) [44] |
8.5 × 10−10 cm2 s−1 (CVA) [26] Max – 2 × 10−7 cm2 s−1 (EIS) [36] Max – 7.5 × 10−8 cm2 s−1 (EIS) [44] 3 × 10−9–5.8 × 10−7 mol cm−2 s−1/2 (CVA) [19] |
‒ |
| Poly-[Ni(Salphen)] | 85 F g−1 (GCD test at 0.05 mA cm−2) [49] | 4.3 × 10−7 cm2 s−1 (EIS) [44] | 69 S cm−1 (in dry state by a four probe method) [49] |
Effective charge diffusion coefficient, DCT (cm2 s−1), is the parameter that describes the rate of charge carriers transport in the polymer. Due to the uncertainties in the determination of the concentration of redox sites in the polymer films, the parameter cDCT 1/2 (mol cm−2 s−1/2) has been quite often determined from the experimental data, instead of DCT. Metal-salen polymers have also been shown to possess electronic conductivity like other conventional conducting polymers. It has been measured in the dry state [5], [7], [25] and very recently, during redox conversion of the polymers deposited onto interdigitated array (IDA) electrodes [105]. Experimentally determined values of DCT and conductivity were found to be potential dependent (minima and maxima correspond to different doping levels of the polymer); examples are shown in Table 1.
All conducted investigations demonstrated that the energy storage related performance parameters of metal-salen polymers could be easily tuned by varying the metal center, the ligand substituents, the polymerization conditions, and the conditions for redox transformations in an electrochemical system. As follows from Table 1, poly-[M(salen)] films exhibit high specific capacity and high conductivity that can lead to high rates of charge transfer. Coupled with versatile and reversible redox properties, it makes them promising materials for energy storage. Yet, no polymer films presented a winning performance in all the parameters that characterize their functioning in supercapacitors and lithium-ion batteries. At the same time, the maximum values of key charge transfer and storage parameters of the studied polymeric complexes are among the top values determined elsewhere for conventional conducting polymers [44].
Electrochemical stability of metal-salen polymers
While specific capacitance, charge transfer rate, and conductivity are crucial parameters that should be considered in the first place when selecting a metal-salen polymer for the application in energy storage devices, the final choice should also be based on other performance parameters, such as, for example, long-term electrochemical stability of the polymers, which would directly influence the cycle life and lifetime of the final device. Many metal-salen polymers exhibit very good stability in non-aqueous solutions but their cyclability in aqueous electrolytes is not satisfactory [39], [47], [55], [56], [118]. At the same time, non-aqueous electrolyte-based energy storage devices can contain trace amounts of moisture (up to 300 ppm) that could be detrimental to the polymer performance.
We have recently unveiled two potential mechanisms for poly-[M(salen)] electrochemical activity decay in the presence of water [55], [56]. The first mechanism involves the axial coordination of water molecules (or its oxidation products) to the metal ions during polymer oxidation. In the case of poly-[Ni(salen)] films, it leads to the stabilization of nickel ions in the +3 oxidation state and disruption of the metal-bridged charge transfer pathways [55], [56]. The second proposed mechanism involves an attack of water molecules on the carbon atoms of the imine (−C=N−) fragments of the ligands, which ultimately leads to the polymer decomposition and dissolution from the electrode [56]. The results of electrochemical quartz crystal microbalance (EQCM) studies, which show that a decrease in the redox activity of the polymers during cycling in water-containing electrolytes is accompanied by an increase in the polymer mass [55], provide support for the first mechanism so we currently consider it the most probable. The metal-centered mechanism gives a rational explanation to the experimentally observed phenomenon of high electrochemical stability of metal polymers of Saltmen ligands [15], [55], [56]: four methyl groups of the imine bridge impose steric hindrance to the coordination of water molecules to the nickel ions. The recently synthesized stacked metal-salen material, [Ni(Phazosalen)] n (Fig. 3), exhibits an unusually high stability in aqueous electrolytes: it can be cycled in 0.1 M LiClO4 aqueous solution in the potential range from 0 to 1.2 V without any loss of redox activity [47]. The superior stability of [Ni(Phazosalen)] n can also be explained from the standpoint of the metal-centered degradation: metal ions in the stacked structure are effectively shielded from the environment, which precludes possible attacks on them by the water molecules.
Repeated volumetric and structural changes induced by the ingress and egress of charge compensating counterions and solvent during redox processes are one on the main reasons for undesirable deterioration of electrochemical properties of many conducting polymers [119]. The stability of poly-[M(salen)] films upon electrochemical charging and discharging has also been directly linked to the extent of reversibility of concomitant changes in their viscoelastic and mechanical properties [57]. Generating enough space between polymer chains for efficient counterion movement by introducing substituents in the imine bridge that provide steric hindrance between polymer layers and help repel them one from the other proved to be an effective strategy for electrochemical stability enhancement [58].
Metal-salen polymers in supercapacitors
Electrochemical capacitors (also called “supercapacitors” or “ultracapacitors”) have attracted considerable attention as an intermediate power source between conventional capacitors and rechargeable batteries. Supercapacitors fill the gap by providing higher specific power than batteries and higher specific energy than dielectric capacitors [120], [121]. The overwhelming majority of commercially available electrochemical capacitors are electrical double layer capacitors (EDLCs) utilizing the phenomenon of potential-induced separation of ionic and electronic charges at the electrode/electrolyte interface for energy storage. Due to the fact that the double-layer capacitance is directly proportional to the accessible surface area of an electrode, activated carbon-based materials with high specific surface areas (1500–2500 m2 g−1) are most commonly used as the electrode materials for EDLCs. Electrical double layer capacitors offer excellent performance: they have very good reversibility, relatively low temperature coefficient, ability to generate high specific power, and very long cycle life of up to 106 cycles. The only characteristic of carbon-based supercapacitors that still requires significant improvement is specific energy. One of the most powerful tools to increase specific energy of an ultracapacitor is to increase the capacitance of its electrodes, for example, by modifying high surface area carbons with a pseudocapacitive (electrochemically active) material. We have explored the concept of using metal-salen polymers as such pseudocapacitive material [88], [89], [90], [91], [92]; the results are discussed below.
