Electrochemical synthesis and amidation of benzoin: benzamides from benzaldehydes
-
Daniele Rocco


Abstract
The benzoin condensation starting from benzaldehyde and the subsequent benzoin amidation to benzamide can be efficiently carried out under very mild conditions in an electrolysis cell. Among the advantages of using electrochemistry to generate our active reagents, the use of the easily dosed and non pollutant electron, instead of stoichiometric amounts of redox reagents or bases, usually renders the electrochemical methodology “greener” than classical organic reactions. Benzoin is obtained in good yield (85 %) carrying out the reaction in the room temperature ionic liquid BMIm-BF4. In this electrochemical reaction this liquid salt assumes the double role of solvent-supporting electrolyte system and precatalyst, yielding the corresponding N-heterocyclic carbene. The subsequent benzoin amidation is carried out by electrochemically generated superoxide anion, in the presence of an aliphatic primary or secondary amine. In this case the system superoxide/molecular oxygen acts as base and oxidant, yielding very good yields of benzamides (up to 89 %).
Introduction
Electrochemistry can be considered a “green” methodology, due to the use of the electron, a green reagent. In fact, electrochemistry allows to carry out reductions and oxidations in the absence of stoichiometric redox reagents, thus avoiding the production of stoichiometric waste substances; the solid electrode behaves as “electron collector” or “electron donor”, the redox reaction being in heterogeneous phase, and at the end of electrolysis it can be easily removed from the reaction mixture. Moreover, the amount of reagent can be easily dosed, simply interrupting the current flow through the cell [1], [2], [3]. Electroorganic synthesis (i.e. organic synthesis in which redox reactions are carried out by electrolysis) is thus gaining popularity among organic chemists and nowadays it is not unusual to find this kind of equipment in organic synthesis laboratories.
The chemical requirements for an electroorganic synthesis are quite simple: a solvent-supporting electrolyte system (in order to ensure the current flow through the cell), at least two electrodes (three if a controlled potential configuration is needed) and one or more electroactive species. In most cases, the supporting electrolyte can be recovered and reused after ethereal extraction of the crude reaction mixture.
In some cases the outcome of an electrochemical reaction is different from that of a classical one [1], but in the majority of cases the same product is obtained using milder conditions [4], [5], [6].
In this context, the use of room temperature ionic liquids (RTILs) as solvent-supporting electrolyte system has some advantages. Ionic liquids are salts formed by a large, non-coordinating organic cation and an inorganic or organic anion, liquid at or near room temperature [7]. They show good solvent ability towards a lot of organic compounds and most used gases; moreover it is possible to choose the cation-anion couple in order to render the isolation of products simple (e.g. by ethereal extraction). Being liquid and constituted of ions, RTILs can assume both solvent and supporting electrolyte roles, and sometimes also reagents [8], [9], [10]. The almost null vapour pressure of RTILs allows to recover and recycle them quite easily [11], rendering their use advantageous in organic chemistry.
Amide functional group is present in a large number of natural products and in pharmaceutical products, and amide bond formation is one of the most important reactions in pharmaceutical industry [12]. Although the chemical literature reports plenty of efficient syntheses of amides starting from the most varied reagents, this topic is still current and many new syntheses are reported every month, demonstrating the importance of this kind of reaction. In particular, benzaldehydes are convenient starting materials, due to their abundancy, cheapness and variety of possible substituents.
Nonetheless, the direct amidation of benzaldehyde by its reaction with an amine suffers from the concurrent and more favourite formation of the corresponding imine or enamine, leading to very low yields in desired product [13]. The “activation” of benzaldehyde as benzoin allows to avoid the formation of imine, leading to good yields of benzamides.
In the past years we reported the electrochemical synthesis of benzoin from the corresponding aromatic aldehyde using imidazolium ionic liquids as precursors of NHCs and as solvent-supporting electrolyte systems [14], [15], and the transformation of benzoin into the corresponding amide using electrogenerated superoxide anion [16]; herein, we summarize our results on the two-step electrochemical synthesis of arylamides starting from the corresponding benzaldehyde and an aliphatic amine.
Results and discussion
Benzoin (2a, Scheme 1) is the product of benzaldehyde dimerization, usually catalyzed by cyanide ion or an organocatalyst [17].
