Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
-
Dipannita Kalyani


Abstract
This manuscript describes the development of Pd- and Ni-catalyzed arylations of unactivated arene C–H bonds using C–O electrophiles. A method for Pd-catalyzed intramolecular C–H arylation is accomplished using inexpensive and readily available tosylates and mesylates as electrophiles. This transformation is efficient for the synthesis of various heterocyclic motifs including furans, carbazoles, indoles, and lactams. Additionally, a protocol for a one-pot sequential tosylation/arylation of phenol derivatives is presented. The use of earth-abundant and inexpensive Ni catalysts for an intramolecular C–H arylation using aryl pivalates as electrophiles is described. Preliminary mechanistic studies for both the Pd- and Ni-catalyzed arylations are discussed.
Introduction
Direct arylation reactions are now well established for the construction of C–C bonds [1]. A vast majority of these reactions utilize aryl halides and their derivatives as electrophiles. Organohalide substrates and the byproducts of their reactions are generally undesirable from an environmental standpoint. Additionally, aryl halides are not naturally abundant, and their synthesis often involves tedious steps, harsh reaction conditions, and waste production. As such, recently there has been an increasing demand for the use of phenolic electrophiles in place of aryl halides [2].
Early examples of transition-metal-catalyzed C–H arylations using C–O electrophiles involved the use of aryl triflates [3]. While this represents a significant advancement, these reactions are not ideal for use in long synthetic sequences due to the sensitive and expensive nature of triflates [4]. More recently, the accomplishment of direct arylations using fluorine free phenol-derived electrophiles, such as tosylates, mesylates, carbamates, carbonates, and esters, has been reported [5]. The vast majority of these reports involve the arylation of activated arene and heteroarene (azoles, N-oxides, fluoroaromatics, etc.) C–H bonds [6, 7]. In contrast, the methods for the arylation of unactivated arene C–H bonds remain limited. Specifically, Ackermann and co-workers have accomplished the Pd- or Ru-catalyzed ligand-directed intermolecular arylation of aromatic C–H bonds using tosylates and mesylates [8]. Furthermore, the first example of Co-catalyzed direct arylation using carbamates, sulfamates, and phosphates was reported recently [9]. These being the only examples, C–H arylations of unactivated arenes using phenolic electrophiles remains a relatively unexplored field.
Results and discussion
Pd-catalyzed direct arylation
We began our efforts with the exploration of Pd-catalyzed intramolecular arylation using substrate 1-OTs. Upon significant optimization we discovered that the desired benzofuran product 1 could be obtained in excellent yields using the Pd(OAc)2/dcype catalyst system (Scheme 1) [10]. Importantly, ligands such as XPhos and RuPhos that had been previously used for Pd-catalyzed arylations using sulfonates were not efficient for this transformation [6, 8].
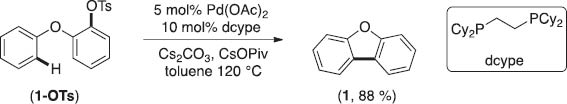
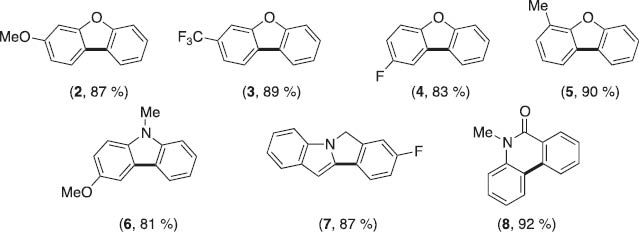
The reaction is general with respect to varied substitution patterns on both aryl rings. Electron-donating and -withdrawing groups were well tolerated on both the C–H and the tosylate bearing aryl rings. These reactions could be expanded to the synthesis of a variety of heterocyclic motifs such as carbazoles and amides, which are prevalent in bioactive molecules (Scheme 2).
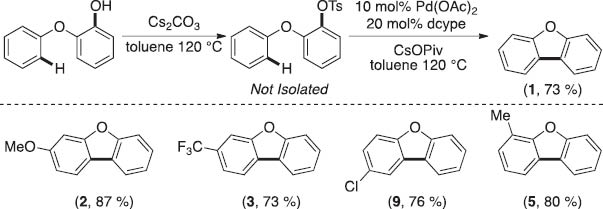
We have also shown a preliminary scope for the sequential tosylation/arylation of phenolic substrates (Scheme 3). The formation of the desired benzofuran products using this protocol obviates the isolation and purification of the intermediate tosylates, thereby increasing the step economy of the process.