In order to maximize the benefits of using an electrochemically active polymer for EDLC electrode modification, the polymer layer should not limit or restrict the ability of the nano- and meso-porous carbon materials to store charge by the double layer capacitive mechanism. However, when a conventional electrochemical polymerization technique is used for a metal-salen polymer-modified electrode fabrication, the polymer starts growing mainly on the outer surface of the porous carbon and may eventually restrict electrolyte access to a significant portion of pores thus limiting the electrode capacity (Fig. 10a). We have proposed two approaches that can help effectively overcome this hurdle [90], [91]. Both methods involve soaking the porous carbon material in a highly concentrated metal-salen monomer solution in order to adsorb the complex onto the inner surface of the substrate. After that, the electrode can be transferred to a low concentration monomer solution [90] or a monomer-free solution [91] for electrochemical polymerization. In the former case, a pulse polymerization technique [88], [90] has proven successful, especially for the modification of activated carbon fiber materials. Short anodic potential pulses ensure gradual polymerization of the adsorbed and solution monomers whereas pauses between pulses allow restoring monomer concentration near the electrode surface and prevent potential slowing of the polymerization rate due to monomer depletion in the inner pores of the carbon material. As a result, the substrate becomes uniformly coated with a metal-salen polymer layer (Fig. 10b). The efficiency of this approach has also been confirmed in other studies [94].
![Fig. 10:
SEM images of the Kynol™ ACC 710-25 carbon cloth modified with poly-[Ni(SaltmEn)] by: (a) conventional potentiostatic polymerization ((1) ‒ inner filaments of the carbon material and (2) ‒ polymer on the electrode surface); (b) pulse polymerization [90].](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_010.jpg)
SEM images of the Kynol™ ACC 710-25 carbon cloth modified with poly-[Ni(SaltmEn)] by: (a) conventional potentiostatic polymerization ((1) ‒ inner filaments of the carbon material and (2) ‒ polymer on the electrode surface); (b) pulse polymerization [90].
When the porous carbon material containing adsorbed monomers is immersed in a monomer-free electrolyte, the adsorbed species are the only ones that undergo polymerization. This approach referred to as “adsorption-electrochemical modification” [91] usually involves anodic galvanostatic charging of the electrode to complete the polymer formation. The resulting polymer-modified electrode features uniform polymer content across the electrode thickness (Fig. 11). The polymer resides in the pores of the electrode material instead of its surface thus ensuring optimum electrode performance. The extent of pore filling can be easily controlled at the adsorption step. The described electrode fabrication method has proven effective for a wide range of activated carbon powder-based electrodes. After the adsorption step, the carbon material can be dried and then withstand long storage times without losing the monomeric metal-salen component. This has opened up an exciting opportunity to use the monomer-containing carbon for an EDLC electrode fabrication and perform the polymerization in situ, after the device is assembled and sealed [89].
![Fig. 11:
(a) – SEM of the cross-section of a YP-17D activated carbon-based electrode modified with poly-[Ni(Saltmen)]; (b) – distribution of Ni atoms obtained by energy-dispersive X-ray spectroscopy, which reflects the polymer distribution (carbon layer thickness corresponds to the layer on one side of the Al foil).](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_011.jpg)
(a) – SEM of the cross-section of a YP-17D activated carbon-based electrode modified with poly-[Ni(Saltmen)]; (b) – distribution of Ni atoms obtained by energy-dispersive X-ray spectroscopy, which reflects the polymer distribution (carbon layer thickness corresponds to the layer on one side of the Al foil).
CV curves of poly-[M(salen)]-modified electrodes prepared according to the above described procedures display faradaic currents due to the oxidation-reduction of the redox-active polymer (Fig. 12a). Their charge-discharge curves deviate from a classic triangular shape of an EDLC electrode and have two distinctive regions: at lower potentials, the polymer-modified electrode capacitance is largely determined by the double-layer capacitance of the activated carbon, and at higher potentials, the electrode capacitance is the sum of the double-layer capacitance and the electrochemical (pseudo-) capacitance of the deposited polymer (Fig. 12b).
![Fig. 12:
Electrochemical behavior of Kynol™ ACC 710-25 carbon cloth electrodes (1 – non-modified; 2 – modified with poly-[Ni(SaltmEn)]) in 1 M Et4NBF4/AN: (a) – cyclic voltammograms; (b) – galvanostatic charge-discharge curves [90].](/document/doi/10.1515/pac-2019-1218/asset/graphic/j_pac-2019-1218_fig_012.jpg)
Electrochemical behavior of Kynol™ ACC 710-25 carbon cloth electrodes (1 – non-modified; 2 – modified with poly-[Ni(SaltmEn)]) in 1 M Et4NBF4/AN: (a) – cyclic voltammograms; (b) – galvanostatic charge-discharge curves [90].
The experimentally observed improvement of the EDLC electrode capacitance after its modification with metal-salen polymers depended on the type of porous carbon, the monomer, and the deposition technique, and was in the range 40–120% [88], [89], [90], [91], [92]. Improved charge storage characteristics of the polymer-modified electrodes preserved when they were used as positive electrodes in small-volume hermetically sealed non-aqueous hybrid (asymmetric) supercapacitor prototype cells that also contained non-modified negative electrodes. A significant improvement in the energy density of the hybrid supercapacitor vs. a standard EDLC was achieved without noticeable deterioration of the specific power and cycle life of the device [89], [90]. Similar positive effect on performance was observed when the polymer-modified electrode was used as a positive electrode in a lithium-ion supercapacitor cell [92].
Other types of carbon-based materials (multiwalled carbon nanotubes [93], [94], [95], [96], [97], [98], nitrogen-doped graphene [99], reduced graphene oxide [96], [98], [100], and reduced graphene oxide/multiwalled carbon nanotubes composite [98]) have been also successfully modified with nickel-salen polymers by other researchers to produce advanced electrodes potentially suitable for supercapacitor applications. In all studies, nickel-salen polymer-based composite electrodes featured improved specific capacitance vs. their unmodified counterparts. Some of them also showed enhanced charge transfer ability [93], [96]. Impressive cycle life of polymer-containing supercapacitor prototypes (for example, 91% capacitance retention after 10000 charge-discharge cycles [100]) has been demonstrated.