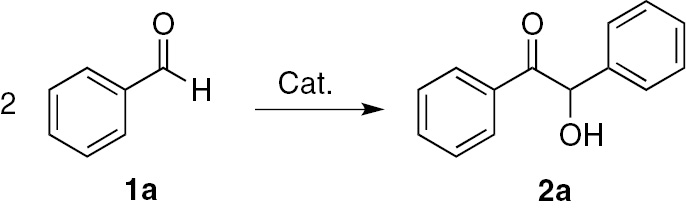
Benzoin 2a from benzaldehyde 1a.
The core of this reaction is the “umpolung” of the carbonyl carbon atom, from electrophilic to nucleophilic, due to the interaction with the catalyst, which frequently is a N-heterocyclic carbene NHC, e.g. an imidazole-2-ylidene 3 (Scheme 2). In fact, the addition of the nucleophilic NHC to the carbonyl carbon atom yields, after a proton shift, an intermediate (the Breslow intermediate) in which the carbonyl carbon atom has now a nucleophilic character. The addition of a second benzaldehyde molecule to this site and the following rearrangement yield benzoin 2a and regenerate the NHC catalyst 3 (Scheme 2) [14].
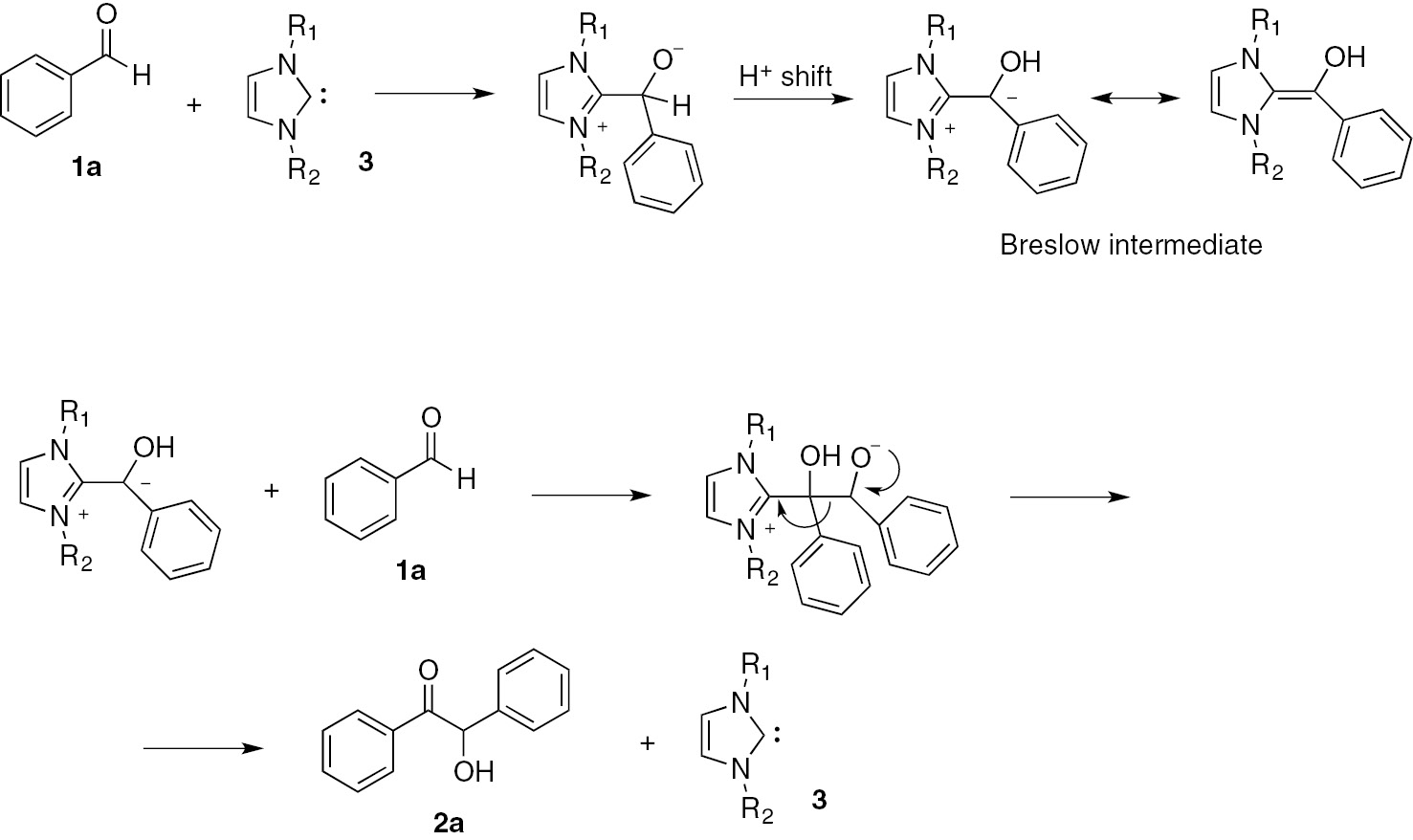
Benzoin condensation mechanism.