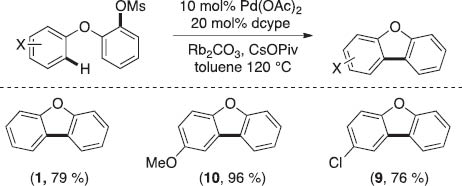
We next turned toward developing analogous transformations employing more atom economical mesylate electrophiles. As shown in Scheme 4, mesylates can act as competent electrophiles for the intramolecular C–H arylation to afford the products in good yields. Notably, the results illustrated in Schemes 1–4 represent the first examples of efficient intramolecular direct arylation using tosylates and mesylates as electrophiles.
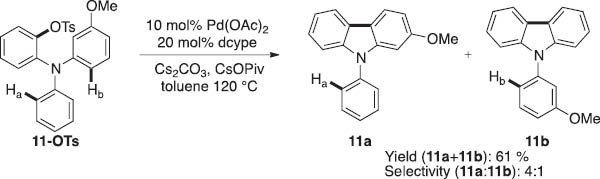
Having explored a preliminary scope for the Pd-catalyzed intramolecular arylations, we next turned toward investigating the electronic requirements for the C–H activation step. As such, we studied the site selectivity for the reaction of substrate 11-OTs. Tosylate 11-OTs bears two electronically different C–H bonds, Ha and Hb, which could undergo functionalization to afford products 11b and 11a, respectively. As depicted in Scheme 5, the reaction exhibits modest selectivity for the formation of 11a via preferential arylation of the electron-rich aromatic ring. The observed electronic effect is similar to that documented for similar Pd-catalyzed C–H arylations using aryl halides [11].
Ni-catalyzed direct arylation
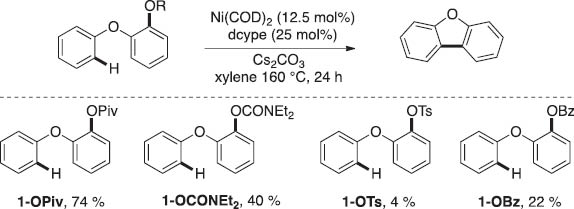
As detailed above, Pd catalysis affects the arylation of unactivated arenes using tosylates and mesylates. Our next efforts turned toward accomplishing similar transformations under Ni catalysis. The utilization of inexpensive earth abundant Ni catalysts in place of precious metals such as Pd has the potential to impact the economic viability and the widespread applicability of such transformations [1, 7]. Our initial studies toward this goal have focused on the optimization of reactions with substrates shown in Scheme 6. Importantly, these substrates only differ in the nature of the electrophile-bearing ring. As shown in Scheme 6, the use of dcype as the ligand, allows for the Ni-catalyzed C–H arylation with a number of C–O electrophiles. However, pivalates (1-OPiv) and carbamates (1-OCONEt2) are the most effective electrophiles, affording the desired product in 74 and 40 % yields, respectively [12].
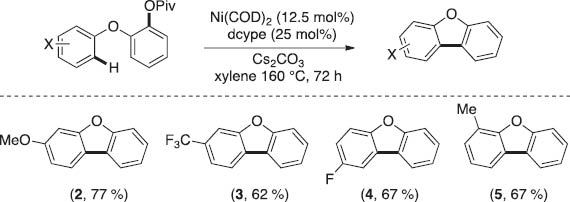
The efficiency for the intramolecular coupling of aryl pivalates with arenes was not significantly influenced by the electronic nature of the C–H bearing ring. Electronically diverse benzofuran products could be obtained in modest-to-good yields (Scheme 7).
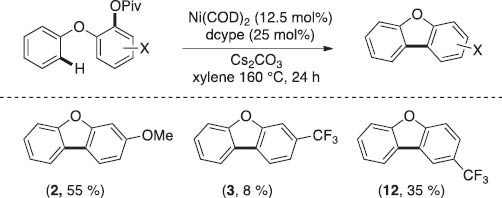
Interestingly however, the electronics of the pivalate-bearing ring did impact the reaction efficiency. While electron-rich pivalates led to products (2) in modest yields, the electron-poor pivalate bearing substrates afforded the desired products (3 and 12) in significantly diminished yield (Scheme 8).
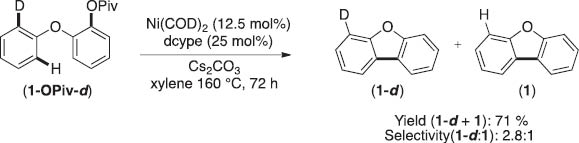
The results illustrated in Schemes 6 and 8 represent promising leads toward the development of general Ni-catalyzed arylation of unactivated arenes. Further optimization and broadening of the scope of such transformations will require an understanding of the C–H activation step. As such we have conducted preliminary studies to explore the mechanism of C–H activation at Ni centers. Specifically, we have obtained the intramolecular K.I.E from the reaction of the mono-deuterated substrate 1-OPiv-d (Scheme 9). Although further studies are needed, the observed primary kH/kD of 2.8 is similar to that observed for Pd-catalyzed C–H arylations, thought to proceed via a concerted metallation–deprotonation sequence [11].