The vast majority of described studies have been devoted to the development of electrode materials for supercapacitors filled with non-aqueous electrolytes because of the known sensitivity of metal-salen polymers to the presence of water (see Section 6). We have recently demonstrated a poly-[Ni(CH3-Salen]-based composite material that can efficiently function in aqueous electrolytes [101]. The second component of the electrodeposited composite was a redox polymer bearing stable nitroxyl radical groups, poly-TEMPO-methacrylate (PTMA) (TEMPO = a 2,2,6,6-tetramethylpiperidin-1-oxyl-4-yl fragment). The nickel-salen polymer served as a conducting and capacitive matrix to the PTMA, a highly capacitive material (theoretical specific capacity = 111 mAh g−1), which however is rarely used in energy storage because of low electron conductivity and high solubility in many electrolytes. The cyclic voltammograms of the composite material were indicative of the redox activity of both the PTMA and salen components. Its conductivity surpassed the conductivity of the pristine PTMA film by approximately two orders of magnitude and was on par with the conductivity of a PTMA-carbon black composite. When used as a positive electrode in an asymmetrical supercapacitor prototype filled with 1 M aqueous LiClO4 solution, the composite electrode demonstrated high specific capacity (83 mAh g−1 at 1C, 53 mAh g−1 at 5C and 47 mAh g−1 at 10C), high rate capability as well as cycling stability (55% capacity retention after 1000 cycles). Surprisingly high material stability in the aqueous electrolyte likely resulted from the stabilizing effect of PTMA chains that created steric hindrance to the approach of nucleophiles to the active sites of poly-[Ni(CH3-Salen)]. The demonstrated strategy of electrochemical co-polymerization of metal-salen complexes and other redox active monomers could potentially be used for obtaining next generation electrochemically active composites for energy storage devices, including but not limited to supercapacitors.
Metal-salen polymers in lithium-ion batteries
Lithium ion batteries are a relatively new but probably the most popular battery storage technology, especially in portable consumer electronics. Recently emerged applications of lithium-ion batteries include electric vehicles, backup power, and stationary energy storage. Their widespread use is caused by multiple advantages such as high specific energy, reasonably fast charge-discharge capability, and high capacity utilization even during fast charge and discharge [122], [123]. Unfortunately, there are still some concerns related to their safety and relatively short lifetime. A search continues for new electrode and electrolyte materials that could help further improve energy density and power density and overcome above hurdles to their large-scale applications.
A conventional Li-ion battery cathode consists of an active material, a conductive additive (carbon black), and a binder, which form an active layer on a current collector (typically an Al foil). Below we show how metal-salen polymers could potentially be used in Li-ion battery cathodes as (a) an active material, (b) a multifunctional conductive binder, and (c) a buffer interlayer for overcharge protection.
Monocomponent electrodes were obtained by electrochemical galvanostatic polymerization of [Ni(Salen)], [Ni(CH3-Salen)], and [Ni(CH3O-Salen)] on carbon paper and evaluated in half-cells with lithium metal as an anode [102]. The cells were filled with 1 M LiPF6 in 1:1 ethylene carbonate (EC)/diethyl carbonate (DEC) and tested in the 2.1–4.2 V (vs. Li/Li+) range. Poly-[Ni(CH3-Salen)] demonstrated the highest specific capacity (50 mA g−1) and power capability, which was mainly attributed to the favorable polymer morphology. All three polymers showed 98–99% coulombic efficiency at a 1C rate and stability sufficient for use as cathode active materials in organic Li-ion batteries.
A novel polymeric material based on a nickel(II) bis(salicylideneiminate) complex with a carboxyethylene group as a substituent in the imine bridge has recently been synthesized and proposed for a potential application in lithium-ion batteries [103]. A distinctive feature of this polymer is that its neutral (non-charged) form exists as an ionic pair with a lithium cation. When the polymer is oxidized the cations exit to maintain the electroneutrality of the system and the polymer film becomes self-doped: positive charges on the monomer units are compensated by a negative charge of the carboxyl group. Such unconventional charge transfer mechanism has been confirmed by EQCM studies.
In another recent R&D effort, we used poly-[Ni(CH3-Salen)] as a multifunctional cathode additive that could replace electrochemically inactive electrode components (a conductive additive and portion of the binder) that not only reduce the energy density of conventional Li-ion cells by occupying volume and mass without contributing to energy storage, but are also a potential source of side reactions at the interface between the components and the electrolyte that decrease the battery life [104]. A unique in situ electrode fabrication method was developed, which involved mixing an active material (LiFePO4), [Ni(CH3-Salen)], and a binder (PVdF) according to the electrode formulation of 93:5.5:1.5 wt ratio, casting it onto the aluminum current collector, and drying. The monomer-containing electrodes were then assembled in half-cells with lithium metal as an anode and 1 M LiPF6 in EC/DEC (1:1 vol ratio) as an electrolyte. The polymerization of the monomer was accomplished during the first charging cycle. Poly[Ni(CH3-Salen)]‒LiFePO4 cathodes delivered promising battery performances that surpassed the conventional cathodes (i.e., consisting of an active material, carbon conductor, and PVdF binder) in a voltage range of 3−4 V (vs. Li/Li+). That included higher specific discharge capacity and coulombic efficiency combined with improved high rate capability and long term cycling. Observed performance improvement was a result of multiple positive roles of the nickel-salen polymer in the system. The polymer formed a three-dimensional conducting web that provided electronic communication between all LiFePO4 particles, helped boost up their effective capacity, and provided facile electron transfer through the large surface contact between conductive polymer and LiFePO4 surfaces. The poly-[Ni(CH3-Salen)] additive also ensured mechanical stability of the cathode by efficiently binding all active material particles and helped maintain the mechanical integrity of the electrode during repeated cycling. The redox processes in the polymer contributed to an increase in the electrode specific capacity. The polymer also passivated the top surficial layer of LiFePO4, which largely prevented unwanted interfacial reactions during repeated electrochemical charge-discharge cycling. The improved specific capacity of the poly-[Ni(CH3-Salen)]‒LiFePO4 cathode over the carbon‒LiFePO4 cathode was also confirmed in full cells, using the graphite anodes.
Preparing a buffer interlayer between the aluminum current collector and the cathode active layer to provide protection against both internal and external short-circuiting and overcharge is one of potential solutions to the safety issues of lithium-ion batteries. Such buffer interlayers could be made of conducting polymers exhibiting potential-dependent conductivity: it should remain sufficiently high in the “potential window” of the battery cathode and sharply decrease at potentials corresponding to the battery overcharge. In our recent study [105], a series of nickel-salen polymers were electrodeposited onto interdigitated platinum electrodes and their conductivities were measured during an electrochemically induced redox switching in 1 M LiPF6 solution in EC:DEC (1:1). All polymers exhibited potential-dependent conductivities when cycled in the 2.8–5.0 V (vs. Li/Li+) range. Among them, poly-[Ni(CH3-Salen)] showed the highest maximum conductivity, the widest conductivity window, and a high overcharge stability, with the upper limit of 4.6 V. The first indication of suitability of metal-salen polymers as buffer interlayer materials has thus been obtained.
