Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
-
Yulia H. Budnikova

 and
Vera V. Khrizanforova
and
Vera V. Khrizanforova

Abstract
Nowadays, hydrogen has become not only an extremely important chemical product but also a promising clean energy carrier for replacing fossil fuels. Production of molecular H2 through electrochemical hydrogen evolution reactions is crucial for the development of clean-energy technologies. The development of economically viable and efficient H2 production/oxidation catalysts is a key step in the creation of H2-based renewable energy infrastructure. Intrinsic limitations of both natural enzymes and synthetic materials have led researchers to explore enzyme-induced catalysts to realize a high current density at a low overpotential. In recent times, highly active widespread numerous electrocatalysts, both homogeneous or heterogeneous (immobilized on the electrode), such as transition metal complexes, heteroatom- or metal-doped nanocarbons, metal-organic frameworks, and other metal derivatives (calix [4] resorcinols, pectates, etc.), which are, to one extent or another, structural or functional analogs of hydrogenases, have been extensively studied as alternatives for Pt-based catalysts, demonstrating prospects for the development of a “hydrogen economy”. This mini-review generalizes some achievements in the field of development of new electrocatalysts for H2 production/oxidation and their application for fuel cells, mainly focuses on the consideration of the catalytic activity of M[P2N2]2 2+ (M = Ni, Fe) complexes and other nickel structures which have been recently obtained.
Introduction
Hydrogen is a perfect energy-yielding material for future applications, while water decomposition via solar/electric power use is one of the methods for hydrogen production [1], [2], [3], [4], [5], [6], [7], [8]. Importantly, hydrogen has the highest gravimetric energy density among all known substances, i.e., a lower heating value (LHV) of ∼120 kJ/g [5]. It is a pure source of energy, since during its burning out only water is formed. Water and heat are the only by-products of hydrogen-based fuel cells, the so-called H2/O2 fuel cells (fuel cell technologies) [9], [10], [11], [12], [13]. Fuel cells are considered as very prospective ones in the global energy market.
Many automobile companies (Hyundai, Toyota, Honda, BMW, Mazda) have already developed vehicles operating on hydrogen fuel, which allows reducing the emissions of gases down to 90%. The viability of such car models is evident. Toyota Mirai was recognized as a modern no-emission car. One re-fueling of such cars provides haulage of 800 km (Hyundai Nexo). Nexo passes crush tests with excellent results; in other words, it is an absolutely safe vehicle. The cost per one km will be several times less as compared to a gasoline similar motorcar, provided that the technologies and a network of filling stations are developed. Herewith the environmental harm is absent, while the fuel reserves are inexhaustible [9].
The road map for hydrogen energy production was developed in Europe [14]. Following the forecasts for the year of 2050, the hydrogen share in the energy consumption structure will amount to 24%, while a reduction of vehicle emissions will amount down to 15%, a huge decrease in CO2 emissions will be realized, hydrogen use will provide incomes in the amount of 820 billion Euro, 5.4 million work positions (maintenance, supply, control) will be created. In Hamburg port, Siemens Company supposes to construct the worldwide largest plant for hydrogen production by way of electrolysis with a capacity of 100 MW. It will produce nearly 2 tons of hydrogen per hour at the expense of electric power generated by North Sea wind farms. In Russia by 2035, electric power generation based on renewable sources should be increased by 20 times. In general, the hydrogen technologies primarily may and should appear as the supporting technologies in those fields, where it is economically viable, for instance, in faraway regions and isolated electricity systems, where access to the central supply system is not available. Within the context of the recent Fifth Eastern Economic Forum, Rosatom supposes to launch the project of trains operating on hydrogen fuel cells, in Sakhalin first of all.
All the mentioned items require the creation of more effective, leaner processes of hydrogen production and its oxidation in fuel cells (FCs), which, in turn, will require new effective and inexpensive catalysts as part of the newly formed “green” field of chemistry. Simple calculations show that cars equipped with H2 fuel cells with pure platinum membrane-electrode blocks are not viable since Pt reserves within the Earth are not enough in order to provide their long-term production. Besides, such extensive use of fuel cells for vehicles will result in environmental pollution with platinum caused by the unavoidable decomposition of the catalyst agent during the catalytic reaction and its excretion in the form of a solution in wastewater. This stimulates intensive research aimed at developing unconventional catalysts primarily based on the widespread first-row transition metals, such as nickel (Ni), cobalt (Co) and, in the first instance, iron (Fe), one of the least expensive and the most widespread metals on Earth [1], [4], [7], [15].
Herein the researchers called to memory hydrogenases, that is, the enzymes (biocatalysts), which effectively catalyze both the production and oxidation of hydrogen with the use of widespread metals (nickel and iron) [15], [16], [17], [18]. There exist several types of hydrogenases. Enzymes of two classes – [FeFe]-hydrogenase and [NiFe]-hydrogenase – have bimetallic active areas containing iron atoms, on the active centers of which H2/H+ transformations occur. The third class of hydrogenases, which is called [Fe]-hydrogenases, was found only in methanogens, where H2 decomposes on the mononuclear iron-carbonyl center, thus being transformed into a proton, and transmitting the hydride to the transmitter N5, N10 – methenyl-tetrahydromethanopterin [15].
Hydrogenases are active both in the reaction of H2 oxidation and in the reaction of its production. The catalytic reactivity of [NiFe] hydrogenases is generally lower. In perfect conditions, one enzyme of hydrogenase may generate 9000 molecules of hydrogen per second. In natural conditions, a catalyst has been created, which is extremely active even without the use of any uncommon noble metals. Unfortunately, oxygen sensitivity heretofore constrained the large-scale use of hydrogenases [15], [16], [17], [18]. Thus, the natural hydrogenases, which were recognized as the most effective catalysts for the transformation of protons and electrons into hydrogen (and for the counter hydrogen oxidation reaction), have inspired chemists to try to develop the simplest synthetic catalysts, being their structural and functional models, with the use of widespread metals (Fig. 1). The development of bioinspired homogeneous metal catalysts includes the following stages: detailed description of the metalloenzyme and of its active center (structure and mechanism); bio-inspired design of the coordination/organometallic compound based on the conception of structural/chemical principles, being the basis for the creation of the active metal area and its neighborhood, which have been determined at the first stage; synthesis of new ligands and their coordination with the metallic centers, creation of metal complexes (organic and inorganic synthesis); evaluation of the catalytic characteristics; selection of the most effective structures and their tests within the FC content. Certainly, the macro environment of the active center and molecular channels hardly may be reconstructed and there were attempts to apply provisions of supramolecular chemistry for these purposes through immobilizing the molecular catalyst within the space of organometallic polymer, on the conducting substrate, via planting to protein, etc. (Fig. 1).
![Fig. 1:
Prototypes of natural enzyme – hydrogenase – catalytic systems (molecular [Ni–Fe] complexes, FenSm clusters, complexes embedded within a various matrix, etc.).](/document/doi/10.1515/pac-2019-1207/asset/graphic/j_pac-2019-1207_fig_001.jpg)
Prototypes of natural enzyme – hydrogenase – catalytic systems (molecular [Ni–Fe] complexes, FenSm clusters, complexes embedded within a various matrix, etc.).
X-ray studies made it possible to establish the structures of [NiFe] and [FeFe] hydrogenases, the characteristics of crystals [19], while a better understanding of the functions of these enzymes, which has been realized via electrochemistry methods and theoretical calculations, opened up the new horizons for the development of synthetic catalysts used for the hydrogen evolution reaction (HER). The detailed information on the catalytic reactions of enzymes was received from voltammetric studies of protein films; however, it was difficult to produce enzymes in the required quantities making it possible to use these enzymes for commercial proposals, while their stability was often limited by the native surrounding limits. The important role of the suspended amine (or thiolate) group was established; this group belongs to the second coordination sphere of the ligand, which performs as if it was suspended over the metal center, approaching to it, and due to such location it may operate as the proton relay, transmit hydrogen to the metal and vice versa, attach it in the form of a proton [15], [17], [20], [21]. The availability of such a group appeared to be very important for producing effective synthetic hydrogenases. The supposed mechanism of H2 oxidation includes H2 joining with the Fe center and subsequent heterolytic decomposition of the hydrogen molecule with the formation of iron hydride and the migration of the proton to the nitrogen atom. Electron transfer is realized through the Fe4S4 cluster system, proton/hydrogen are delivered to the required active center through the channels, which provide the exact delivery of the reagent and the elimination of products from the catalytically active metal center [15], [17], [20], [21].
Based on the knowledge of the structures of metalloenzymes, it was supposed that the design of the new biomimetic catalysts, which imitate the operation of these metalloenzymes, and of biological nanoreactors should be based on framework structures containing coordinating P,N,S-atoms, which are connected with atoms of Ni or other metal and which form certain spaces in the second coordination sphere from macrocycles at the expense of channels within metal-organic frameworks (MOFs), etc. On the one hand, copying such an entire complex enzyme seems preposterous. It looks like impossible, too expensive and completely contradicting the unmitigated perceptions of biochemical/bioinspired chemistry, which primarily seeks to receive simple and inexpensive enzyme analogs, preserving reactivity, though substantially a lower one, at the expense of the structure simplification. On the other hand, it is necessary to understand the molecular factors, which provide high selectivity and reactivity, while synthetic chemistry may substantially improve the functional properties of the bioinspired catalysts through a change of the substituent agents and architectonic of their molecules.
In current mini-review we describe some recent examples of electrocatalysts for HER developed in our group. Mainly, we focused on the catalytic activity of nickel complexes with cyclic aminomethylphosphines and thiophorylated calixarenes in hydrogen evolution/oxidation reactions and their application in fuel cell. However, some recent examples of Fe catalysts and a few Ni, Co and Zn MOFs for HER also will be discussed. It is important to note that the ligand environment in synthetic analogs of hydrogenases must be selected to stabilize low oxidation states of the metal, to promote proton and hydrogen transfers, and rapid catalyst regeneration on the electrode, to ensure long-term performance (here, MOFs and pectin derivatives are especially interesting).
Molecular bioinspired artificial hydrogenases
It should be noted that though substantial information on the structure and mechanisms pertinent to important hydrogenase items has been obtained during the current studies, unfortunately by no means all structural analogs are operable, i.e., there are appreciably less functional analogs corresponding to effective electrocatalysts for H2 production [15], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31]. The high catalytic rates are impressive, but all synthetic mimics reported to date still require a large overpotential to drive proton reduction catalysis. The fact that an overall majority of investigators tried to simulate the active center of the natural [FeFe]-hydrogenases should not disregard possible new decisions, in which the active complex center will substantially or completely differ from the natural iron environment. In other words, as it has been shown already, other iron or nickel complexes, the structures of which differ from each other, or which resemble natural enzymes only partly, may have more effective electrocatalytic properties. For instance, there are many iron complexes, which contain conventional organometallic ligands, such as cyclopentadienyl (η5-C5H5, Cp), bridged and/or terminal CO and bridged ligands (such as alkyl, alkinyl, alkenyl, allyl, allenyl, etc.), some redox-active ligands, clathrochelates, which have properties of electrocatalysts for H2 evolution [15], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31] (Fig. 2). The new ligands, which are characterized by other ligand and acceptor properties, steric and other factors, may play a decisive role during the investigation of catalytic reactivity in the model systems. Scientists’ target is to create synthetic analogs of hydrogenases, capable to operate at room temperature, in aqueous or weak acidic media, at near-zero or low overvoltages of the hydrogen evolution, however characterized by comparable or even higher values of TOF and TON catalytic characteristics, while being more stable and available than the natural hydrogenases.