Summary
In summary, this paper describes the development of Pd- and Ni-catalyzed intramolecular C–H arylation of arenes using phenolic electrophiles. The transformation could be applied toward the synthesis a variety of heterocyclic motifs including benzofurans, carbazoles, indoles, and lactams. Further studies will focus on broadening the scope of the current transformations. Additionally, detailed mechanistic studies will be conducted and reported in due course.
Acknowledgments
We thank the donors of the American Chemical Society Petroleum Research Fund for partial support of this work. The authors would also like to thank the CURI (Collaborative Undergraduate Research and Inquiry) program at St. Olaf College for providing financial support for this work.
References
[1] For representative reviews on C–H arylation, see: (a) D. Alberico, M. E. Scott, M. Lautens. Chem. Rev. 107, 174 (2007); (b) F. Kakiuchi, T. Kochi. Synthesis 3013 (2008); (c) G. P. McGlacken, L. M. Bateman. Chem. Soc. Rev. 38, 2447 (2009); (d) X. Chen, K. M. Engle, D.-H. Wang, J.-Q. Yu. Angew. Chem., Int. Ed. 48, 5094 (2009); (e) L. Ackermann, R. Vincente, A. R. Kapdi. Angew. Chem., Int. Ed. 48, 9792 (2009); (f) F. Bellina, R. Rossi. Tetrahedron 65, 10269 (2009); (g) T. W. Lyons, M. S. Sanford. Chem. Rev. 110, 1147 (2010); (h) O. Daugulis. Top. Curr. Chem. 292, 57 (2010); (i) G. P. Chiusoli, M. Catellani, M. Costa, E. Motti, N. Della Ca’, G. Maestri. Coord. Chem. Rev. 254, 456 (2010); (j) J. Yamaguchi, K. Muto, K. Itami. Eur. J. Org. Chem. 19 (2013).Suche in Google Scholar
[2] (a) D.-G. Yu, B.-J. Li, Z.-J. Shi. Acc. Chem. Res. 43, 1486 (2010); (b) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg, V. Percec. Chem. Rev. 111, 1346 (2011); (c) B.-J. Li, D.-G. Yu, C.-L. Sun, Z.-J. Shi. Chem.–Eur. J. 17, 1728 (2011).Suche in Google Scholar
[3] For some representative recent reports, see: (a) M. Brenner, G. Mayer, A. Terpin, W. Steglich. Chem.—Eur. J. 3, 70 (1997); (b) K. Kametani, T. Satoh, M. Miura, M. Nomura. Tetrahedron Lett. 41, 2655 (2000); (c) O. Hara, T. Nakamura, F. Sato, K. Makino, Y. Hamada. Heterocycles 68, 1 (2006); (d) A. C. F. Cruz, N. D. Miller, M. C. Willis. Org. Lett. 9, 4391 (2007); (e) J. Roger, H. Doucet. Org. Biomol. Chem. 6, 169 (2008); (f) D. J. Schipper, M. El-Salfiti, C. J. Whipp, K. Fagnou. Tetrahedron 65, 4977 (2009).Suche in Google Scholar
[4] L. J. Gooβen, N. Rodriguez, P. P. Lange, C. Linder. Angew. Chem., Int. Ed. 49, 1111 (2010).Suche in Google Scholar
[5] (a) C. Ming So, F. Y. Kwong. Chem. Soc. Rev. 40, 4963 (2011); (b) S. I. Kozhushkov, H. K. Potukuchi, L. Ackermann. Catal. Sci. Technol. 3, 562 (2013).Suche in Google Scholar
[6] (a) L. Ackermann, M. Mulzer. Org. Lett. 10, 5043 (2008); (b) L. Ackermann, A. Althammer, S. Fenner. Angew. Chem., Int. Ed. 48, 201 (2009); (c) L. Ackermann, S. Barfuesser, J. Pospech. Org. Lett. 12, 724 (2010); (d) L. Ackermann, S. Fenner. Chem. Commun. 47, 430 (2011); (e) S. Fan, J. Yang, X. Zhang. Org. Lett. 13, 4374 (2011); (f) L. Ackermann, J. Pospech, H. K. Potukuchi. Org. Lett. 14, 2146 (2012); (g) J. W. W. Chang, E. Y. Chia, C. L. Chai, J. Seayad. Org. Biomol. Chem. 10, 2289 (2012).Suche in Google Scholar
[7] (a) K. Muto, J. Yamaguchi, K. Itami. J. Am. Chem. Soc. 134, 169 (2012); (b) K. Amaike, K. Muto, J. Yamaguchi, K. Itami. J. Am. Chem. Soc. 134, 13573 (2012).Suche in Google Scholar
[8] L. Ackermann, A. Althammer, R. Born. Angew. Chem., Int. Ed. 45, 2619 (2006).Suche in Google Scholar
[9] W. F. Song, L. Ackermann. Angew. Chem., Int. Ed. 51, 8251 (2012).Suche in Google Scholar
[10] C. S. Nervig, P. J. Waller, D. Kalyani. Org. Lett. 14, 4838 (2012).Suche in Google Scholar