NHCs are usually generated in situ by deprotonation of the corresponding azolium cation with a strong base (n-BuLi, DBU, DABCO, etc.). Electrochemistry can be an alternative for the generation of NHC from the azolium cation, under very mild conditions and avoiding the production of waste molecules (as the strong bases byproducts). In fact, the cathodic reduction of an imidazolium cation leads to the cleavage of the C–H bond between the two nitrogen atoms (C-2), yielding the corresponding NHC and molecular hydrogen (Scheme 3) [18], [19].
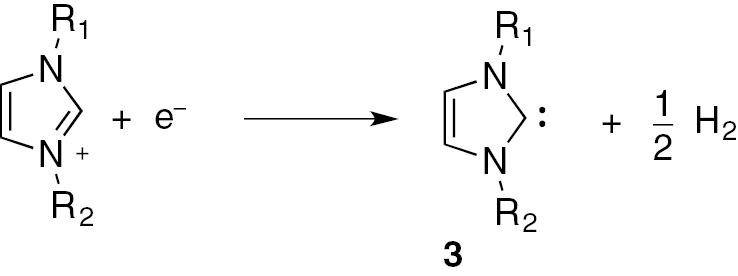
Electrochemical generation of NHC 3.
Moreover, it is possible to electrochemically generate NHC 3 starting from the corresponding imidazolium salt using this ionic liquid (RTIL) also as solvent-supporting electrolyte system [20]. Using an RTIL (with a virtually null vapour pressure) as solvent-supporting electrolyte system allows to avoid a volatile organic solvent, thus minimizing air pollution. Moreover, RTILs can be easily recovered from the reaction mixture and reused in a subsequent electrolysis. Last, the use of a very polar medium such as a RTIL could favour those reactions in which the intermediates are very polar or charged (as in the benzoin condensation).
This last consideration is well demonstrated by the electrochemical synthesis of benzoin 2a, carried out under the same experimental conditions, in an ionic liquid and in a classical organic solvent (Table 1) [15].
Electrochemically induced benzoin condensation: effect of the solvent.a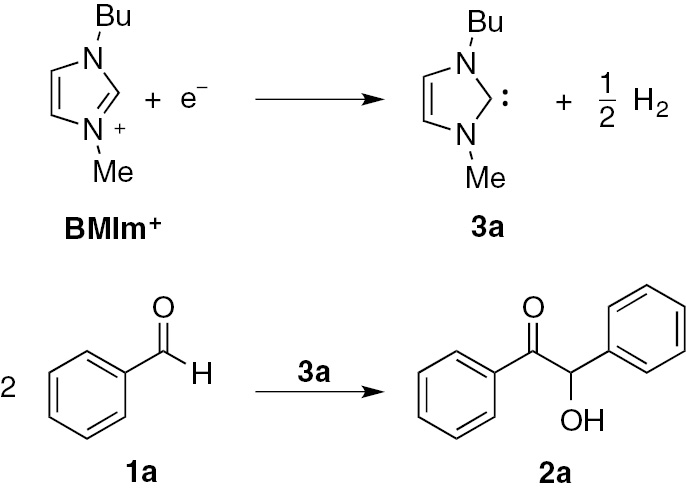
| Solvent/supporting electrolyte | Q (Faradays)b | 2a, yield |
|---|---|---|
| DMF/BMIm-BF4 | 0.2 | Traces |
| DMF/BMIm-BF4 | 2.0 | 30% |
| BMIm-BF4 | 0.2 | 85% |
aExperimental conditions: electrolyses carried out under galvanostatic conditions, in a divided cell, using Pt electrodes, under inert atmosphere. At the end of electrolysis, stopped the current flow, benzaldehyde was added to the catholyte and the mixture was kept at 65°C for 2 h. bAmount of charge referred to benzaldehyde, proportional to NHC amount.
Carrying out this electrochemical synthesis in BMIm-BF4, very good yields in benzoin were obtained starting from benzaldehydes with various substituents [15].
The subsequent step was the transformation of benzoin into a benzamide by reaction with an aliphatic primary or secondary amine in the presence of electrogenerated superoxide anion and molecular oxygen. Unfortunately, this reaction could not be carried out in BMIm-BF4, due to the reactivity of the corresponding NHC with molecular oxygen (Scheme 4) [21], [22].