![Fig. 2:
Representative synthetic [FeFe] models with redox-non-innocent ligands.](/document/doi/10.1515/pac-2019-1207/asset/graphic/j_pac-2019-1207_fig_002.jpg)
Representative synthetic [FeFe] models with redox-non-innocent ligands.
HER catalytic characteristics in the presence of transition metal complexes are usually evaluated by means of cyclic voltammetry (CVA), in which the electrode delivers electrons and the added acid is a donor of protons [32], [33]. Catalytic reactivity is determined by changes in CVA response with an increase in acid concentration. The principal criterion is increasing the reduction current at the potential value near the one corresponding to the reduction of the complex or of its protonated form. Large electrocatalytic current may be received at the expense of catalyst regeneration at the electrode. CVA may be used for determining the key parameters of catalytic reactivity, such as overvoltage and the turn-over frequency of the cycle (TOF or TON), as well as determining the catalytic mechanism at different experimental conditions.
Among numerous described structural and functional analogs of Fe-Fe hydrogenases, the leading role for a long time belonged to the one from the row of biferrous Rauchfuss complexes with the amidic proton relay of 58 000 s−1 TOF, however operating at rather high overvoltages (up to 0.78–1 V) [15], [34]. Nickel (and other metals) complexes with cyclic diaza-diphosphacyclooctane (and other cyclic) ligands characterized by different substituents attached to phosphorous and nitrogen [15], [35], [36], [37], [38], [39], [40], [41], which have been obtained for the first time by researchers from Arbuzov Institute for Organic and Physical Chemistry of RAS [41], [42], [43], [44], [45], [46], [47], [48], are of special interest. Various cyclic and macrocyclic aminomethylphosphines with a variety of properties have been obtained using diverse primary or secondary phosphines, aldehydes and primary or secondary amines in a Mannich-like condensation reaction [42], [43], [44], [45], [46], [47], [48]. However, their first investigations as the biomimetics-hydrogenases have been performed by Daniel DuBois and were continued by scientists from different countries [35], [36], [37], [38], [39], [40]. It has been reported that [Ni(PR 2NR′2)2]2+ complexes are highly active catalysts for H2 production and oxidation and bidirectional catalysts active for both H2 production and oxidation, where PR 2NR′2 is a cyclic 1,5-diaza-3,7-diphosphacyclooctane ligand. A key feature of these catalysts is a positioned pendant amine, which operates as a proton relay in close proximity to a vacant coordination site of a redox-active metal center, which permits bifunctional activation of H2 during the heterolytic cleavage or the formation of the H−H bond, so this amine group can significantly accelerate the rate of intra- and intermolecular proton transfers. Similar iron complexes did not exhibit catalytic reactivity under the studied conditions.
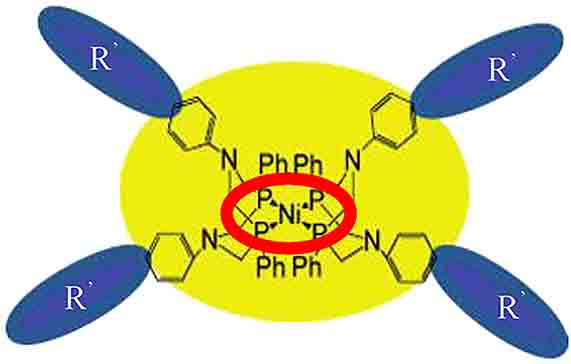
A lot of such ligands with different substituents and complexes based on these ligands have been synthesized [49], [50], [51] with a view to establish definite trends in their reactive capacity; some examples are presented in Fig. 3. In aqueous and weakly acidic media, these complexes are active in hydrogen evolution reaction; substantial catalytic currents were observed, using which the characteristics of these catalysts were determined. Some examples of catalytic data obtained by different groups of researchers (DuBois, Helm, Bullock, Karasik-Budnikova), which characterize the reactivity of nickel complexes with different substituents in the second and third coordination spheres of the complex, are summarized in Table 1 [52], [53], [54], [55], [56], [57], [58], [59], [60]. It is of interest that these catalytic structures are rather mobile with quick response to changes both in the second (substituents attached to phosphorous and nitrogen) and third coordination spheres. From the presented data, several trends in ligand structure modification may be emphasized, which result in a substantial increase in the catalytic reactivity of the complex as a whole. An increase in the number of the principal centers, for example, via introducing a pyridyl or amino-acid fragment, exerts a favorable effect on catalytic reactivity (Table 1). The presence of multiple protonation sites in the [Ni(PR 2NR′2)2]2+ systems, endo versus exo protonation, causes branching in the H2 production mechanism. The formation of exo protonated isomers is detrimental to the catalytic production of H2 because the exo isomers should be isomerized to endo isomers, where the protons can interact with the metal center for H2 production to occur. For this purpose, the volumetric benzhydryl substituent attached to nitrogen atoms has been introduced [56]. Actually, it appeared that the catalytic reactivity of [Ni(PPh 2NPh(CH)Ph 2)2]2+ was 250 times as high as compared with [Ni(PPh 2NBn 2)2]2+ (where Bn = benzyl) (TOFs 100 vs. 0.4 s−1). HER is also accelerated via introducing the high-chain substituents in the third coordination sphere, where TOF reaches the value of 1.5 mln s−1, which exceeds the corresponding values for natural hydrogenases [53].
![Fig. 3:
Selected examples of P,N-cyclic ligands for nickel complexes investigations in electrocatalytic processes [49], [50], [51].](/document/doi/10.1515/pac-2019-1207/asset/graphic/j_pac-2019-1207_fig_003.jpg)
The effect of the substituent nature on the catalytic efficiency, the TOF value of nickel complexes of diazadiphosphacyclooctanes in the reaction of hydrogen evolution [52], [53], [54], [55], [56], [57], [58], [59], [60]. *Trifluoroacetic acid was used as proton source.
| Substituents at the phosphorus atom (Ph at nitrogen) | TOF, s−1 | Substituents at the nitrogen atom (Ph at phosphorus) | TOF, s−1 | |
|---|---|---|---|---|
| Ph | 590 |
|
||
| Bn | 7 | Ph | 590 | |
| Me | 1540 | Bn | 5 | |
| n-Bu | 46 | C6H4Me | 590 | |
| 2-phenylethyl | 31 | C6H4OMe | 310 | |
| 2,4,4-trimethylpentyl | <1 | C6H4Br | 740 | |
| Cy | 720 | C6H4CF3 | 95 | |
| R ’ (Ph at phosphorus) | TOF, s −1 | CH2P(O)(OEt) | 500 | |
| Lys | 3350 | Substituents at the nitrogen atom (o-Py at phosphorus) | TOF, s −1 | |
| Glu | 3100 | |||
| Asp | 2600 | p-Tolyl | 3050* | |
| C6 | 740–7700 | Benzyl | 5200* | |
| C14 | 98 000–1 500 000 (various solvents) | Benzhydryl | 15 200 |
Immobilized heterogeneous catalysts with a metal catalytic center modelling hydrogenase
Among disadvantages of the homogeneous catalysts are difficulties of their separation and reuse, time instability (especially in an acidic medium), as well as problems appearing during subsequent scaling and tests in actual fuel cells, where catalyst immobilization in the membrane-electrode block is required. However, despite the advantages, many diiron or nickel complexes are limited by their low solubility in aqueous solutions, thus requiring efforts to solubilize the catalyst by either introducing a hydrophilic moiety to the ligand of catalyst or embedding the catalyst into a supramolecular cage, such as an amphiphilic polymer or a protein matrix, or immobilization onto or on a solid support with a high surface area [carbon nanotubes, metal-organic framework (MOF) or a mesoporous silica (MS)] [61], [62], [63], [64], [65], [66], [67], [68], [69], [70], [71]. Usually, these immobilized catalysts have limited turnover numbers (TONs) for H2 evolution (TONs were ∼6 for MOF and ∼18 for MS), although the TONs were better than those for the corresponding homogeneous catalysts [61], [62], [63], [64], [65], [66], [67], [68], [69], [70], [71]. Based on the abovementioned Ni complexes with diazadiphosphocyclooctane, the researchers from Fontecave and Artero group have developed the electrocatalyst material via its grafting on the nanotube surface (Figs. 1 and 4) [72], [73], [74]. Unexpectedly, it was found that the grafting of the bioinspired catalyst to the carbon nanotube substantially improved its stability and thus its reactivity in both reactions, though the reasons of this phenomenon are not clear yet. It may be supposed that carbon-conducting nanotubes somehow imitate the linear electric circuit with electron-transport iron-sulfur clusters, which link the deepened active areas within the enzymes with the protein surface. The mentioned researchers have manufactured the MEB via the deposition on the Nafion membrane of multiwall carbon nanotubes (MCNTs) with subsequent grafting of nickel complexes with a diazadiphosphine ligand to the deposited nanotubes. Measurements were performed only in electrochemical conditions, in the medium of 0.5 M solution of H2SO4. The current density of 3 mA/cm2 at an overvoltage of 300 mV was realized [72]. Subsequently, these studies reported on the similar scheme, in which the catalyst was connected with the nanotube not via a covalent link, but via π-stacking interaction of pyrene with the carbon nanotube [73]. In the performed electrochemical tests, this catalyst also demonstrated improved effectiveness; however, tests in the fuel cell were not performed.
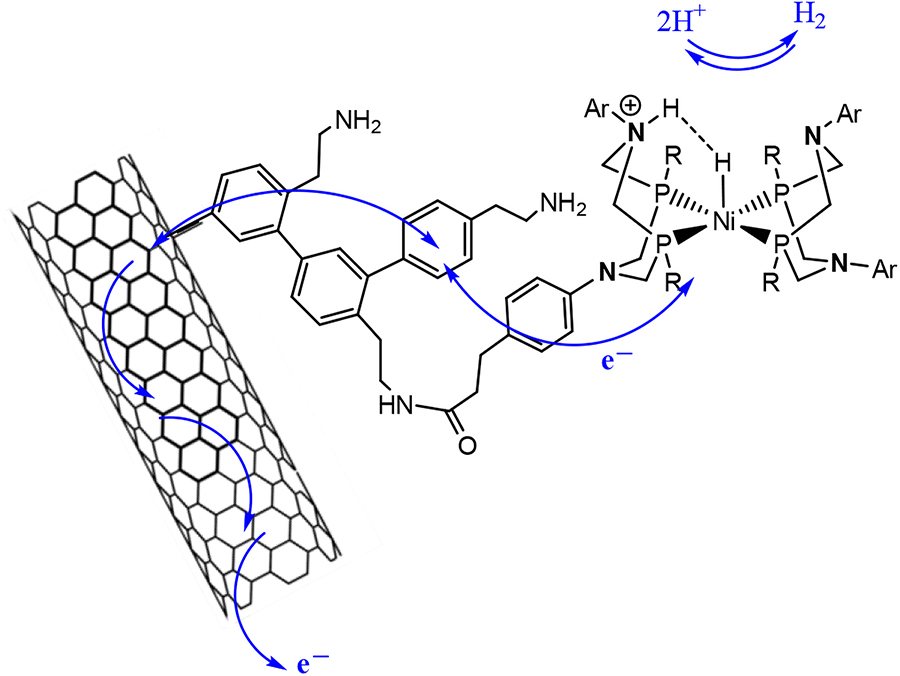
Schematic representation of the structure and reactivity of the bioinspired nickel catalyst grafted on a carbon nanotube. Electrons are exchanged between the carbon nanotube and the Ni centers where H+ is reduced to H2 or H2 oxidized to H+.