[11] (a) A. D. Ryabov, I. K. Sakodinskaya, A. K. Yatsimirsky. J. Chem. Soc., Dalton Trans. 2629 (1985); (b) C. Jia, W. Lu, J. Oyamada, T. Kitamura, K. Matsuda, M. Irie, Y. Fujiwara. J. Am. Chem. Soc. 122, 7252 (2000); (c) J. A. Tunge, L. N. Foresee. Organometallics 24, 6440 (2005); (d) D. L. Davies, S. M. A. Donald, S. A. Macgregor. J. Am. Chem. Soc. 127, 13754 (2005); (e) M. Lafrance, K. Fagnou. J. Am. Chem. Soc. 128, 16496 (2006); (f) D. Garcia-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras, A. M. Echavarren. J. Am. Chem. Soc. 129, 6880 (2007); (g) L. Ackermann. Chem. Rev. 111, 1315 (2011); (h) S. I. Gorelsky, D. Lapointe, K. Fagnou. J. Org. Chem. 77, 658 (2012).Suche in Google Scholar
[12] J. Wang, D. M. Ferguson, D. Kalyani. Tetrahedron 69, 5780 (2013).10.1016/j.tet.2013.04.096Suche in Google Scholar
Article note
A collection of invited papers based on presentations at the 17th International IUPAC Conference on Organometallic Chemistry Directed Towards Organic Synthesis (OMCOS-17), Fort Collins, Colorado, USA, 28 July–1 August 2013.
©2014 by IUPAC & De Gruyter
Artikel in diesem Heft
- Masthead
- Masthead
- Conference papers
- International Union of Pure and Applied Chemistry
- Enantioselective palladium(0)-catalyzed C–H arylation strategy for chiral heterocycles
- Organometallic catalysis for applications in radical chemistry and asymmetric synthesis
- Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
- Copper-catalyzed aminoboration and hydroamination of alkenes with electrophilic amination reagents
- Activation of stable σ-bonds for organic synthesis
- Pd-catalyzed reactions of unactivated Csp3−I bonds
- Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
- Nickel-catalyzed oxidative cross-coupling of arylboronic acids with olefins
- Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents
- Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
- Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions
- Beyond C–H and C–O activation: the evolution of components in cross-coupling reactions
- The effect of acceptor-substituted alkynes in gold-catalyzed intermolecular reactions
- Chemoselective silver-catalyzed nitrene insertion reactions
- The strategic generation and interception of palladium-hydrides for use in alkene functionalization reactions
- 3-Acyloxy-1,4-enyne: A new five-carbon synthon for rhodium-catalyzed [5 + 2] cycloadditions
- Cobalt-catalyzed directed alkylation of arenes with primary and secondary alkyl halides
- IUPAC Technical Report
- Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)
Artikel in diesem Heft
- Masthead
- Masthead
- Conference papers
- International Union of Pure and Applied Chemistry
- Enantioselective palladium(0)-catalyzed C–H arylation strategy for chiral heterocycles
- Organometallic catalysis for applications in radical chemistry and asymmetric synthesis
- Rhenium(I)-catalyzed reaction of terminal alkynes with imines leading to allylamine derivatives
- Copper-catalyzed aminoboration and hydroamination of alkenes with electrophilic amination reagents
- Activation of stable σ-bonds for organic synthesis
- Pd-catalyzed reactions of unactivated Csp3−I bonds
- Pd- and Ni-catalyzed C–H arylations using C–O electrophiles
- Nickel-catalyzed oxidative cross-coupling of arylboronic acids with olefins
- Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents
- Transition-metal clusters as catalysts for chemoselective transesterification of alcohols in the presence of amines
- Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions
- Beyond C–H and C–O activation: the evolution of components in cross-coupling reactions
- The effect of acceptor-substituted alkynes in gold-catalyzed intermolecular reactions
- Chemoselective silver-catalyzed nitrene insertion reactions
- The strategic generation and interception of palladium-hydrides for use in alkene functionalization reactions
- 3-Acyloxy-1,4-enyne: A new five-carbon synthon for rhodium-catalyzed [5 + 2] cycloadditions
- Cobalt-catalyzed directed alkylation of arenes with primary and secondary alkyl halides
- IUPAC Technical Report
- Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report)