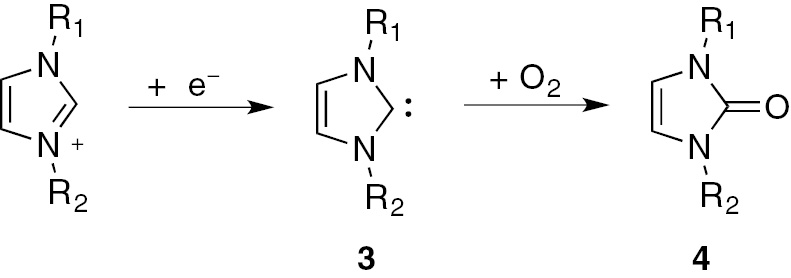
Electrochemical synthesis of imidazole-2-one 4.
The electrochemical reduction of molecular oxygen to superoxide anion was thus carried out in DMF/Et4N-BF4, in which superoxide anion is quite stable in the absence of a protic reagents (Scheme 5).

Electrogeneration of superoxide anion.
The main advantage in using electrogenerated superoxide anion lies in its counterion, the tetraethylammonium cation, non coordinated, which renders superoxide anion a “naked” anion, highly reactive, opposite to commercial potassium superoxide, whose ion pair is really unreactive and which needs large amounts of crown ethers to dissolve into the solvent and react [23].
The voltammetric analysis of DMF-Et4NBF4 solutions of molecular oxygen or benzoin showed that it is possible to reduce selectively molecular oxygen in the presence of benzoin; in fact their first reduction potentials [glassy carbon (GC) electrode, saturated calomel reference electrode (SCE)] are −0.96 V (molecular oxygen) and −1.84 V (benzoin). So if the electrolysis of a solution containing both benzoin and molecular oxygen is carried out at −0.96 V, only oxygen is reduced at the cathode. Moreover, the voltammetric analysis shows that there is not an electron exchange between reduced oxygen (superoxide anion) and benzoin (Fig. 1); in fact the first peak current of molecular oxygen is the same in the absence and in the presence of benzoin, excluding a redox reaction between them. On the contrary, the disappearance of the first anodic peak of oxygen in the return scan demonstrates a reaction between superoxide anion (electrogenerated during the direct scan) and benzoin, leading to the consumption of superoxide anion, thus no more available for the oxidation at the electrode.
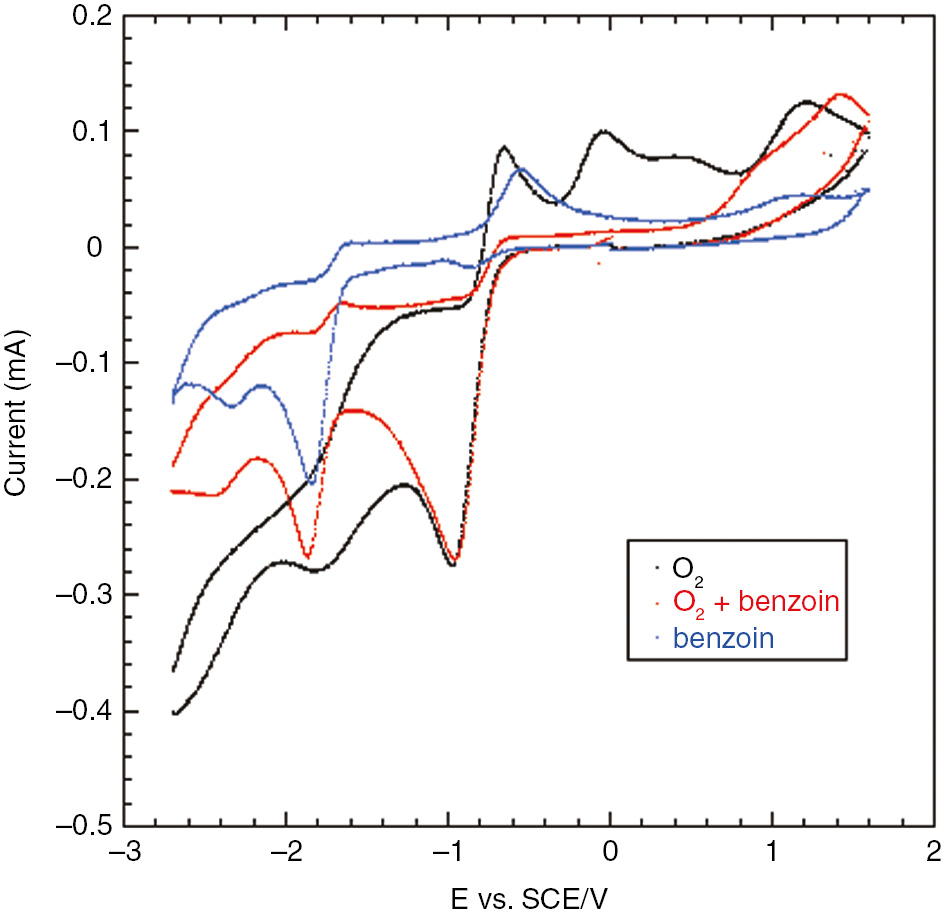
Superimposed cyclic voltammetries of DMF-0.1 M Et4NBF4 solutions of O2 (≈2·10−3 M), benzoin (1·10−2 M) and O2+benzoin (same concentrations). GC electrode, rt, SCE reference electrode, scan rate: 0.2 V s−1, scan potential: 0.0 to −2.7 to +1.6 to 0.0 V.