To the best of our knowledge, the highlighted studies represent the first and only examples of the potential application of environmentally benign metal-salen polymers in lithium-ion batteries.
Concluding remarks
In this contribution, we presented an overview of metal-salen polymers, focusing on the topics that have long been debated matters among scientists: molecular and supramolecular structure, charge transfer mechanism, location of redox centers and multielectron reactions. All these characteristics of poly-[M(salen)] networks are of outmost importance for their practical applications in electrochemical energy storage. Over the past years, we demonstrated that the metal-salen polymer structure could be modulated by varying metal centres, peripheral substituents in the ligand, and altering polymerization conditions. The combination of structural characterization methods, DFT calculations, electrochemical, spectroscopic, and in situ spectroelectrochemical techniques employed in our studies enabled us to begin uncovering key structure-property relationships in poly-[M(salen)] materials and use this knowledge for the fine tuning of polymer properties relevant to energy storage applications (range of potentials for oxidation-reduction processes, specific capacity, charge transfer rate, stability). In our studies we have gone beyond pure research: advanced polymer-modified electrodes for the next generations of supercapacitors and lithium ion batteries have been designed and tested in laboratory scale prototypes. The success of these initial efforts encourages us to continue to explore the fascinating world of metal-salen polymeric materials.
Funding source: Russian Foundation for Basic Research
Award Identifier / Grant number: 18-29-04058
Acknowledgments
The authors would like to gratefully acknowledge the contributions of former and current members of the research teams, especially Dr. A.M. Timonov, a pioneer and a leading figure in the investigation of metal-salen polymers in the Russian Federation, and Dr. V.V. Malev. This work was partially supported by the Russian Foundation for Basic Research (grant 18-29-04058). Researches were performed with the aid of Centre for X-ray Diffraction Studies, Chemistry Educational Centre, Magnetic Resonance Research Centre, Centre for Applied Aerodynamics, Centre for Microscopy and Microanalysis, Centre for Optical and Laser Materials Research, Computing Centre, Interdisciplinary Resource Centre for Nanotechnology, Thermogravimetric and Calorimetric Research Centre and Chemical Analysis and Materials Research Centre of the Research Park of Saint Petersburg State University.
-
Funding: This work was partially supported by the Russian Foundation for Basic Research (grant 18-29-04058).
References
[1] L. A. Hoferkamp, K. A. Goldsby. Chem. Mater. 1, 348 (1989).10.1021/cm00003a015Search in Google Scholar
[2] K. A. Goldsby, J. K. Blaho, L. A. Hoferkamp. Polyhedron. 8, 113 (1989).10.1016/S0277-5387(00)86388-3Search in Google Scholar
[3] F. Bedioui, E. Labbe, S. Gutierrez-Granados, J. Devynck. J. Electroanal. Chem. Interfacial Electrochem. 301, 267 (1991).10.1016/0022-0728(91)85477-7Search in Google Scholar
[4] P. Audebert, P. Hapiot, P. Capdevielle, M. Maumy. J. Electroanal. Chem. 338, 269 (1992).10.1016/0022-0728(92)80428-7Search in Google Scholar
[5] P. Audebert, P. Capdevielle, M. Maumy. New J. Chem. 16, 697 (1992).Search in Google Scholar
[6] C. E. Dahm, D. G. Peters, J. Simonet. J. Electroanal. Chem. 410, 163 (1996).10.1016/0022-0728(95)04520-1Search in Google Scholar
[7] P. Audebert, P. Capdevielle, M. Maumy. Synth. Met. 43, 3049 (1991).10.1016/0379-6779(91)91235-3Search in Google Scholar
[8] I. A. Orlova, A. M. Timonov, G. A. Shagisultanova. Russ. J. Appl. Chem. 68, 406 (1995).Search in Google Scholar
[9] C. P. Horwitz, R. W. Murray. Mol. Cryst. Liq. Cryst. Inc. Nonlinear Opt. 160, 389 (1988).10.1080/15421408808083034Search in Google Scholar
[10] J. L. Reddinger, J. R. Reynolds. Macromolecules 30, 673 (1997).10.1021/ma961689oSearch in Google Scholar
[11] J. L. Reddinger, J. R. Reynolds. Synth. Met. 84, 225 (1997).10.1016/S0379-6779(97)80724-2Search in Google Scholar
[12] R. P. Kingsborough, T. M. Swager. Adv. Mater. 10, 1100 (1998).10.1002/(SICI)1521-4095(199810)10:14<1100::AID-ADMA1100>3.0.CO;2-2Search in Google Scholar
[13] R. P. Kingsborough, T. M. Swager. J. Am. Chem. Soc. 121, 8825 (1999).10.1021/ja991285mSearch in Google Scholar
[14] M. Vilas-Boas, C. Freire, B. de Castro, P. A. Christensen, A. R. Hillman. Inorg. Chem. 36, 4919 (1997).10.1021/ic970467jSearch in Google Scholar
[15] M. Vilas-Boas, C. Freire, B. de Castro, A. R. Hillman. J. Phys. Chem. B. 102, 8533 (1998).10.1021/jp982160rSearch in Google Scholar
[16] S. V. Vasil’eva, K. Balashev, A. Timonov. Russ. J. Electrochem. 34, 978 (1998).Search in Google Scholar