Recently, the [Ni(PPh 2NPhCH2COOH 2)2](BF4)2 complex has been studied under heterogeneous electrocatalysis in an aqueous solution by grafting the nickel complex onto the surface of mesoporous TiO2. The obtained results show that this compound displays good catalytic properties under homogeneous conditions and undergoes rapid deactivation (via [Ni(PPh 2NPhCH2COOH 2)2](BF4)2 outwashing from the electrode surface modified by TiO2), leading to inefficient catalysis under heterogeneous conditions [71].
Incorporation of the synthetic catalyst into protein or another macromolecule with a view to create the desired structure of the active center is a difficult task. In one of the cases within this approach, the Fe2S2 complex was incorporated into the nitrobindin protein (Fig. 1) [64]. Nitrobindin represents the β-barrel-protein, which usually links gem within its large internal space. The Fe2S2 complex was incorporated as a substituent of gem by covalent link via the maleimide fragment on the catalyst with the introduced Cys residue (Fig. 1). In the reaction of H2 evolution induced by photosensitizer, improved reactivity was realized (TON 130) as compared with the Fe2S2 complex [64]. Further attempts to obtain the effective and convenient heterogeneous biomimetic catalyst via different immobilizing techniques and on different substrates are in progress [75–82].
Mechanism of hydrogen transformations
In view of the high catalytic reactivity of [Ni(PR 2NR′2)2]2+ complexes in hydrogen evolution reaction and in the counter hydrogen oxidation reaction, the mechanism of such reactions has been studied in details via the application of experimental and theoretical approaches. In the proposed mechanism (Scheme 1), electrocatalysis is initiated by the reduction of NiII to NiI, which subsequently may be protonated at a pendant amine or nickel center. Thus, the creation of the NiI state is the key stage of the catalytic reaction. In order to establish the NiI state protonation area of the complex in situ, electrochemical synthesis of NiI complexes was performed [59], [60]. The protonation reaction of Ni1+(PPh 2NR 2)2 in the presence of DMFH+ as a proton source has been investigated by cyclic voltammetry, ESR, UV/Vis, and NMR spectroscopy. The results of these studies have shown that the subsequent stage of the catalytic cycle strongly depends on the nature of substituents within the principal ligand center (Scheme 1). For the case of ligands with aromatic substituents attached to nitrogen atoms, it was shown that the attachment of a proton to Ni1+(PPh 2NR 2)2 occurred according to the ECCE-type process (where E – electrochemical, C – chemical pathways). In other words, during the catalytic reaction, progressive reduction of the Ni(I) complex with Ni0 formation occurs, after which double protonated and hydrogen evolution stages are realized. However, in the case of complexes with benzyl substituents, this process is complicated by the chemical stage of interaction between protonated and unprotonated NiI species in the acidic medium. This interaction enables the formation of NiII-NH+-species from NiI, as well as the regeneration of the Ni(II) form of the catalyst through intermolecular electron transfer in the presence of DMFH+. In the case of such complexes, the proposed catalytic cycle belongs to the CEEC type [59], [60]. In turn, the replacement of the phenyl substituent for the pyridyl one in the cyclooctane ligand exerts a strong effect on the appearance of cyclic voltammograms of complexes in the presence of the proton donor and thus on the catalytic reaction mechanism. In the case of [Ni(Po−Py 2NR 2)2]2+ complexes, in the presence of a proton donor, a substantial anodic shift of the reduction potential for the Ni(II/I) redox couple is observed in CVs, which is caused by the connection of the proton with the nitrogen pyridyl atom, while a substantial increase in the catalytic current is observed at potential corresponding to the peak reduction of the Ni(I/0) couple, which results in a substantial increase in the overvoltage value for the hydrogen evolution reaction as compared with the [Ni(PPh 2NR 2)2]2+ complex. As a result, after an increase in TOFs by more than 150 times, an undesirable rise of the overvoltage value almost by 600 mV occurs [41], [56].
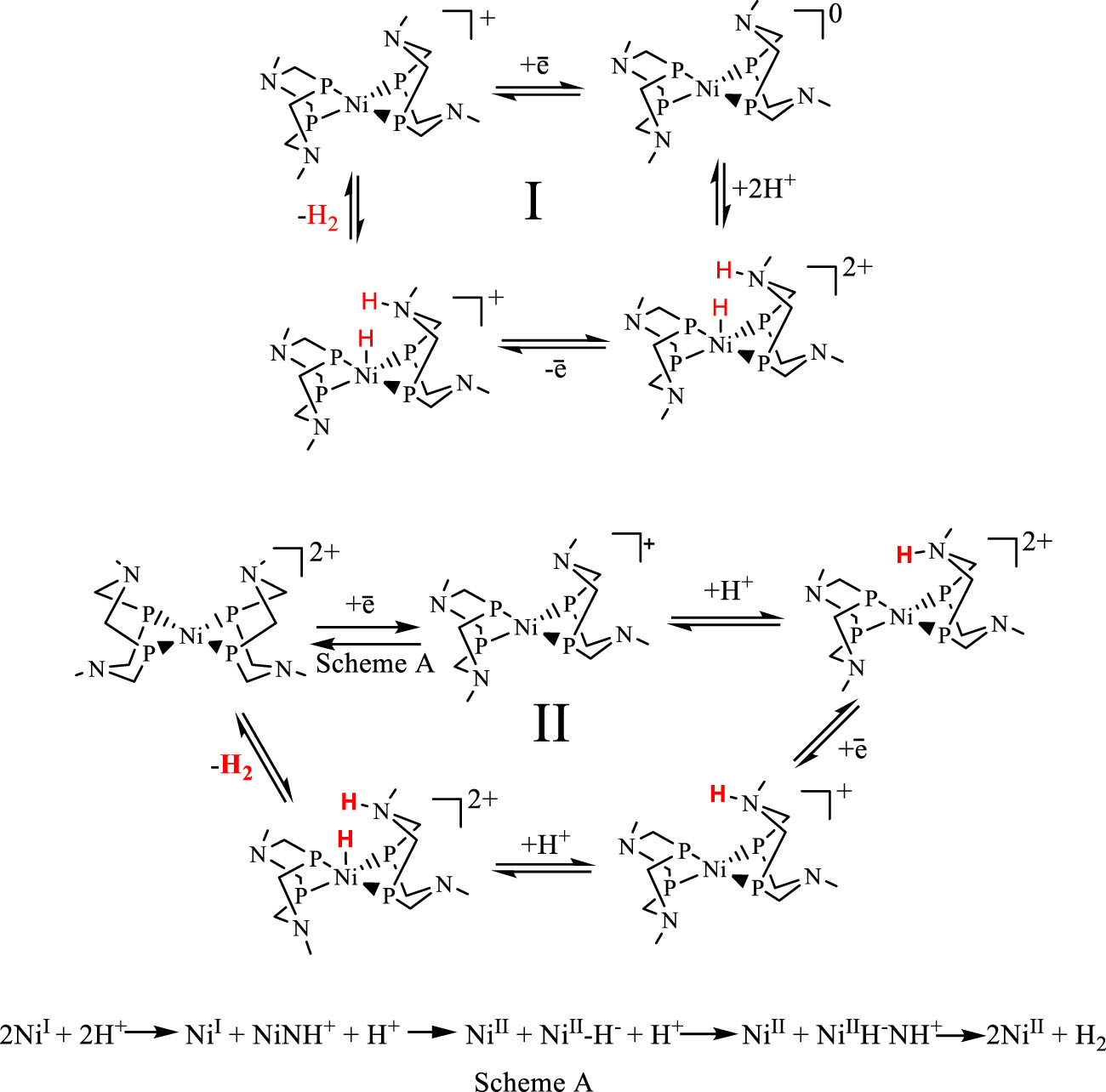
The proposed catalytic cycles by in situ generated Ni1+(PPh 2NR 2)2 (where R = p-Tolyl for I, R = benzyl for II).
It is of interest that some Ni complexes with diazadiphosphocyclooctane ligands may oxidize hydrogen in their own coordination sphere (Fig. 5). In other words, these are chemical reaction chameleons. The CVA method appeared to be the convenient express-method for testing hydrogen interaction reaction with nickel complexes [58]. During hydrogen passing through the solutions of different complexes, all other conditions being equal, it was found that some complexes actively reacted with hydrogen, which reduced the catalyst in such a way that the peak of the latter in the voltammogram completely disappeared. Some complexes do not react with hydrogen during a long period of time (up to several hours). The EPR method made it possible to find the formation of Ni(I) complexes in these reactions. At that, the lower the reduction potential and the less the electrochemical gap, the higher the catalytic reactivity. From the authors’ investigations of Ni complexes with diazadiphosphocyclooctane ligands, it follows that similarly to the case with hydrogenases, the possibility of creation of the stable Ni(I) state is of paramount importance for catalytic reactivity. These are the structures stabilizing the Ni(I) state, which are able to activate molecular hydrogen. In Fig. 5, the most effective complexes in hydrogen oxidation reaction are presented.