The reaction of benzoin with an aliphatic amine (primary or secondary) in the presence of electrogenerated superoxide ion and molecular oxygen leads to the formation of the corresponding benzamide and ammonium benzoate (Scheme 6). Benzamide yields strongly depend on the amount of current, and using benzylamine, the corresponding benzylbenzamide is obtained in the highest yield (68%) after 0.3 F (with respect to benzoin) (Table 2).
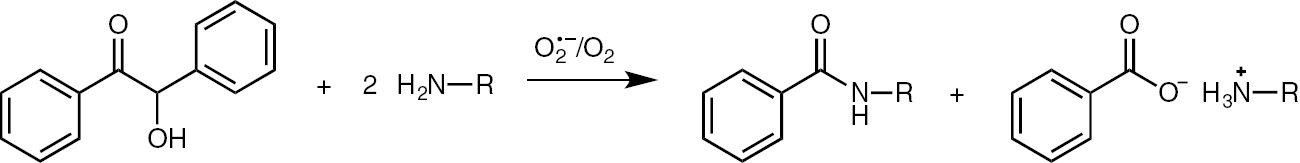
Amidation of benzoin.
Electrochemical synthesis of benzyl benzamide with superoxide anion. Dependence of the yield from the number of Faradays.a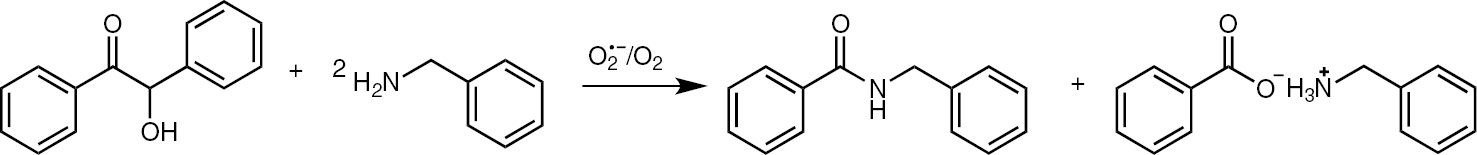
| Faradaysb | Benzamide, yield |
|---|---|
| 1.0 | 30% |
| 0.6 | 65% |
| 0.3 | 68% |
aExperimental conditions: electrolyses carried out under galvanostatic conditions, in a divided cell, using Pt electrodes, under O2 atmosphere, on DMF-0.1 M Et4NBF4 solutions of benzoin and benzylamine in 1:2 mol ratio. At the end of electrolysis, stopped the current flow, the mixture was kept at rt for 12 h. bAmount of charge referred to benzoin.
This reaction leads to very good yields in benzamides both starting from aliphatic primary amines (66–89%) and from secondary ones (51–89%) [16].
As can be seen from Table 2, a catalytic amount of electricity is sufficient to carry out this reaction; moreover, benzil (Ph–CO–CO–Ph) was isolated in low yields from the reaction mixture. A similar experiment starting from benzil allowed to isolate the benzamide in 42% yield (the lower yield is attributable to the co-reduction of benzil at the cathode under our experimental conditions).
From all these results, it was possible to propose a reaction mechanism, in which both electrogenerated superoxide anion and molecular oxygen are involved, and in which benzil is a reaction intermediate [24] (Scheme 7).