[17] M. Vilas-Boas, M. J. Henderson, C. Freire, A. R. Hillman, E. Vieil. Chem. Eur. J. 6, 1160 (2000).10.1002/(SICI)1521-3765(20000403)6:7<1160::AID-CHEM1160>3.0.CO;2-2Search in Google Scholar
[18] M. Vilas-Boas, C. Freire, B. de Castro, P. A. Christensen, A. R. Hillman. Chem. Eur. J. 7, 139 (2001).10.1002/1521-3765(20010105)7:1<139::AID-CHEM139>3.0.CO;2-QSearch in Google Scholar
[19] M. Vilas-Boas, I. C. Santos, M. J. Henderson, C. Freire, A. R. Hillman, E. Vieil. Langmuir 19, 7460 (2003).10.1021/la034525rSearch in Google Scholar
[20] A. Hamnett, J. Abel, J. Eameaim, P. Christensen, A. Timonov, S. Vasilyeva. Phys. Chem. Chem. Phys. 1, 5147 (1999).10.1039/a906632bSearch in Google Scholar
[21] S. V. Vasil’eva, K. P. Balashev, A. M. Timonov. Russ. J. Electrochem. 36, 75 (2000).10.1007/BF02757801Search in Google Scholar
[22] S. V. Vasil’eva, N. A. German, P. V. Gaman’kov, A. M. Timonov. Russ. J. Electrochem. 37, 317 (2001).10.1023/A:1009093801521Search in Google Scholar
[23] S. V. Vasil’eva, I. A. Chepurnaya, S. A. Logvinov, P. V. Gaman’kov, A. M. Timonov. Russ. J. Electrochem. 39, 310 (2003).10.1023/A:1022836113222Search in Google Scholar
[24] I. A. Chepurnaya, P. V. Gaman’kov, T. Y. Rodyagina, S. V. Vasil’eva, A. M. Timonov. Russ. J. Electrochem. 39, 314 (2003).10.1023/A:1022888130060Search in Google Scholar
[25] I. Tchepournaya, S. Vasilieva, S. Logvinov, A. Timonov, R. Amadelli, D. Bartak. Langmuir 19, 9005 (2003).10.1021/la030060tSearch in Google Scholar
[26] T. Y. Rodyagina, P. V. Gaman’kov, E. A. Dmitrieva, I. A. Chepurnaya, S. V. Vasil’eva, A. M. Timonov. Russ. J. Electrochem. 41, 1101 (2005).10.1007/s11175-005-0188-7Search in Google Scholar
[27] G. A. Shagisultanova, L. P. Ardasheva. Russ. J. Appl. Chem. 76, 1626 (2003).10.1023/B:RJAC.0000015726.55537.c1Search in Google Scholar
[28] E. A. Dmitrieva, S. A. Logvinov, V. V. Kurdakova, V. V. Kondrat’ev, V. V. Malev, A. M. Timonov. Russ. J. Electrochem. 41, 381 (2005).10.1007/s11175-005-0079-ySearch in Google Scholar
[29] J. Tarabek, P. Rapta, M. Kalbac, L. Dunsch. Anal Chem. 76, 5918 (2004).10.1021/ac049283uSearch in Google Scholar PubMed
[30] M. Martins, M. V. Boas, B. de Castro, A. R. Hillman, C. Freire. Electrochim. Acta. 51, 304 (2005).10.1016/j.electacta.2005.04.026Search in Google Scholar
[31] A. N. Borisov, A. V. Shchukarev, G. A. Shagisultanova. Russ. J. Appl. Chem. 82, 1242 (2009).10.1134/S1070427209070167Search in Google Scholar
[32] S. A. Krasikova, M. A. Besedina, M. P. Karushev, E. A. Dmitrieva, A. M. Timonov. Russ. J. Electrochem. 46, 218 (2010).10.1134/S102319351002014XSearch in Google Scholar
[33] J. Fonseca, J. Tedim, K. Biernacki, A. L. Magalhães, S. J. Gurman, C. Freire, A. R. Hillman. Electrochim. Acta. 55, 7726 (2010).10.1016/j.electacta.2010.01.073Search in Google Scholar
[34] B. N. Afanas’ev, Y. A. Polozhentseva, A. M. Timonov. Russ. J. Phys. Chem. A. 84, 2148 (2010).10.1134/S0036024410120241Search in Google Scholar
[35] B. N. Afanas’ev, Y. A. Polozhentseva, A. M. Timonov. Russ. J. Appl. Chem. 84, 1341 (2011).10.1134/S1070427211080088Search in Google Scholar
[36] V. V. Malev, O. V. Levin, A. M. Timonov. Electrochim. Acta. 108, 313 (2013).10.1016/j.electacta.2013.06.101Search in Google Scholar
[37] O. V. Levin, M. P. Karushev, A. M. Timonov, E. V. Alekseeva, S. Zhang, V. V. Malev. Electrochim. Acta. 109, 153 (2013).10.1016/j.electacta.2013.07.070Search in Google Scholar
[38] V. V. Sizov, M. V. Novozhilova, E. V. Alekseeva, M. P. Karushev, A. M. Timonov, S. N. Eliseeva, A. A. Vanin, V. V. Malev, O. V. Levin. J. Solid State Electr. 19, 453 (2014).10.1007/s10008-014-2619-4Search in Google Scholar
[39] C. S. Martin, C. Gouveia-Caridade, F. N. Crespilho, C. J. L. Constantino, C. M. A. Brett. Electrochim. Acta. 178, 80 (2015).10.1016/j.electacta.2015.07.123Search in Google Scholar
[40] C. R. Peverari, D. N. David-Parra, M. M. Barsan, M. F. S. Teixeira. Polyhedron 117, 415 (2016).10.1016/j.poly.2016.06.021Search in Google Scholar
[41] C. Chen, X. Li, F. Deng, J. Li. RSC Adv. 6, 79894 (2016).10.1039/C6RA17794HSearch in Google Scholar
[42] D. V. Anishchenko, O. V. Levin, V. V. Malev. Electrochim. Acta. 188, 480 (2016).10.1016/j.electacta.2015.11.044Search in Google Scholar
[43] K. Łępicka, P. Pieta, A. Shkurenko, P. Borowicz, M. Majewska, M. Rosenkranz, S. Avdoshenko, A. A. Popov, W. Kutner. J. Phys. Chem. C 121, 16710 (2017).10.1021/acs.jpcc.7b04700Search in Google Scholar
[44] E. V. Alekseeva, I. A. Chepurnaya, V. V. Malev, A. M. Timonov, O. V. Levin. Electrochim. Acta. 225, 378 (2017).10.1016/j.electacta.2016.12.135Search in Google Scholar
[45] D. Tomczyk, W. Bukowski, K. Bester, P. Urbaniak, P. Seliger, G. Andrijewski, S. Skrzypek. New J. Chem. 41, 2112 (2017).10.1039/C6NJ03635JSearch in Google Scholar
[46] M. T. Nguyen, R. A. Jones, B. J. Holliday. Macromolecules 50, 872 (2017).10.1021/acs.macromol.6b02349Search in Google Scholar