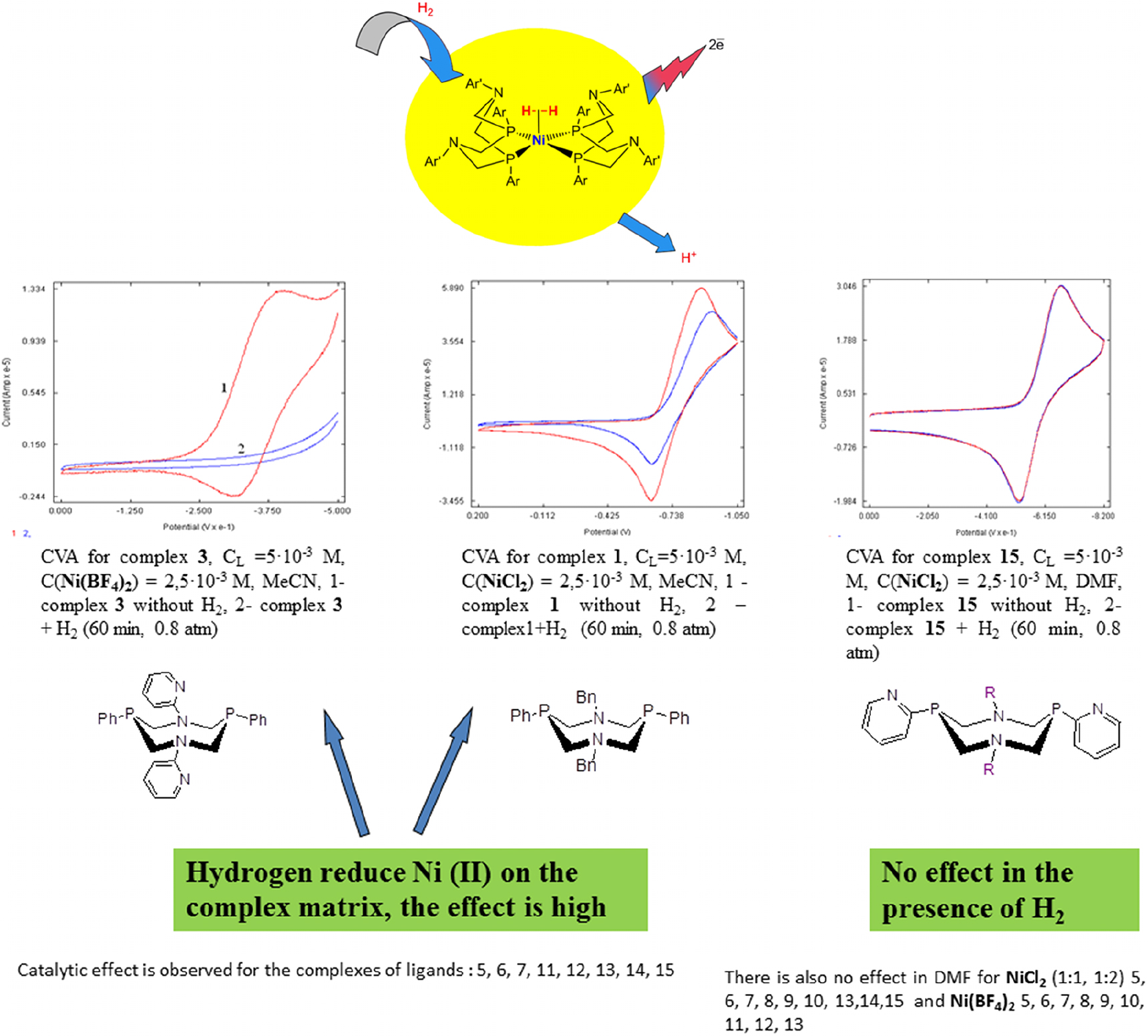
Testing of Ni complexes with P, N-cyclic ligands (from Fig. 3) in the hydrogen H2 oxidation.
The supposed mechanism includes the stages of nickel reduction, as well as nickel hydride and protonated nitrogen ligand formation. At that electrons are emitted, that is, the current is generated. It means that the system will operate and generate electric power.
Because of high abundance and low toxicity, iron is a particularly attractive metal for incorporation into synthetic catalysts for the production and oxidation of H2. The investigation of mono- and binuclear iron complexes within a nearby diphosphine environment for the hydrogen oxidation reaction made it possible to establish that the pendant amines could play an important role in relaying protons between the metal and the solution, in coupling proton and electron transfers and in activating dihydrogen. Nevertheless, as it was demonstrated for iron derivatives with pendant amines, the electrocatalytic reactivity for H2 formation is often limited by low rates, low stability, and/or high overpotentials [15].
The electrocatalytic properties for hydrogen evolution have been demonstrated for iron complexes with a framework of redox non-innocent bis(imino)acenaphthenes (BIANs) [83]. Catalysis occurs at FeIII/II reduction couple potential. In the presence of strong acid (DMFH+), the overpotential value for hydrogen evolution is very low and is equal to 0.17–0.19 V at TOF values of 37–460 s−1. The best catalytic reactivity for the complex with methoxy substituents on BIAN was observed.
The authors assumed it would be possible to involve the iron complexes with diazadiphosphocyclooctane ligands in hydrogen catalysis by entering the additional redox active ligand into the iron coordination sphere. Iron complexes bearing diphosphine ligands with positioned pendant amines along with a non-innocent dpp-BIAN ligand prepared in situ were investigated by cyclic voltammetry, which is the usual way to demonstrate electrocatalytic ability for hydrogen evolution [84]. Cyclic voltammograms (CVs) in CH3CN were recorded in the presence of increasing amounts of acids (Fig. 6). The catalytic reactivity of these new mixed ligands iron complexes at low overpotential was demonstrated. The catalysis potential for all complexes is around −0.5 V ref Fc+/Fc in THF, so it is very low overpotential of hydrogen evolution [84].
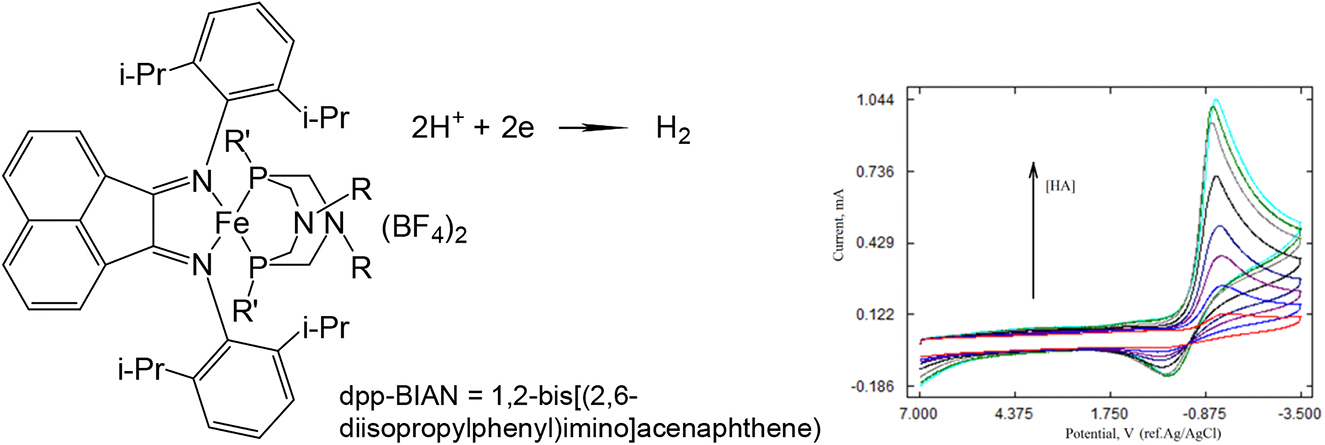
Structure of iron complexes bearing diphosphine ligands with positioned pendant amines along with non-innocent dpp-BIAN ligand and CVs of complex in the presence of increasing acid concentration.
Ni complexes of thiophosphorylated calixarenes for catalytic HER
Calixarenes are universal platforms for designing and synthesizing molecular receptors and multivalent ligands, which may imitate biological functions. It was shown that complexes of thiophosphorylated calixarenes and nickel (II) could also act as a model of the active center of the hydrogenase due to the availability of four P = S enzymes at the low rim of the molecule. Catalytic reactivity in the reaction of hydrogen evolution from acids has been determined for these complexes and their effectiveness and factors determining this effectiveness have been evaluated [87].
Calixarenes modified by thiophosphoryl fragments were selected as ligands, since they are characterized by the required ability for complex formation and are stable in the conditions of hydrolysis and electrochemical oxidation. Cyclic voltammograms of nickel complexes surrounded by such resorcinol fragments in the presence of a proton source show a catalytic increase in current and a linear dependence of the peak catalytic current versus acid concentration [87]. It was established that the catalytic pattern of the reduction of protons at CVA depends on calixarene conformation. Cone conformation creates point particles of catalyst and probably creates more favorable conditions for catalysis; energy gain exceeds 1.4 V. The use of such nickel complexes as hydrogen evolution catalysts (trifluoroacetic acid was used as a proton source) provides a decrease in the potential of direct reduction of acid on electrode, thus facilitating hydrogen reduction by 0.7–1.2 V [87]. The value of catalytic efficiency (as an alternative of the TOF value for such complexes) was evaluated. Since catalytic efficiency in some cases exceeds 0.75, these catalysts are considered as strong ones for hydrogen evolution [87]. The further modification of the upper rim of thiphosphorylated ligands by amino groups or the functionalization by triazole-containing derivatives provides catalytic hydrogen evolution at more positive potentials (as compared to the unmodified precursors), which corresponds to a higher energy gain for the potential as compared to the heterogeneous acid reduction on the electrode.
Heterogeneous organometallic polymer catalysts for hydrogen catalysis
Metal-organic frameworks (MOFs) represent the class of porous materials with the unique properties, which in recent years attract increasing attention to their practical application connected with hydrogen generation (in the course of water decomposition or reduction of proton donor sources) due to the remarkable flexibility of their design, the super large surface/volume ratios and adjustable porous channels (similar to actual hydrogenases) [89]. Especial interest is attracted by bimetallic coordination polymers similar to bimetallic hydrogenase functions. Based on the original redox-active organometallic coordination polymers designed on the basis of ferrocenyl-, arylphosphine and metal (Co, Zn and others)-bipyridine block, the new prospective catalysts for hydrogen energetics have been designed [90] (Fig. 7). These compounds are characterized by the reversible redox-active reactions connected with the ferrocenyl fragment. It was first determined that these coordination polymers (CPs) were effective as electrocatalysts for the reduction of protons to hydrogen. It was established that Co 1D-coordination polymer (2) is characterized by the catalyst regeneration rate of 300 s−1 in the case of N,N-dimethyl formamide ([DMF(H)+]) use as the acid in the acetonitrile solution, which corresponds to one of the highest rates registered for any electrocatalyst from CP in this solvent. Such a high catalytic regeneration rate is achieved at the expense of the overvoltage of 820–840 mV at catalysis potential. As the catalyst for hydrogen evolution reaction (HER), CP in 0.5 M H2SO4 had the following characteristics: η10 overvoltage equal to 340 or 450 mV, onset overvoltage of 220 or 300 mV (rel. RHE), Tafel slope of 110 or 120 mV/decade for 1 and 2, respectively, and substantial time stability during hydrogen evolution reaction (HER). These CPs may continuously operate during 1000 cycles with a minor loss of reactivity. TOFs were evaluated in 0.5 M H2SO4. TOFs (at 300 mV) for 1 and 2 were equal to 4.5 10−3 and 1.5 10−3 s−1, respectively [90]. Though these values do not correspond to the highest reactivity in aqueous solutions of H2SO4, such reactivity is rather satisfactory as compared to the known organometallic structures. For the redox-active coordination polymers of Zn and Co based on the ferrocene-containing diphosphinate ligand, such reactivity was observed for the first time. In general, these encouraging functional characteristics allow relying on the subsequent improvement of catalysts and their application in actual fuel cells. 3D Ni and Co redox-active metal-organic frameworks based on ferrocenyl diphosphinate and 4,4′-bipyridine ligands are efficient electrocatalysts for hydrogen evolution reaction [91]. An additional protonation-capable 4,4′-bpy linker significantly changes the catalytic properties in the HER in organic and aqueous acidic media compared with 1D polymers based on ferrocene-containing diphosphinate [90]. Therefore, Ni-based polymer surpasses the previously described polymers in terms of stability and long-term durability at low overvoltages of 350 mV and Tafel slope 60 mV dec−1. There was a rare opportunity to compare 1D and 3D polymers, differing in one linker. For Co-based 3D polymer, there is a significant decrease in overvoltage by ∼440 mV in comparison with the 1D CofcdHp polymer in an organic medium and by 50 mV in an aqueous acidic medium. The Tafel slope changes from 120 to 65 mV dec−1 when moving from 1D CofcdHp to a 3D structure. Thus, these new 3D Ni and Co redox-active MOFs based on ferrocenyl diphosphinate and 4,4′-bipyridine ligands demonstrate excellent catalytic properties in HER and are quite stable catalysts [91].