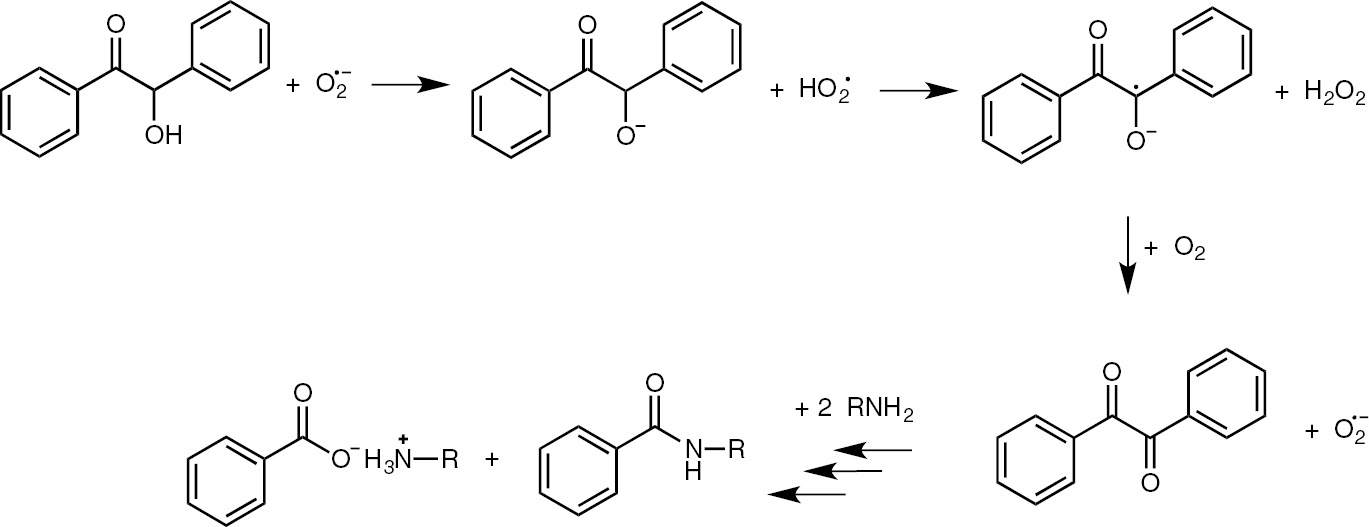
Proposed mechanism for the amidation of benzoin.
Conclusions
A two-step electrochemical synthesis of benzamides, starting from benzaldehyde and an aliphatic amine, can be efficiently carried out, obtaining good yields in valuable products. The use of the electrochemical methodology allows to carry out this synthesis under very mild conditions and in a “green” way, mainly due to the use of the “green” electron, massless and non pollutant, instead of stoichiometric amounts of bases or redox chemical reagents (which imply a stoichiometric production of waste). Moreover, it is possible to carry out the first step in an ionic liquid, which acts both as solvent and as precatalyst; this renders easy the recycling of the solvent in subsequent runs.
Experimental section
Methods
Constant current electrolyses were performed at 25°C, using an Amel Model 552 potentiostat equipped with an Amel Model 731 integrator. All the experiments were carried out in a divided glass cell separated through a porous glass plug, filled up with a layer of gel (i.e. methyl cellulose 0.5% vol dissolved in DMF-Et4NBF4 1.0 mol dm−3) in case DMF was used as solvent; Pt spirals (apparent area 0.8 cm2) were used as both cathode and anode.
Typical electrosynthesis of benzoin in ionic liquid [15]
Anolyte (1.0 cm3 of BMIm-BF4) and catholyte (2.0 cm3 of BMIm-BF4) were separated through a G-3 glass septum. The electrolysis was carried out, under nitrogen atmosphere at room temperature, at constant current (20 mA cm−2) and stopped after 194°C. At the end of the electrolysis, 10 mmol of freshly distilled benzaldehyde were added to the catholyte, the temperature was increased up to 65°C and the mixture was stirred for 2 h. The reaction mixture was then extracted with diethyl ether (3×10 cm3). After the removal of the solvent from the combined ethereal layers under reduced pressure, the crude reaction mixture was analyzed by 1H-NMR and TLC. Benzoin was purified by flash chromatography (n-hexane-ethyl acetate 8:2) and characterised by NMR spectroscopy. Benzoin spectral data are in accordance with those reported in the literature.
Typical electroamidation of benzoin in DMF-Et4NBF4 [16]
Anolyte (5 cm3 DMF-Et4NBF4 0.1 mol dm−3) and catholyte (10 cm3 DMF-Et4NBF4 0.1 mol dm−3) were separated through a G-3 glass septum filled up with a layer of gel. The electrolysis was carried out, under oxygen atmosphere at room temperature, at constant current (20 mA cm−2).