[47] A. A. Vereschagin, V. V. Sizov, P. S. Vlasov, E. V. Alekseeva, A. S. Konev, O. V. Levin. New J. Chem. 41, 13918 (2017).10.1039/C7NJ03526HSearch in Google Scholar
[48] D. Tomczyk, W. Bukowski, K. Bester. Electrochim. Acta. 267, 181 (2018).10.1016/j.electacta.2018.02.051Search in Google Scholar
[49] F. Deng, X. Li, F. Ding, B. Niu, J. Li. J. Phys. Chem. C 122, 5325 (2018).10.1021/acs.jpcc.8b00434Search in Google Scholar
[50] D. S. Kurchavov, M. P. Karushev, A. M. Timonov. Russ. J. Gen. Chem. 88, 1553 (2018).10.1134/S1070363218070332Search in Google Scholar
[51] M. V. Novozhilova, E. A. Smirnova, J. A. Polozhentseva, J. A. Danilova, I. A. Chepurnaya, M. P. Karushev, V. V. Malev, A. M. Timonov. Electrochim. Acta. 282, 105 (2018).10.1016/j.electacta.2018.06.030Search in Google Scholar
[52] E. Dmitrieva, M. Rosenkranz, J. S. Danilova, E. A. Smirnova, M. P. Karushev, I. A. Chepurnaya, A. M. Timonov. Electrochim. Acta. 283, 1742 (2018).10.1016/j.electacta.2018.07.131Search in Google Scholar
[53] E. A. Dmitrieva, I. A. Chepurnaya, M. P. Karushev, A. M. Timonov. Russ. J. Electrochem. 55, 1039 (2019).10.1134/S1023193519110041Search in Google Scholar
[54] D. Tomczyk, W. Bukowski, K. Bester. J. Electrochem. Soc. 166, H194 (2019).10.1149/2.0961906jesSearch in Google Scholar
[55] E. V. Alekseeva, V. A. Ershov, A. S. Konev, O. V. Levin. ECS Trans. 87, 167 (2018).10.1149/08701.0167ecstSearch in Google Scholar
[56] V. A. Ershov, E. V. Alekseeva, A. S. Konev, N. S. Chirkov, T. A. Stelmashuk, O. V. Levin. Russ. J. Gen. Chem. 88, 277 (2018).10.1134/S1070363218020147Search in Google Scholar
[57] K. Łępicka, M. Majewska, R. Nowakowski, W. Kutner, P. Pieta. Electrochim. Acta. 297, 94 (2019).10.1016/j.electacta.2018.11.147Search in Google Scholar
[58] K. Łępicka, P. Pieta, G. Francius, A. Walcarius, W. Kutner. Electrochim. Acta. 315, 75 (2019).10.1016/j.electacta.2019.05.075Search in Google Scholar
[59] C. E. Dahm, D. G. Peters. Anal. Chem. 66, 3117 (1994).10.1021/ac00091a020Search in Google Scholar
[60] C. E. Dahm, D. G. Peters. J. Electroanal. Chem. 406, 119 (1996).10.1016/0022-0728(95)04453-1Search in Google Scholar
[61] M. Vilas-Boas, E. M. Pereira, C. Freire, A. R. Hillman. J. Electroanal. Chem. 538-539, 47 (2002).10.1016/S0022-0728(02)01097-5Search in Google Scholar
[62] A. Olean-Oliveira, C. F. Pereira, D. N. David-Parra, M. F. S. Teixeira. ChemElectroChem 5, 3557 (2018).10.1002/celc.201800532Search in Google Scholar
[63] G. D. Liu, Z. Q. Li, S. S. Huan, G. L. Shen, R. Q. Yu. Anal. Lett. 33, 175 (2000).10.1080/00032710008543045Search in Google Scholar
[64] D. Deletioğlu, S. Yalçınkaya, C. Demetgül, M. Timur, S. Serin. Mater. Chem. Phys. 128, 500 (2011).10.1016/j.matchemphys.2011.03.050Search in Google Scholar
[65] F. Miomandre, P. Audebert, M. Maumy, L. Uhl. J. Electroanal. Chem. 516, 66 (2001).10.1016/S0022-0728(01)00657-XSearch in Google Scholar
[66] J. Tedim, A. Carneiro, R. Bessada, S. Patrício, A. L. Magalhães, C. Freire, S. J. Gurman, A. R. Hillman. J. Electroanal. Chem. 610, 46 (2007).10.1016/j.jelechem.2007.06.025Search in Google Scholar
[67] J. Tedim, R. Bessada, S. Patricio, A. L. Magalhaes, C. Freire, S. J. Gurman, A. R. Hillman. Langmuir 24, 8998 (2008).10.1021/la801424kSearch in Google Scholar PubMed
[68] J. Tedim, C. Freire, A. R. Hillman. Soft Matter 5, 2603 (2009).10.1039/b818245kSearch in Google Scholar
[69] M. F. S. Teixeira, T. R. L. Dadamos. Procedia Chem. 1, 297 (2009).10.1016/j.proche.2009.07.074Search in Google Scholar
[70] S. Patricio, A. I. Cruz, K. Biernacki, J. Ventura, P. Eaton, A. L. Magalhaes, C. Moura, A. R. Hillman, C. Freire. Langmuir 26, 10842 (2010).10.1021/la1006956Search in Google Scholar PubMed
[71] C. S. Martin, T. R. L. Dadamos, M. F. S. Teixeira. Sens. Actuators B Chem. 175, 111 (2012).10.1016/j.snb.2011.12.098Search in Google Scholar
[72] C. Sousa, P. Gameiro, C. Freire, B. de Castro. Polyhedron 23, 1401 (2004).10.1016/j.poly.2004.02.014Search in Google Scholar
[73] T. R. L. Dadamos, M. F. S. Teixeira. Electrochim. Acta. 54, 4552 (2009).10.1016/j.electacta.2009.03.045Search in Google Scholar
[74] P. K. Sonkar, V. Ganesan, A. Prajapati. Ionics 22, 1741 (2016).10.1007/s11581-016-1699-9Search in Google Scholar
[75] C. F. Pereira, A. Olean-Oliveira, D. N. David-Parra, M. F. S. Teixeira. Talanta. 190, 119 (2018).10.1016/j.talanta.2018.07.080Search in Google Scholar PubMed
[76] M. T. Nguyen, R. A. Jones, B. J. Holliday. Chem. Commun. 52, 13112 (2016).10.1039/C6CC07193GSearch in Google Scholar
[77] B. de Castro, R. Ferreira, C. Freire, H. García, E. J. Palomares, M. J. Sabater. New J. Chem. 26, 405 (2002).10.1039/b108436dSearch in Google Scholar
[78] J. Tedim, S. Patrício, J. Fonseca, A. L. Magalhães, C. Moura, A. R. Hillman, C. Freire. Synth. Met. 161, 680 (2011).10.1016/j.synthmet.2011.01.014Search in Google Scholar