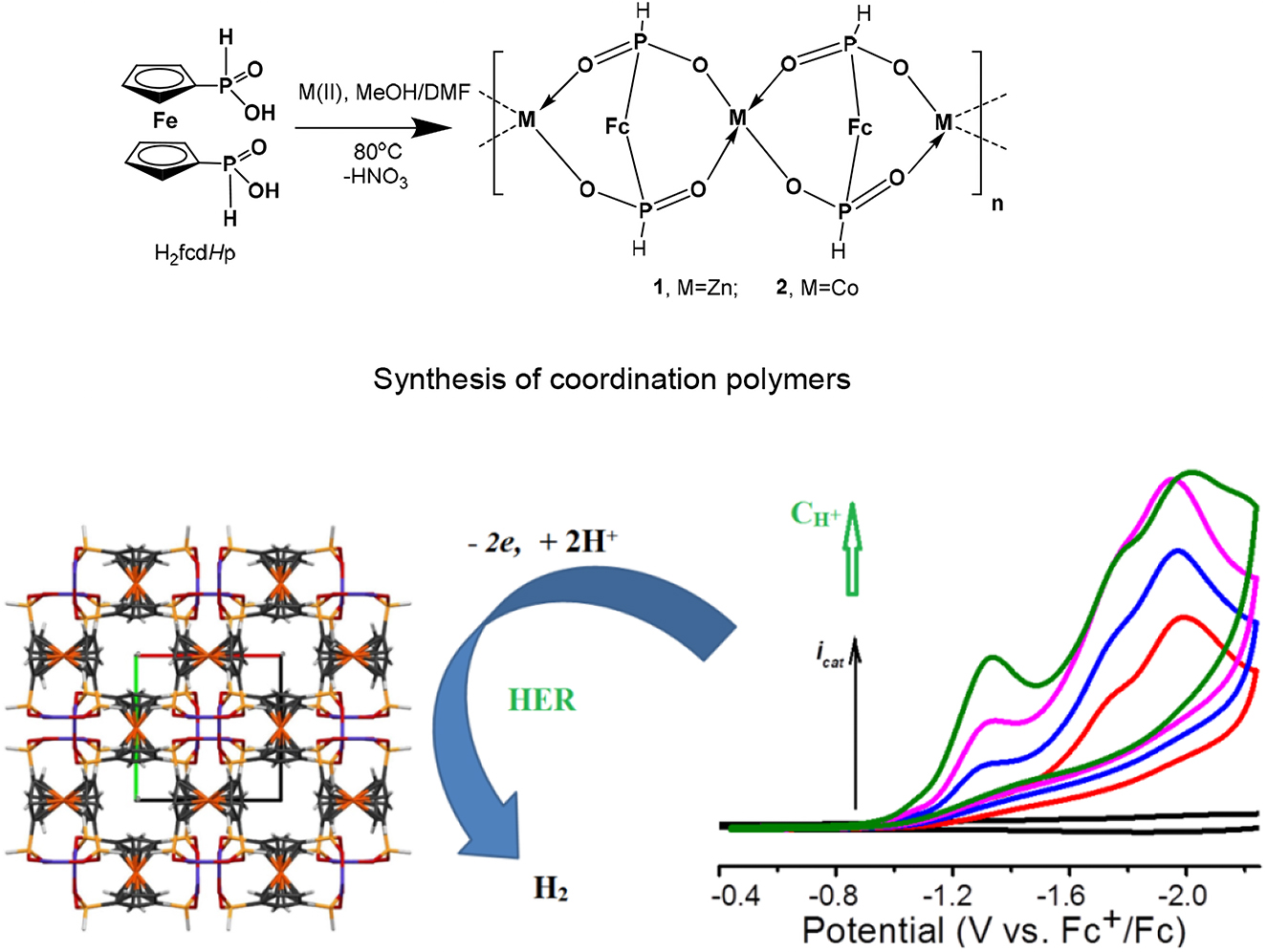
Zn and Co redox active coordination polymers based on a ferrocene-containing diphosphinate ligand as efficient electrocatalysts for the hydrogen evolution reaction.
Fuel cell based on hydrogenase mimics
The next stage of investigations includes tests of catalysts in actual fuel cells, as both the cathode and anode catalysts in a polymer-electrolytic cell element with a membrane-electrode block (a membrane with gas-diffusion and catalytic layers on both sides). Currently, the catalyst cost amounts to 40% of the FC cost. Generally, platinum is used. As it was noticed earlier, besides high prices of platinum, there exists a problem pertinent to the limited reserves of this precious metal. Therefore, it is relevant to either completely or at least partly replace Pt for the widespread transition metals, such as metals of the Ni subgroup.
It was found that the Ni(PPh 2N p−Tol 2)2](BF4)2 complex was operative on the cathode side [85], where the maximum current density of 16 mA cm−2 and the maximum power density of 0.664 mW cm−2 were obtained.
Ni(PPh 2N p−Tol 2)2](BF4)2 proved to be more effective both on the cathodic and anodic side (Fig. 8) [86]. Its application as the anode catalyst in the conditions of Pt/C use as the cathode allows realizing the power density of PEFC equal to 14.66 mW cm-2, which exceeds the characteristics of many similar devices all around the world, containing only basic metals. The maximum power density for Ni(PPh 2NR 2)2](BF4)2 is equal to 11.84 mW cm-2. It was established that the Ni(I) state in this case was also the key one for the reaction development and it was fixed by the EPR method. The mechanism of hydrogen oxidation in the fuel cell was proposed by [86].
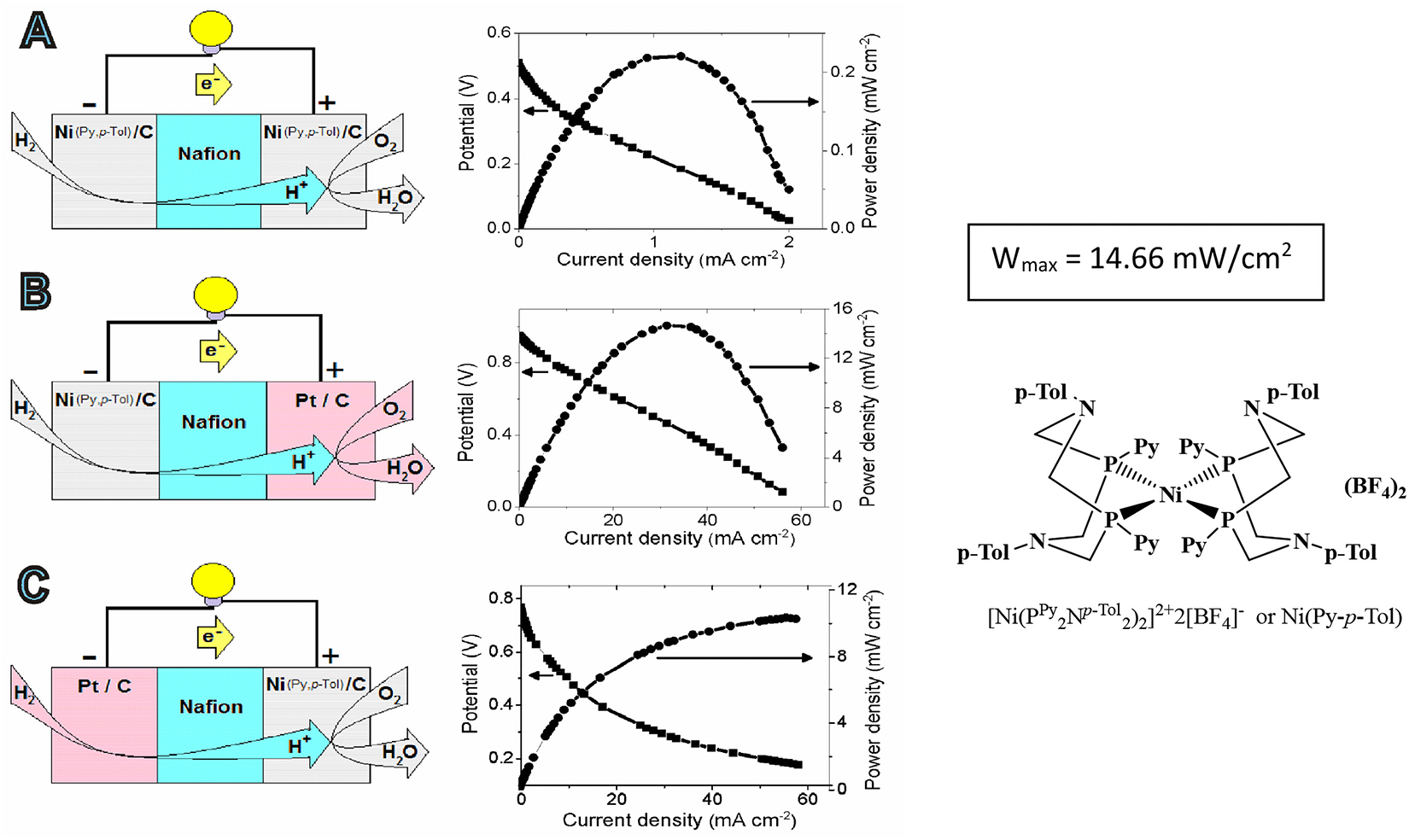
Representation of the H2/O2 PEMFC and polarization and power curves at 80 °C for A) Ni(Py-p-Tol)/Nf/Ni(Py-p-Tol), B) Ni(Py-p-Tol)/Nf/Pt, and C) Pt/Nf/Ni(Py-p-Tol).