In the catholyte 0.5 mmol of benzoin and 1.0 mmol of aliphatic amine were present, with continuous O2 bubbling. The electrolysis was stopped after 25°C, the oxygen bubbling was stopped and the catholyte was kept under stirring at rt for 12 h. Usual workup gave the benzamide, whose spectral data are identical to those reported in the literature.
Article note
A collection of invited papers based on presentations at the15th Eurasia Conference on Chemical Sciences (EuAsC2S-15) held at Sapienza University of Rome, Italy, 5–8 September 2018.
Acknowledgments
This work was financially supported by Sapienza University of Rome. The authors want to thank Prof. Achille Inesi and Mr Marco Di Pilato, for their willingness whenever needed.
References
[1] H. Lund, O. Hammerich. Organic Electrochemistry. Marcel Dekker Inc., New York (2001).10.1201/9781420029659Search in Google Scholar
[2] D. Kyriacou. Modern Electroorganic Chemistry. Springer-Verlag, Berlin (1994).10.1007/978-3-642-78677-8Search in Google Scholar
[3] J. Volke, F. Liška. Electrochemistry in Organic Synthesis. Springer-Verlag, Berlin (1994).10.1007/978-3-642-78699-0Search in Google Scholar
[4] A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes, S. R. Waldvogel. Angew. Chem. Int. Ed.57, 5594 (2018).10.1002/anie.201711060Search in Google Scholar PubMed PubMed Central
[5] M. Yan, Y. Kawamata, P. S. Baran. Chem. Rev.117, 13230 (2017).10.1021/acs.chemrev.7b00397Search in Google Scholar PubMed PubMed Central
[6] S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe, S. R. Waldvogel. Angew. Chem. Int. Ed.57, 6018 (2018).10.1002/anie.201712732Search in Google Scholar PubMed PubMed Central
[7] P. Wasserscheid, T. Welton. Ionic Liquids in Synthesis. Wiley VCH, Weinheim (2008).10.1002/9783527621194Search in Google Scholar
[8] M. Feroci, M. Orsini, L. Rossi, A. Inesi. Curr. Org. Synth. 9, 40 (2012).10.2174/157017912798889206Search in Google Scholar
[9] M. Feroci, M. Orsini, A. Inesi. in Green Solvents II. Properties and Applications of Ionic Liquids. A. Mohammad, Inammudin (Eds.), pp. 435–471, Springer, Dordrecht (2012).10.1007/978-94-007-2891-2_16Search in Google Scholar
[10] M. Feroci, I. Chiarotto, A. Inesi. Curr. Org. Chem. 17, 204 (2013).10.2174/1385272811317030003Search in Google Scholar
[11] I. Chiarotto, M. Feroci, G. Sotgiu, A. Inesi. Eur. J. Org. Chem.2013, 326 (2013).10.1002/ejoc.201201023Search in Google Scholar
[12] J. R. Dunetz, J. Magano, G. A. Weisenburger. Org. Process Res. Dev.20, 140 (2016).10.1021/op500305sSearch in Google Scholar
[13] M. Feroci, I. Chiarotto, A. Inesi. Electrochim. Acta89, 692 (2013).10.1016/j.electacta.2012.11.061Search in Google Scholar
[14] M. Orsini, I. Chiarotto, M. N. Elinson, G. Sotgiu, A. Inesi. Electrochem. Commun.11, 1013 (2009).10.1016/j.elecom.2009.02.045Search in Google Scholar
[15] I. Chiarotto, M. Feroci, M. Orsini, M. M. M. Feeney, A. Inesi. Adv. Synth. Catal.352, 3287 (2010).10.1002/adsc.201000555Search in Google Scholar
[16] F. Pandolfi, I. Chiarotto, D. Rocco, M. Feroci. Electrochim. Acta254, 358 (2017).10.1016/j.electacta.2017.09.135Search in Google Scholar
[17] R. S. Menon, A. T. Biju, V. Nair. Beilstein J. Org. Chem.12, 444 (2016).10.3762/bjoc.12.47Search in Google Scholar PubMed PubMed Central
[18] M. Feroci, I. Chiarotto, S. Vecchio Ciprioti, A. Inesi. Electrochim. Acta109, 95 (2013).10.1016/j.electacta.2013.07.115Search in Google Scholar
[19] M. Feroci, I. Chiarotto, A. Inesi. Catalysts6, 178 (2016).10.3390/catal6110178Search in Google Scholar
[20] M. Feroci, I. Chiarotto, F. D’Anna, G. Forte, R. Noto, A. Inesi. Electrochim. Acta153, 122 (2015).10.1016/j.electacta.2014.11.135Search in Google Scholar
[21] M. Feroci, I. Chiarotto, M. Orsini, G. Sotgiu, A. Inesi. Electrochim. Acta56, 5823 (2011).10.1016/j.electacta.2011.04.067Search in Google Scholar