[79] M. Nunes, M. Araujo, J. Fonseca, C. Moura, R. Hillman, C. Freire. ACS Appl. Mater. Inter. 8, 14231 (2016).10.1021/acsami.6b01977Search in Google Scholar PubMed
[80] M. Nunes, C. Moura, A. R. Hillman, C. Freire. Langmuir 33, 6826 (2017).10.1021/acs.langmuir.7b01380Search in Google Scholar PubMed
[81] M. Nunes, C. Moura, A. R. Hillman, C. Freire. Electrochim. Acta. 238, 142 (2017).10.1016/j.electacta.2017.04.010Search in Google Scholar
[82] M. Nunes, M. Araújo, R. Bacsa, R. V. Ferreira, E. Castillejos, P. Serp, A. R. Hillman, C. Freire. Carbon 120, 32 (2017).10.1016/j.carbon.2017.05.001Search in Google Scholar
[83] M. P. Araujo, M. Nunes, J. Fonseca, C. Moura, R. Hillman, C. Freire. J. Colloid Interf. Sci. 504, 790 (2017).10.1016/j.jcis.2017.06.023Search in Google Scholar PubMed
[84] C. Pinheiro, A. J. Parola, F. Pina, J. Fonseca, C. Freire. Sol. Energy Mater. Sol. Cells 92, 980 (2008).10.1016/j.solmat.2008.03.001Search in Google Scholar
[85] A. Branco, C. Pinheiro, J. Fonseca, J. o. Tedim, A. Carneiro, A. J. Parola, C. Freire, F. Pina. Electrochem. Solid-State Lett. 13, J114 (2010).10.1149/1.3457474Search in Google Scholar
[86] E. A. Smirnova, M. A. Besedina, M. P. Karushev, V. V. Vasil’ev, A. M. Timonov. Russ. J. Phys. Chem. A 90, 1088 (2016).10.1134/S0036024416050319Search in Google Scholar
[87] A. S. Konev, M. Y. Kayumov, M. P. Karushev, Y. V. Novoselova, D. A. Lukyanov, E. V. Alekseeva, O. V. Levin. ChemElectroChem. 5, 3138 (2018).10.1002/celc.201800846Search in Google Scholar
[88] A. Timonov, S. Logvinov, I. Chepurnaya, V. Kuznetsov. in The 15th International Seminar on Double Layer Capacitors and Hybrid Energy Storage Devices December 5 - 7, 2005, Embassy Suites Deerfield Beach Resort, N. Marincic (Ed.), pp. 258–272, Deerfield Beach, Florida, USA (2005).Search in Google Scholar
[89] N. Shkolnik, A. Timonov, S. Logvinov, V. Kuznetsov, I. Chepurnaya. in 2nd International Symposium on Large Ultracapacitor Technology and Application (UCAP). Baltimore’s Inner Harbor USA, pp. 136–162 (2006).Search in Google Scholar
[90] I. A. Chepurnaya, S. A. Logvinov, M. P. Karushev, A. M. Timonov, V. V. Malev. Russ. J. Electrochem. 48, 538 (2012).10.1134/S1023193512040040Search in Google Scholar
[91] M. P. Karushev, A. M. Timonov. Russ. J. Appl. Chem. 85, 914 (2012).10.1134/S1070427212050134Search in Google Scholar
[92] Y. A. Polozhentseva, M. P. Karushev, A. M. Rumyantsev, I. A. Chepurnaya, A. M. Timonov. Tech. Phys. Lett. 46, 196 (2020).10.1134/S106378502002025XSearch in Google Scholar
[93] F. Gao, J. Li, F. Kang, Y. Zhang, X. Wang, F. Ye, J. Yang. J. Phys. Chem. C 115, 11822 (2011).10.1021/jp111831ySearch in Google Scholar
[94] J. Li, F. Gao, Y. Zhang, F. Kang, X. Wang, F. Ye, J. Yang. Sci. China Chem. 55, 1338 (2012).10.1007/s11426-012-4585-ySearch in Google Scholar
[95] Y. Zhang, J. Li, F. Kang, X. Wang, F. Ye, J. Yang. B. Korean Chem. Soc. 33, 1972 (2012).10.5012/bkcs.2012.33.6.1972Search in Google Scholar
[96] G. Yan, J. Li, Y. Zhang, F. Gao, F. Kang. J. Phys. Chem. C 118, 9911 (2014).10.1021/jp500249tSearch in Google Scholar
[97] Z. Zhu, J. Lu, X. Li, G. Xu, C. Chen, J. Li. Electrochemistry 84, 427 (2016).10.5796/electrochemistry.84.427Search in Google Scholar
[98] X. Li, J. Li, F. Deng, F. Kang. Ionics. 24, 3143 (2018).10.1007/s11581-018-2472-zSearch in Google Scholar
[99] X. Li, F. Deng, J. Li, Z. Li, F. Kang. Electrochim. Acta. 284, 355 (2018).10.1016/j.electacta.2018.07.065Search in Google Scholar
[100] K. Łępicka, P. Pieta, R. Gupta, M. Dabrowski, W. Kutner. Electrochim. Acta. 268, 111 (2018).10.1016/j.electacta.2018.02.085Search in Google Scholar
[101] A. A. Vereshchagin, P. S. Vlasov, A. S. Konev, P. Yang, G. A. Grechishnikova, O. V. Levin. Electrochim. Acta. 295, 1075 (2019).10.1016/j.electacta.2018.11.149Search in Google Scholar
[102] S. N. Eliseeva, E. V. Alekseeva, A. A. Vereshchagin, A. I. Volkov, P. S. Vlasov, A. S. Konev, O. V. Levin. Macromol. Chem. Phys. 218, 1700361 (2017).10.1002/macp.201700361Search in Google Scholar
[103] S. A. Samokhvalova, V. A. Ershov, D. A. Lukyanov, P. S. Vlasov, O. V. Levin. Russ. J. Gen. Chem. 89, 852 (2019).10.1134/S1070363219040352Search in Google Scholar
[104] C. O’Meara, M. P. Karushev, I. A. Polozhentceva, S. Dharmasena, H. Cho, B. J. Yurkovich, S. Kogan, J. H. Kim. ACS Appl. Mater. Interfaces 11, 525 (2019).10.1021/acsami.8b13742Search in Google Scholar PubMed
[105] E. V. Beletskii, Y. A. Volosatova, S. N. Eliseeva, O. V. Levin. Russ. J. Electrochem. 55, 339 (2019).10.1134/S1023193519030030Search in Google Scholar
[106] J. Zhang, L. Xu, W. Y. Wong. Coord. Chem. Rev. 355, 180 (2018).10.1016/j.ccr.2017.08.007Search in Google Scholar
[107] C. Freire, M. Nunes, C. Pereira, D. M. Fernandes, A. F. Peixoto, M. Rocha. Coord. Chem. Rev. 394, 104 (2019).10.1016/j.ccr.2019.05.014Search in Google Scholar