For similar conditions, nickel complexes of thiophosphorylated calix[4]resorcines exhibited catalytic reactivity in the oxygen reduction reaction within the fuel cell with polymeric electrolyte (Fig. 9) [88]. It proved to be that conformation of the macrocyclic ligand also determines morphology and catalytic properties. The power density of complex with chair conformation, immobilized in membrane-electrode ensemble, in the oxygen reduction reaction within the fuel cell on anode is more than 5 times as larger. FC diagnostic characteristics with cathode represented by organometallic catalyst with chair conformation are substantially better as compared to the characteristics of catalyst with cone conformation. It was found that the number of electrons transmitted to ORR was equal to 2.1; this reaction amounted to 95% and was realized following the two-electron mechanism with the creation of peroxide. At that, these complexes do not catalyze hydrogen reduction [88].
![Fig. 9:
Oxygen reduction reaction catalyzed by nickel complexes based on thiophosphorylated calix[4]resorcinols and immobilized in the membrane electrode assembly of fuel cells.](/document/doi/10.1515/pac-2019-1207/asset/graphic/j_pac-2019-1207_fig_009.jpg)
Oxygen reduction reaction catalyzed by nickel complexes based on thiophosphorylated calix[4]resorcinols and immobilized in the membrane electrode assembly of fuel cells.
In search of non-platinum catalysts for FCs, used both in hydrogen oxidation reaction (HOR) and in oxygen reduction reaction (ORR), nickel complexes of pectin polysaccharides – stable natural polymers have been studied (Fig. 10) [92]. These complexes poorly dissolved in water, where they only plumped and were immobilized on the membrane, thereby providing the stability of diagnostic characteristics of these catalysts within the membrane-electrode block (MEB) of the FC. The basic carbohydrate chain of pectin polysaccharides is composed of 1,4-bonded residues of α-D-galacturonic acid, complexes of which with ions of transition metals form a three-dimensional network with a dense location of metal ions, being the catalytic nodes of ORR and HOR. Pectin was received from orange peels. The scheme illustrating the creation of polymeric structures of the complex, having the eggbox form, is presented in Fig. 10. The authors have tested the effectiveness of the catalyst represented by the complex of nickel sodium pectate PG-NaNi with 25% replacement of sodium by nickel on the anode of H2/O2 FCPE in the membrane-electrode block (MEB). The diagnostic characteristics of this PG-NaNi/Nf/Pt FC on the anode and with the commercial Pt catalyst on the cathode distinctly depended on temperature – increasing the cell temperature resulted in their rise. The maximum current density of 59 mA cm−2 and the maximum power density of 5.9 mW cm−2 were obtained; though these values are somewhat lower than the similar characteristics for the completely platinum FC, nevertheless the development of environmentally suitable and stable cathodic catalysts manufactured from pectin, which is an inexpensive and readily available biologic material, may be continued.
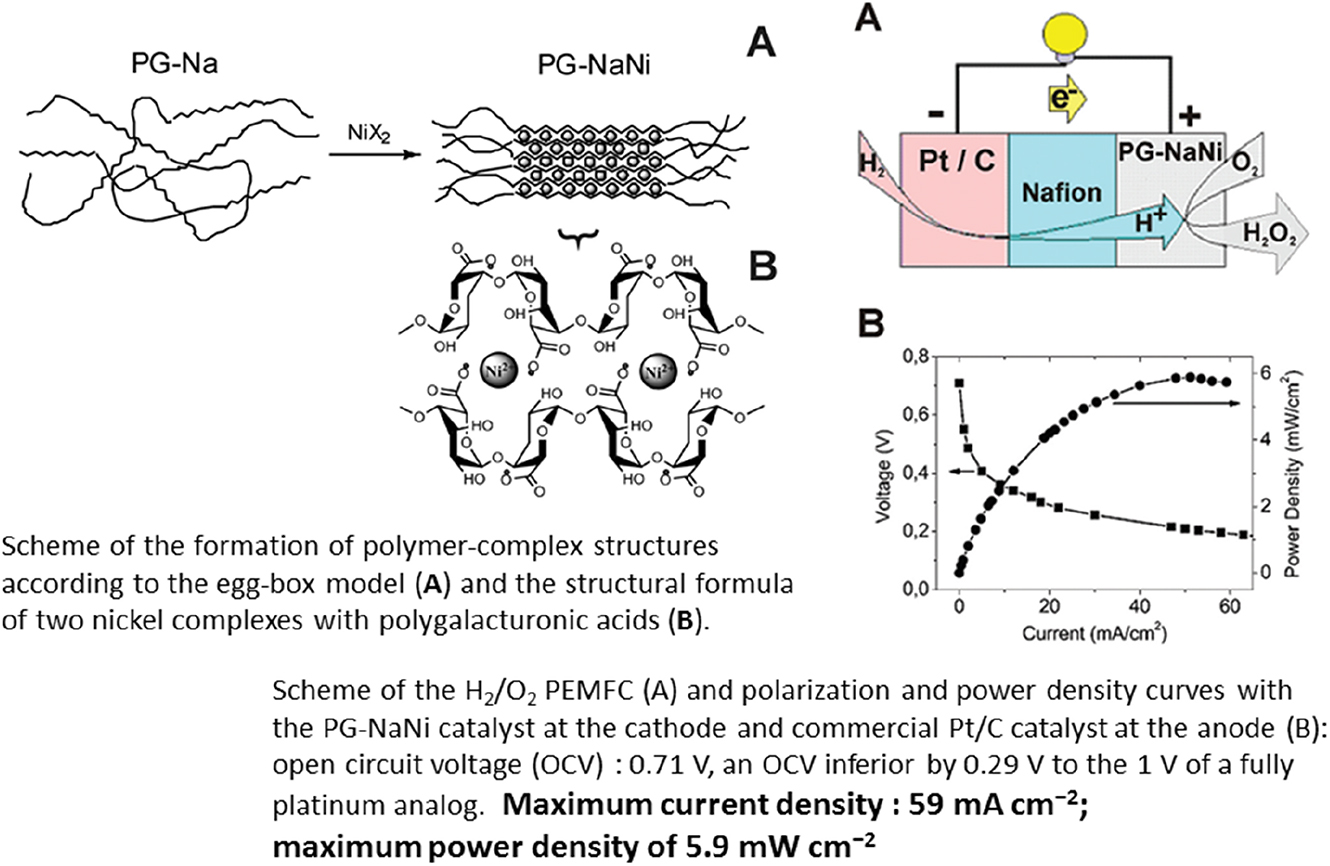
Nickel-based pectin coordination polymer as an oxygen reduction reaction catalyst for proton exchange membrane fuel cells.
Conclusions
To conclude, the need in new effective and inexpensive chemical catalysts as part of the newly formed “green” field of chemistry is increased. Living organisms provide a lot of different enzymes with high catalytic reactivity, which may be either directly used in biotechnological synthetic systems or may be imitated by chemists. One of the important approaches used for the creation of synthetic catalytic systems is based on the simulation of the active sites of a biological enzyme. This may be demonstrated by the example of a molecular electrode, i.e., a constructed matrix including a redox-active metal center and a suitable ligand environment, at which electrocatalytic processes may occur and which are imitating the operation of hydrogenases and metalloenzymes involved in hydrogen metabolism; this molecular electrode is characterized by extraordinary electrocatalytic properties, being important for hydrogen production and oxidation, as well as for other complex oxidation-reduction processes. The studied bioinspired catalysts, which are operating as nanoreactors, are suitable for application in fuel cells and redox-process technologies. Catalysts, which have been included in this review, were based on the basic widely occurring metals, while such an approach is very important for hydrogen energetics problems.
Funding source: Russian Foundation for Basic Research
Award Identifier / Grant number: 19-03-00084
Acknowledgments
The authors gratefully acknowledge the CSF-SAC FRC KSC RAS for the spectral (NMR, ESR, UV, etc.) studies.
-
Funding: The reported study was funded partially by RFBR according to the research project№ 19-03-00084, namely homogeneous catalysis section.
References
[1] E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel, E. Reisner. Chem. Rev. 119, 2752, 2019, https://doi.org/10.1021/acs.chemrev.8b00392.Search in Google Scholar PubMed PubMed Central
[2] Z. Wang, C. Li, K. Domen. Chem. Soc. Rev. 48, 2109 (2019), https://doi.org/10.1039/c8cs00542g.Search in Google Scholar PubMed
[3] D. A. J. Rand, R. M. Dell. Hydrogen Energy: Challenges and Prospects, Royal Society of Chemistry: Cambridge, UK (2007).Search in Google Scholar
[4] T. B. Liu, D. L. DuBois, R. M. Bullock. Nat. Chem. 5, 228 (2013), https://doi.org/10.1038/nchem.1571.Search in Google Scholar PubMed
[5] K. T. Møller, T. R. Jensen, E. Akiba, H. Li. Progr. Nat. Sci. Mater. Int. 27, 34 (2017).10.1016/j.pnsc.2016.12.014Search in Google Scholar
[6] W. Zhang, W. Z. Lai, R. Cao. Chem. Rev. 117, 3717 (2017), https://doi.org/10.1021/acs.chemrev.6b00299.Search in Google Scholar PubMed
[7] J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler, H. B. Gray. Chem. Sci. 5, 865 (2014), https://doi.org/10.1039/C3SC51711J.Search in Google Scholar
[8] O. S. Burheim. In Engineering Energy Storage, chapter 8, pp. 147–192 (2017).10.1016/B978-0-12-814100-7.00008-0Search in Google Scholar
[9] A. J. Martín, A. Hornés, A. Martínez-Arias, L. Daza. Renewable Hydrogen Technologies: Production, Purification, Storage, Applications and Safety, Elsevier: Amsterdam, chapter 15, pp. 361–380 (2013).10.1016/B978-0-444-56352-1.00015-5Search in Google Scholar
[10] P. Millet, S. Grigoriev. Renewable Hydrogen Technologies, Elsevier: Amsterdam, chapter 2, pp. 19–41 (2013).10.1016/B978-0-444-56352-1.00002-7Search in Google Scholar
[11] D. Bessarabov, P. Millet. PEM Water Electrolysis. Hydrogen Energy and Fuel Cells Primers, Elsevier: chapter 4, pp. 75–115 (2018).10.1016/B978-0-12-811145-1.00004-6Search in Google Scholar
[12] A. J. Cruden. Preface Context Hydrogen Fuel Cells 4, 1 (2012).10.1016/B978-0-08-087872-0.00401-7Search in Google Scholar
[13] U. Desideri, S. Ubertini. Appl. Energy 256, 113906 (2019), https://doi.org/10.1016/j.apenergy.2019.113906.Search in Google Scholar
[14] Hydrogen roadmap Europe. A Sustainable Pathway for the European Energy Transition. Luxembourg: Publications Office of the European Union, p. 75 (2019).Search in Google Scholar
[15] V. V. Khrizanforova, A. A. Karasik, Y. H. Budnikova. Russ. Chem. Rev. 86, 298 (2017), https://doi.org/10.1070/RCR4676.Search in Google Scholar
[16] W. Lubitz, H. Ogata, O. Rudiger, E. Reijerse. Chem. Rev. 114, 4081 (2014), https://doi.org/10.1021/cr4005814.Search in Google Scholar PubMed
[17] M. Haumann, S. T. Stripp. Acc. Chem. Res. 51, 1755 (2018), https://doi.org/10.1021/acs.accounts.8b00109.Search in Google Scholar PubMed
[18] J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King, M. W. Adams. Biochim. Biophys. Acta Mol. Cell Res. 1853, 1350 (2015), https://doi.org/10.1016/j.bbamcr.2014.11.021.Search in Google Scholar PubMed
[19] J. W. Peters, W. N. Lanzilotta, B. J. Lemon, L. C. Seefeldt. Science 282, 1853 (1998), https://doi.org/10.1126/science.282.5395.1853.Search in Google Scholar PubMed
[20] M. E. Ahmed, A. Dey. Curr. Opin. Electrochem. 15, 155 (2019).10.1016/j.coelec.2019.05.009Search in Google Scholar