[22] M. Hayyan, F. S. Mjalli, M. A. Hashim, I. M. AlNashef. J. Mol. Liq.181, 44 (2013).10.1016/j.molliq.2013.02.001Search in Google Scholar
[23] M. A. Casadei, S. Cesa, F. Micheletti Moracci, A. Inesi, M. Feroci. J. Org. Chem.61, 380 (1996).10.1021/jo9512057Search in Google Scholar
[24] M. Singh, R. A. Misra. Synthesis1989, 403 (1989).10.1055/s-1989-27268Search in Google Scholar
©2019 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- 15th Eurasia Conference on Chemical Sciences (EuAsC2S-15) – 5th–8th September 2018, Rome, Italy
- Conference papers
- The Jahn-Teller effect in mixed aqueous solution: the solvation of Cu2+ in 18.6% aqueous ammonia obtained from ab initio quantum mechanical charge field molecular dynamics
- Facile synthesis of hydrogel-nickel nanoparticle composites and their applications in adsorption and catalysis
- The effect of pore morphology on the catalytic performance of β-glucosidase immobilized into mesoporous silica
- Competitive pseudo-ELISA based on molecularly imprinted nanoparticles for microcystin-LR detection in water
- Titanium based complexes with melanin precursors as a tool for directing melanogenic pathways
- Stability of PMMA-grafted/Ti hybrid biomaterial interface in corrosive media
- High performance liquid chromatographic profiling of antioxidant and antidiabetic flavonoids purified from Azadirachta indica (neem) leaf ethanolic extract
- Effects mediated by M2 muscarinic orthosteric agonist on cell growth in human neuroblastoma cell lines
- Heterogeneous palladium SALOPHEN onto porous polymeric microspheres as catalysts for heck reaction
- Transfer of chemical elements from milk to dairy products
- Is hydrogen electronegativity higher than Pauling’s value? New clues from the 13C and 29Si NMR chemical shifts of [CHF3] and [SiHF3] molecules
- How alkali-activated Ti surfaces affect the growth of tethered PMMA chains: a close-up study on the PMMA thickness and surface morphology
- Dual inhibitors of urease and carbonic anhydrase-II from Iris species
- Electrochemical synthesis and amidation of benzoin: benzamides from benzaldehydes
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- 15th Eurasia Conference on Chemical Sciences (EuAsC2S-15) – 5th–8th September 2018, Rome, Italy
- Conference papers
- The Jahn-Teller effect in mixed aqueous solution: the solvation of Cu2+ in 18.6% aqueous ammonia obtained from ab initio quantum mechanical charge field molecular dynamics
- Facile synthesis of hydrogel-nickel nanoparticle composites and their applications in adsorption and catalysis
- The effect of pore morphology on the catalytic performance of β-glucosidase immobilized into mesoporous silica
- Competitive pseudo-ELISA based on molecularly imprinted nanoparticles for microcystin-LR detection in water
- Titanium based complexes with melanin precursors as a tool for directing melanogenic pathways
- Stability of PMMA-grafted/Ti hybrid biomaterial interface in corrosive media
- High performance liquid chromatographic profiling of antioxidant and antidiabetic flavonoids purified from Azadirachta indica (neem) leaf ethanolic extract
- Effects mediated by M2 muscarinic orthosteric agonist on cell growth in human neuroblastoma cell lines
- Heterogeneous palladium SALOPHEN onto porous polymeric microspheres as catalysts for heck reaction
- Transfer of chemical elements from milk to dairy products
- Is hydrogen electronegativity higher than Pauling’s value? New clues from the 13C and 29Si NMR chemical shifts of [CHF3] and [SiHF3] molecules
- How alkali-activated Ti surfaces affect the growth of tethered PMMA chains: a close-up study on the PMMA thickness and surface morphology
- Dual inhibitors of urease and carbonic anhydrase-II from Iris species
- Electrochemical synthesis and amidation of benzoin: benzamides from benzaldehydes