[108] A. N. Yankin, D. A. Lukyanov, E. V. Beletskii, O. Y. Bakulina, P. S. Vlasov, O. V. Levin. ChemistrySelect 4, 8886 (2019).10.1002/slct.201902385Search in Google Scholar
[109] C. Freire, B. d. Castro. J. Chem. Soc., Dalton Trans. 1491(1998).10.1039/a708231bSearch in Google Scholar
[110] I. C. Santos, M. Vilas-Boas, M. F. M. Piedade, C. Freire, M. T. Duarte, B. de Castro. Polyhedron 19, 655 (2000).10.1016/S0277-5387(00)00300-4Search in Google Scholar
[111] J. Kim, J. Lee, J. You, M.-S. Park, M. S. A. Hossain, Y. Yamauchi, J. H. Kim. Mater. Horiz. 3, 517 (2016).10.1039/C6MH00165CSearch in Google Scholar
[112] M. V. Novozhilova, E. A. Smirnova, M. P. Karushev, A. M. Timonov, V. V. Malev, O. V. Levin. Russ. J. Electrochem. 52, 1183 (2016).10.1134/S1023193516120107Search in Google Scholar
[113] F. Thomas. Dalton Trans. 45, 10866 (2016).10.1039/C6DT00942ESearch in Google Scholar
[114] R. M. Clarke, K. Hazin, J. R. Thompson, D. Savard, K. E. Prosser, T. Storr. Inorg. Chem. 55, 762 (2016).10.1021/acs.inorgchem.5b02231Search in Google Scholar PubMed
[115] R. M. Clarke, K. Herasymchuk, T. Storr. Coord. Chem. Rev. 352, 67 (2017).10.1016/j.ccr.2017.08.019Search in Google Scholar
[116] T. J. Dunn, M. I. Webb, K. Hazin, P. Verma, E. C. Wasinger, Y. Shimazaki, T. Storr. Dalt. Trans. 42, 3950 (2013).10.1039/c2dt32632aSearch in Google Scholar PubMed
[117] A. Kochem, H. Kanso, B. Baptiste, H. Arora, C. Philouze, O. Jarjayes, H. Vezin, D. Luneau, M. Orio, F. Thomas. Inorg. Chem. 51, 10557 (2012).10.1021/ic300763tSearch in Google Scholar PubMed
[118] N. Kuznetsov, P. Yang, G. Gorislov, Y. Zhukov, V. Bocharov, V. Malev, O. Levin. Electrochim. Acta. 271, 190 (2018).10.1016/j.electacta.2018.03.042Search in Google Scholar
[119] T. Liu, Y. Li. InfoMat. 1(2020).10.1002/inf2.12003Search in Google Scholar
[120] A. González, E. Goikolea, J. A. Barrena, R. Mysyk. Renew. Sust. Energ. Rev. 58, 1189 (2016).10.1016/j.rser.2015.12.249Search in Google Scholar
[121] E. Erdem, S. Najib. Nanoscale Adv. 1, 2817 (2019).10.1039/C9NA00345BSearch in Google Scholar
[122] A. Mauger, C. M. Julien. Ionics 23, 1933 (2017).10.1007/s11581-017-2177-8Search in Google Scholar
[123] A. Manthiram. ACS Cent. Sci. 3, 1063 (2017).10.1021/acscentsci.7b00288Search in Google Scholar PubMed PubMed Central
© 2020 IUPAC & De Gruyter
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Recent achievements in copper catalysis for C–N bond formation
- Novel selective anticancer agents based on Sn and Au complexes. Mini-review
- Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
- Surface chemistry of structural materials subjected to corrosion
- Redox-conducting polymers based on metal-salen complexes for energy storage applications
- Thermodynamic approach for prediction of oxide materials properties at high temperatures
- Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
- Fragment-based approach to novel bioactive purine derivatives
- Reaction of 1,3-dimethylimidazolium dimethylphosphate with elemental sulfur
- Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
- Macrokinetic investigation of the interaction mechanism of the pyrophoric iron nanopowder compacts with air
- Polylactide-based stent coatings: biodegradable polymeric coatings capable of maintaining sustained release of the thrombolytic enzyme streptokinase
- 3D printing in analytical chemistry: current state and future
- High purity substances – prototypes of elements of Periodic Table
- Pd(0)-catalyzed amination in the synthesis of chiral derivatives of BINAM and their evaluation as fluorescent enantioselective detectors
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Recent achievements in copper catalysis for C–N bond formation
- Novel selective anticancer agents based on Sn and Au complexes. Mini-review
- Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
- Surface chemistry of structural materials subjected to corrosion
- Redox-conducting polymers based on metal-salen complexes for energy storage applications
- Thermodynamic approach for prediction of oxide materials properties at high temperatures
- Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
- Fragment-based approach to novel bioactive purine derivatives
- Reaction of 1,3-dimethylimidazolium dimethylphosphate with elemental sulfur
- Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
- Macrokinetic investigation of the interaction mechanism of the pyrophoric iron nanopowder compacts with air
- Polylactide-based stent coatings: biodegradable polymeric coatings capable of maintaining sustained release of the thrombolytic enzyme streptokinase
- 3D printing in analytical chemistry: current state and future
- High purity substances – prototypes of elements of Periodic Table
- Pd(0)-catalyzed amination in the synthesis of chiral derivatives of BINAM and their evaluation as fluorescent enantioselective detectors