[21] A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse, W. Lubitz. Angew. Chem. Int. Ed. 51, 11458 (2012), https://doi.org/10.1002/anie.201204800.Search in Google Scholar PubMed
[22] C. Papini, C. Sommer, L. Pecqueur, D. Pramanik, S. Roy, E. J. Reijerse, F. Wittkamp, V. Artero, W. Lubitz, M. Fontecave. ACS Catal. 95, 4495 (2019).10.1021/acscatal.9b00540Search in Google Scholar
[23] F. Wittkamp, M. Senger, S. T. Stripp, U.-P. Apfel. Chem. Commun. 54, 5934 (2018), https://doi.org/10.1039/C8CC01275J.Search in Google Scholar
[24] J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza, Y. Nicolet. Chem. Rev. 107, 4273 (2007), https://doi.org/10.1021/cr050195z.Search in Google Scholar PubMed
[25] M. Watanabe, Y. Honda, H. Hagiwara, T. Ishihara. J. Photochem. and C. Photobiol. Photochem. Rev. 33, 1 (2017), https://doi.org/10.1016/j.jphotochemrev.2017.09.001.Search in Google Scholar
[26] T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave, V. Artero. Coord. Chem. Rev. 270-271, 127 (2014), https://doi.org/10.1016/j.ccr.2013.12.018.Search in Google Scholar
[27] D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer, T. B. Rauchfuss. Chem. Rev. 116, 8693 (2016), https://doi.org/10.1021/acs.chemrev.6b00180.Search in Google Scholar PubMed PubMed Central
[28] L. -C Song. Acc. Chem. Res. 38, 21 (2005), doi: https://doi.org/10.1021/ar030004j.Search in Google Scholar PubMed
[29] Y. Li, T. B. Rauchfuss. Chem. Rev. 116, 7043 (2016), https://doi.org/10.1021/acs.chemrev.5b00669.Search in Google Scholar PubMed PubMed Central
[30] S. Gao, Q. Liang, Q. Duan, D. Jiang. J. Zhao. Int. J. Hydrogen Energy 43, 7245 (2018), https://doi.org/10.1016/j.ijhydene.2018.03.010.Search in Google Scholar
[31] C. Tard, C. J. Pickett. Chem. Rev. 109, 2245 (2009), https://doi.org/10.1021/cr800542q.Search in Google Scholar PubMed
[32] F. Gloaguen. Inorg. Chem. 55, 390 (2016), https://doi.org/10.1021/acs.inorgchem.5b02245.Search in Google Scholar PubMed
[33] I. Bhugun, D. Lexa, J.-M. Save´ant. J. Am. Chem. Soc. 118, 3982 (1996), https://doi.org/10.1021/ja954326x.Search in Google Scholar
[34] M. E. Carroll, B. E. Barton, T. B. Rauchfuss, P. J. Carroll. J. Am. Chem. Soc. 134, 18843 (2012).10.1021/ja309216vSearch in Google Scholar PubMed PubMed Central
[35] R. M. Bullock, M. L. Helm. Acc. Chem. Res. 48, 2017 (2015).10.1021/acs.accounts.5b00069Search in Google Scholar PubMed
[36] T. Liu, Q. Liao, M. O’Hagan, E. B. Hulley, D. L. DuBois, R. M. Bullock. Organometallics 34, 2747 (2015), https://doi.org/10.1021/om501289f.Search in Google Scholar
[37] T. Liu, S. Chen, M. J. O’Hagan, R. M. DuBois, R. M. Bullock, D. L. DuBois. J. Am. Chem. Soc. 134, 6257 (2012), https://doi.org/10.1021/ja211193j.Search in Google Scholar PubMed
[38] S. Raugei, M. L. Helm, S. Hammes-Schiffer, A. M. Appel, M. O’Hagan, E. S. Wiedner, R. M. Bullock. Inorg. Chem. 55, 445 (2016), https://doi.org/10.1021/acs.inorgchem.5b02262.Search in Google Scholar PubMed
[39] Q. Liao, T. Liu, S. I. Johnson, C. M. Klug, E. S. Wiedner, R. M. Bullock, D. L. DuBois. Dalton Trans. 48, 4867 (2019), https://doi.org/10.1039/C9DT00708C.Search in Google Scholar
[40] M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois, D. L. DuBois. Science 333, 863 (2011), https://doi.org/10.1126/science.1205864.Search in Google Scholar PubMed
[41] E. I. Musina, V. V. Khrizanforova, I. D. Strelnik, M. I. Valitov, Yu. S. Spiridonova, D. B. Krivolapov, I. A. Litvinov, M. K. Kadirov, P. Lönnecke, E. Hey-Hawkins, Yu. H. Budnikova, A. A. Karasik, O. G. Sinyashin. Chem. Eur. J. 20, 3169 (2014), https://doi.org/10.1002/chem.201304234.Search in Google Scholar PubMed
[42] B. A. Arbuzov, O. A. Erastov, G. N. Nikonov, R. P. Arshinova, I. P. Romanova, R. A. Kadyrov. Bull. Acad. Sci. USSR 32, 1672 (1983).10.1007/BF00954289Search in Google Scholar
[43] G. N. Nikonov, A. S. Balueva, O. A. Erastov, B. A. Arbuzov. Bull. Acad. Sci. USSR 38, 1223 (1989).10.1007/BF00957157Search in Google Scholar
[44] A. A. Karasik, R. N. Naumov, O. G. Sinyashin, G. P. Belov, H. V. Novikova, P. Lönnecke, E. Hey-Hawkins. Dalton Trans. 11, 2209 (2003), https://doi.org/10.1039/B300754E.Search in Google Scholar
[45] A. A. Karasik, G. N. Nikonov, B. A. Arbuzov, R. Z. Musin, Y. Ya. Efremov. Bull. Acad. Sci. USSR 40, 633 (1991).10.1007/BF00958010Search in Google Scholar
[46] A. A. Karasik, A. S. Balueva, E. I. Moussina, R. N. Naumov, A. B. Dobrynin, D. B. Krivolapov, I. A. Litvinov, O. G. Sinyashin. Heteroatom.Chem. 9, 125 (2008).10.1002/hc.20397Search in Google Scholar
[47] A. A. Karasik, R. N. Naumov, A. S. Balueva, Y. S. Spiridonova, O. N. Golodkov, H. V. Novikova, G. P. Belov, S. A. Katsyuba, E. E. Vandyukova, P. Lonnecke, E. Hey-Hawkins, O. G. Sinyashin. Heteroatom. Chem. 17, 499 (2006).10.1002/hc.20272Search in Google Scholar
[48] A. A. Karasik, A. S. Balueva, O. G. Sinyashin. Comptes Rendus Chimie 13, 1151 (2010), https://doi.org/10.1016/j.crci.2010.04.006.Search in Google Scholar
[49] E. I. Musina, A. A. Karasik, O. G. Sinyashin, G. N. Nikonov. Adv. Heterocyc. Chem. 117, 83 (2015), https://doi.org/10.1016/bs.aihch.2015.10.001.Search in Google Scholar
[50] A. A. Karasik, E. I. Musina, A. S. Balueva, I. D. Strelnik, O. G. Sinyashin. Pure Appl. Chem. 89, 293 (2017), https://doi.org/10.1515/pac-2016-1022.Search in Google Scholar
[51] E. I. Musina, A. S. Balueva, A. A. Karasik. Phosphines Prepar. React. Applic. RSC Catal. Ser. 37, 1 (2019).10.1039/9781788016988-00001Search in Google Scholar
[52] A. Jain, M. L. Reback, M. L. Lindstrom, C. E. Thogerson, M. L. Helm, A. M. Appel, W. J. Shaw. Inorg. Chem. 51, 6592 (2012), https://doi.org/10.1021/ic300149x.Search in Google Scholar PubMed
[53] C. M. Klug, A. Cardenas, R. M. Bullock, M. O’Hagan, E. S. Wiedner. ACS Catal. 8, 3286 (2018), https://doi.org/10.1021/acscatal.7b04379.Search in Google Scholar
[54] V. V. Khrizanforova, R. R. Fayzullin, E. I. Musina, A. A. Karasik, Y. H. Budnikova. Mendeleev Commun. 30, 302 (2020).10.1016/j.mencom.2020.05.013Search in Google Scholar
[55] U. J. Kilgore, M. P. Stewart, M. L. Helm, W. G. Dougherty, W. S. Kassel, M. R. DuBois, D. L. DuBois, R. M. Bullock. Inorg. Chem. 50, 10908 (2011), https://doi.org/10.1021/ic201461a.Search in Google Scholar PubMed
[56] V. V. Khrizanforova, E. I. Musina, M. N. Khrizanforov, T. P. Gerasimova, S. A. Katsyuba, Yu. S. Spiridonova, D. R. Islamov, O. N. Kataeva, A. A. Karasik, O. G. Sinyashin, Yu. H. Budnikova. J. Organometal. Chem. 789-790, 14 (2015), https://doi.org/10.1016/j.jorganchem.2015.04.044.Search in Google Scholar
[57] V. V. Khrizanforova, Yu. G. Budnikova, I. D. Strelnik, E. I. Musina, M. I. Valitov, M. K. Kadirov, A. A. Karasik, O. G. Sinyashin. Russ. Chem. Bull. 62, 1003 (2013), https://doi.org/10.1007/s11172-013-0131-0.Search in Google Scholar
[58] R. M. Galimullina, M. I. Valitov, Yu. S. Spiridonova, E. I. Musina, S. A. Krasnov, M. K. Kadirov, A. A. Karasik, Yu. G. Budnikova, O. G. Sinyashin. Russ. J. Phys. Chem. 85, 2214 (2011), https://doi.org/10.1134/S0036024411120119.Search in Google Scholar
[59] V. V. Khrizanforova, V. I. Morozov, A. G. Strelnik, Yu. S. Spiridonova, M. N. Khrizanforov, T. Burganov, S. A. Katsyuba, Sh. K. Latypov, M. K. Kadirov, A. A. Karasik, O. G. Sinyashin, Yu. H. Budnikova. Electrochim. Acta 225, 467 (2017), https://doi.org/10.1016/j.electacta.2016.12.081.Search in Google Scholar
[60] A. Kochem, M. O’Hagan, E. S. Wiedner, M. Gastel. Chem. Europ. J. 21, 10338 (2015), https://doi.org/10.1002/chem.201500954.Search in Google Scholar PubMed
[61] T. Himiyama, M. Waki, D. Esquivel, A. Onoda, T. Hayashi, P. Voort, S. Inagaki. ChemCatChem 10, 4894 (2018), https://doi.org/10.1002/cctc.201801257.Search in Google Scholar
[62] W.-N. Cao, F.-W. Wang, H.-Y. Wang, B. Chen, K. Feng, C.-H. Tung, L.-Z. Wu. Chem. Commun. 48, 8081 (2012), https://doi.org/10.1039/C2CC33097K.Search in Google Scholar
[63] F. Wang, M. Wen, K. Feng, W.-J. Liang, X.-B. Li, B. Chen, C. H. Tung, L.-Z. Wu. Chem. Commun. 52, 457 (2016), https://doi.org/10.1039/C5CC07499A.Search in Google Scholar PubMed
[64] A. Onoda, Y. Kihara, K. Fukumoto, Y. Sano, T. Hayashi. ACS Catal. 4, 2645 (2014), https://doi.org/10.1021/cs500392e.Search in Google Scholar
[65] G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero, M. Fontecave. Nature 499, 66 (2013), https://doi.org/10.1038/nature12239.Search in Google Scholar PubMed PubMed Central
[66] J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz, T. Happe. Nat. Chem. Biol. 9, 607 (2013), https://doi.org/10.1038/nchembio.1311.Search in Google Scholar PubMed PubMed Central
[67] J. F. Siegel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse, W. Lubitz. Biochemistry 54, 1474 (2015).10.1021/bi501391dSearch in Google Scholar PubMed
[68] F. Wang, W.-G. Wang, X.-J. Wang, H.-Y. Wang, C.-H. Tung, L.-Z. Wu. Angew. Chem. Int. Ed. 50, 3193 (2011), https://doi.org/10.1002/anie.201006352.Search in Google Scholar PubMed
[69] K. Sasan, Q. Lin, C. Mao, P. Feng. Chem. Comm. 50, 10390 (2014), https://doi.org/10.1039/c4cc03946g.Search in Google Scholar PubMed
[70] W. Wang, T. Yu, Y. Zeng, J. Chen, G. Yang, Y. Li. Photochem. Photobiol. Sci. 13, 1590 (2014), https://doi.org/10.1039/c3pp50446h.Search in Google Scholar PubMed
[71] G. Bergamini, M. Natali. Dalton Trans. 48, 14653 (2019).10.1039/C9DT02846CSearch in Google Scholar PubMed
[72] M. Fontecave, V. Artero. Comptes Rendus Chim. 14, 362 (2011), https://doi.org/10.1016/j.crci.2010.01.013.Search in Google Scholar
[73] P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave, V. Artero. Angew. Chem. Int. Ed. 50, 1371 (2011), https://doi.org/10.1002/anie.201005427.Search in Google Scholar PubMed
[74] E. Ahmed, S. Chattopadhyay, L. Wang, D. Brazzolotto, D. Pramanik, D. Aldakov, J. Fize, A. Morozan, M. Gennari, C. Duboc, A. Dey, V. Artero. Angew. Chem. Int. Ed. 57, 16001 (2018), https://doi.org/10.1002/anie.201808215.Search in Google Scholar PubMed
[75] C. Costentin, J. -M. Savéant. Curr. Opin. Electrochem. 15, 58 (2019).10.1016/j.coelec.2019.03.014Search in Google Scholar
[76] G. Bergamini, M. Natali. Dalton Trans. 48, 14653 (2019), https://doi.org/10.1039/C9DT02846C.Search in Google Scholar PubMed
[77] A. Ruff, S. Janke, J. Szczesny, S. Alsaoub, I. Ruff, W. Lubitz, W. Schuhmann. ACS Appl. Energy Mater. 24, 2921 (2019), https://doi.org/10.1021/acsaem.9b00269.Search in Google Scholar
[78] S. S. Nurttila, R. Becker, J. Hessels, S. Woutersen, J. N. H. Reek. Chem. Eur. J. 24, 16395 (2018), https://doi.org/10.1002/chem.201803351.Search in Google Scholar PubMed PubMed Central
[79] A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin, M. Fontecave. Science 326, 1384 (2009), https://doi.org/10.1126/science.1179773.Search in Google Scholar PubMed
[80] D. Balestri, Y. Roux, M. Mattarozzi, C. Mucchino, L. Heux, D. Brazzolotto, V. Artero, C. Duboc, P. Pelagatti, L. Marchiò, M. Gennari. Inorg. Chem. 56, 14801 (2017), https://doi.org/10.1021/acs.inorgchem.7b01824.Search in Google Scholar PubMed
[81] A. Ruff, F. Conzuelo, W. Schuhmann. Nat. Catal., (2019), https://doi.org/10.1038/s41929-019-0381-9.Search in Google Scholar
[82] V. Artero, J.-M. Saveant. Energy Environ. Sci. 7, 3808 (2014), https://doi.org/10.1039/C4EE01709A.Search in Google Scholar PubMed PubMed Central
[83] V. V. Khrizanforova, V. I. Morozov, M. N. Khrizanforov, A. N. Lukoyanov, O. N. Kataeva, I. L. Fedushkin, Y. H. Budnikova. Polyhedron 154, 77 (2018), https://doi.org/10.1016/j.poly.2018.07.041.Search in Google Scholar
[84] Y. H. Budnikova, V. V. Khrizanforova, I. L. Fedushkin, A. A. Karasik. Phosph. Sulf. Silicon Relat. Elem. 191, 1644 (2016).10.1080/10426507.2016.1223664Search in Google Scholar
[85] M. K. Kadirov, T. I. Ismaev, R. A. Safiullin, I. R. Nizameev, I. D. Strelnik, E. I. Musina, Yu. H. Budnikova, A. A. Karasik, O. G. Sinyashin. Phosph. Sulf. Silicon Relat. Elem. 191, 1488 (2016).10.1080/10426507.2016.1212050Search in Google Scholar
[86] M. Kadirov, A. Karasik, I. Nizameev, I. Strelnik, K. Kholin, D. Kadirov, T. Ismaev, Y. Budnikova, O. Sinyashin. Energy Technol. 6, 1088 (2018), https://doi.org/10.1002/ente.201700711.Search in Google Scholar
[87] V. V. Khrizanforova, I. R. Knyazeva, V. I. Matveeva (Sokolova), I. R. Nizameev, T. V. Gryaznova, M. K. Kadirov, A. R. Burilov, O. G. Sinyashin, Y. H. Budnikova. Electrocatalyst 6, 357 (2015), https://doi.org/10.1007/s12678-015-0251-4.Search in Google Scholar
[88] M. K. Kadirov, I. R. Knyazeva, I. R. Nizameev, R. A. Safiullin, V. I. Matveeva, K. V. Kholin, V. V. Khrizanforova, T. I. Ismaev, A. R. Burilov, Yu. H. Budnikova, O. G. Sinyashin. Dalton Trans. 45, 16157 (2016), https://doi.org/10.1039/C9DT04834K.Search in Google Scholar PubMed
[89] S. Kempahanumakkagari, K. Vellingiri, A. Deep, E. E. Kwon, N. Bolan, K.-H. Kim. Coord. Chem. Rev. 357, 105 (2018), https://doi.org/10.1016/j.ccr.2017.11.028.Search in Google Scholar
[90] R. Shekurov, V. Khrizanforova, L. Gilmanova, M. Khrizanforov, V. Miluykov, O. Kataeva, Z. Yamaleeva, T. Burganov, T. Gerasimova, A. Khamatgalimov, S. Katsyuba, V. Kovalenko, Y. Krupskaya, V. Kataev, B. Büchner, V. Bon, I. Senkovska, S. Kaskel, A. Gubaidullin, O. Sinyashin, Y. Budnikova. Dalton Trans. 48, 3601 (2019), https://doi.org/10.1039/C9DT04834K.Search in Google Scholar
[91] V. Khrizanforova, R. Shekurov, V. Miluykov, M. Khrizanforov, V. Bon, S. Kaskel, A. Gubaidullin, O. Sinyashin, Y. Budnikova. Dalton Trans. 49, 2794 (2020), https://doi.org/10.1039/C9DT04834K.Search in Google Scholar
[92] M. K. Kadirov, S. Minzanova, I. Nizameev, L. G. Mironova, I. Gilmutdinov, M. Khrizanforov, K. V. Kholin, K. Ayrat, V. Semenov, V. Morozov, D. M. Kadirov, A. Mukhametzyanov, Y. H. Budnikova, O. G. Sinyashin. Inorg. Chem. Front. 5, 780 (2018).10.1039/C7QI00770ASearch in Google Scholar
© 2020 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Recent achievements in copper catalysis for C–N bond formation
- Novel selective anticancer agents based on Sn and Au complexes. Mini-review
- Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
- Surface chemistry of structural materials subjected to corrosion
- Redox-conducting polymers based on metal-salen complexes for energy storage applications
- Thermodynamic approach for prediction of oxide materials properties at high temperatures
- Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
- Fragment-based approach to novel bioactive purine derivatives
- Reaction of 1,3-dimethylimidazolium dimethylphosphate with elemental sulfur
- Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
- Macrokinetic investigation of the interaction mechanism of the pyrophoric iron nanopowder compacts with air
- Polylactide-based stent coatings: biodegradable polymeric coatings capable of maintaining sustained release of the thrombolytic enzyme streptokinase
- 3D printing in analytical chemistry: current state and future
- High purity substances – prototypes of elements of Periodic Table
- Pd(0)-catalyzed amination in the synthesis of chiral derivatives of BINAM and their evaluation as fluorescent enantioselective detectors
Articles in the same Issue
- Frontmatter
- In this issue
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Recent achievements in copper catalysis for C–N bond formation
- Novel selective anticancer agents based on Sn and Au complexes. Mini-review
- Novel colchicine conjugate with unusual effect on the microtubules of cancer cells
- Surface chemistry of structural materials subjected to corrosion
- Redox-conducting polymers based on metal-salen complexes for energy storage applications
- Thermodynamic approach for prediction of oxide materials properties at high temperatures
- Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols
- Fragment-based approach to novel bioactive purine derivatives
- Reaction of 1,3-dimethylimidazolium dimethylphosphate with elemental sulfur
- Synthetic models of hydrogenases based on framework structures containing coordinating P, N-atoms as hydrogen energy electrocatalysts – from molecules to materials
- Macrokinetic investigation of the interaction mechanism of the pyrophoric iron nanopowder compacts with air
- Polylactide-based stent coatings: biodegradable polymeric coatings capable of maintaining sustained release of the thrombolytic enzyme streptokinase
- 3D printing in analytical chemistry: current state and future
- High purity substances – prototypes of elements of Periodic Table
- Pd(0)-catalyzed amination in the synthesis of chiral derivatives of BINAM and their evaluation as fluorescent enantioselective detectors