Site-specific genome editing in treatment of inherited diseases: possibility, progress, and perspectives
-
Chao Huang

 ,
Qing Li
und
Jinsong Li
,
Qing Li
und
Jinsong Li

Abstract
Advancements in genome editing enable permanent changes of DNA sequences in a site-specific manner, providing promising approaches for treating human genetic disorders caused by gene mutations. Recently, genome editing has been applied and achieved significant progress in treating inherited genetic disorders that remain incurable by conventional therapy. Here, we present a review of various programmable genome editing systems with their principles, advantages, and limitations. We introduce their recent applications for treating inherited diseases in the clinic, including sickle cell disease (SCD), β-thalassemia, Leber congenital amaurosis (LCA), heterozygous familial hypercholesterolemia (HeFH), etc. We also discuss the paradigm of ex vivo and in vivo editing and highlight the promise of somatic editing and the challenge of germline editing. Finally, we propose future directions in delivery, cutting, and repairing to improve the scope of clinical applications.
Introduction
From the first description of alkaptonuria as an inherited disease in the early 20th century, humans have recognized hereditary disorders for over 100 years [1]. In the late 1940s, sickle cell disease (SCD) was associated with alteration in a protein of red blood cells [2]. As a milestone, this finding was the first time a “genetic disease” was linked to a mutation of a specific protein and was called a “molecular disease” at that time. In the following years, the discovery of the double helix structure of DNA and the central dogma opened the door to understanding the human genome and genetic diseases. Now we know that mutations in coding and non-coding sequences can cause genetic disorders, some of which are inherited from parents’ germline or occur spontaneously before embryonic development and will also pass to the next generations, such as SCD. These diseases are termed inherited diseases or hereditary diseases [3]. Others are caused by mutations that occur in the somatic cells during a person’s life, such as cancers. According to statistics, there are over 6,000 genetic disorders, with only about one-tenth of them treatable [4, 5]. Individual genetic disease is rare. For example, approximately 1 in 2,000 people are affected by cystic fibrosis. The prevalence is 1 in 500 for familial hypercholesterolemia and hypertrophic cardiomyopathy, 1 in 625 for SCD, and 1 in 5,000 for Duchenne muscular dystrophy (DMD) [4]. However, due to a large number of the diseases, approximately 1 in 21 are affected [3].
The treatment of genetic diseases is challenging. As an example, the treatment options for SCD, the most common monogenic blood diseases affecting millions of people around the world, remain limited, and the lifespan of the patients has not improved during the last decades, even though several drugs reducing the complications were approved and used in the last few years [6]. Allogenic hematopoietic stem cell (HSC) transplantation is the only curative treatment available to less than 15% of SCD patients. Recently, gene therapy through lentiviral vector-mediated addition of an antisickling protein, LentiGlobin, has proven to be successful in preclinical and is tested the durability of the safety and efficacy in clinical trials [7]. Also, as a common monogenic disorder, DMD is often on a par with SCD. The patients with DMD first show symptoms before age 5 and usually cannot walk at the age of 12 [8]. Corticosteroids such as prednisolone and deflazacort are the primary treatment currently, aiming to control the symptoms, but their benefits are limited. The estimated annual disease burden/cost of DMD is over 60,000 euros [9]. However, no cure for DMD is available, and the average lifespan is only 20–30 years. In the last few years, the United States Food and Drug Administration (FDA) approved three antisense oligonucleotides (ASOs), Exondys 51, Vyondys 53, and Amondys 45, for treating 14%, 8%, and 8% of DMD patients, respectively [10], [11], [12]. Regarding pathogenesis, genetic disorders, especially heritable diseases, are rooted in alterations in the genome. Correction of disease-causing mutations provides a potential therapy achieving permanent cure compared to other treatments.
Genome editing technology provides a powerful tool for treating genetic diseases by targeting pathogenic mutations in a site-specific manner. This technology has been widely used for basic laboratory research and attracts many efforts to prevent and treat various human diseases. These efforts have made enormous progress in pre-clinical studies, and some of the achievements have been successfully translated to clinical studies, such as chimeric antigen receptor T (CAR-T) cell-mediated immunotherapy [13]. This review focuses on genome editing approaches to treat human hereditary disorders. In the first part, we introduce the principles, representative findings in history, and the features of each genome editing tool. In the second part, we discuss the cutting edge of their ongoing therapeutic applications for hereditary disorders in clinics. Lastly, we also highlight this topic’s current challenges and future opportunities.
Overview of the genome editing tools
Genome editing modifies DNA sequences at target sites to disrupt or regulate the gene of interest or to repair harmful mutations. The last few decades have witnessed the rapid development and application of this technology, especially the discoveries of zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9 (CRISPR-Cas9). To precisely change the DNA sequence, homologous recombination (HR) mediated by nucleases (e.g., meganucleases [MegNs], TALENs, ZFNs, and CRISPR-Cas) and CRISPR-Cas-derived agents (e.g., base editors [BEs], prime editors [PEs], and transposases) are currently invented and applied in experimental systems [14, 15]. External nuclease-induced DNA double-strand breaks (DSBs) can be repaired by non-homologous end-joining (NHEJ) and homology-directed repair (HDR), resulting in gene disruption or precise integration and substitution [14]. Efficient HDR induced by nuclease in mammalian cells or organisms is promising for gene therapy [16]. CRISPR-Cas-derived agents independent of DSBs are another promising approach to rewriting the genome and curing genetic diseases [15]. Here, we will briefly summarize these promising approaches’ history, basic principles, and application prospects (Figure 1).
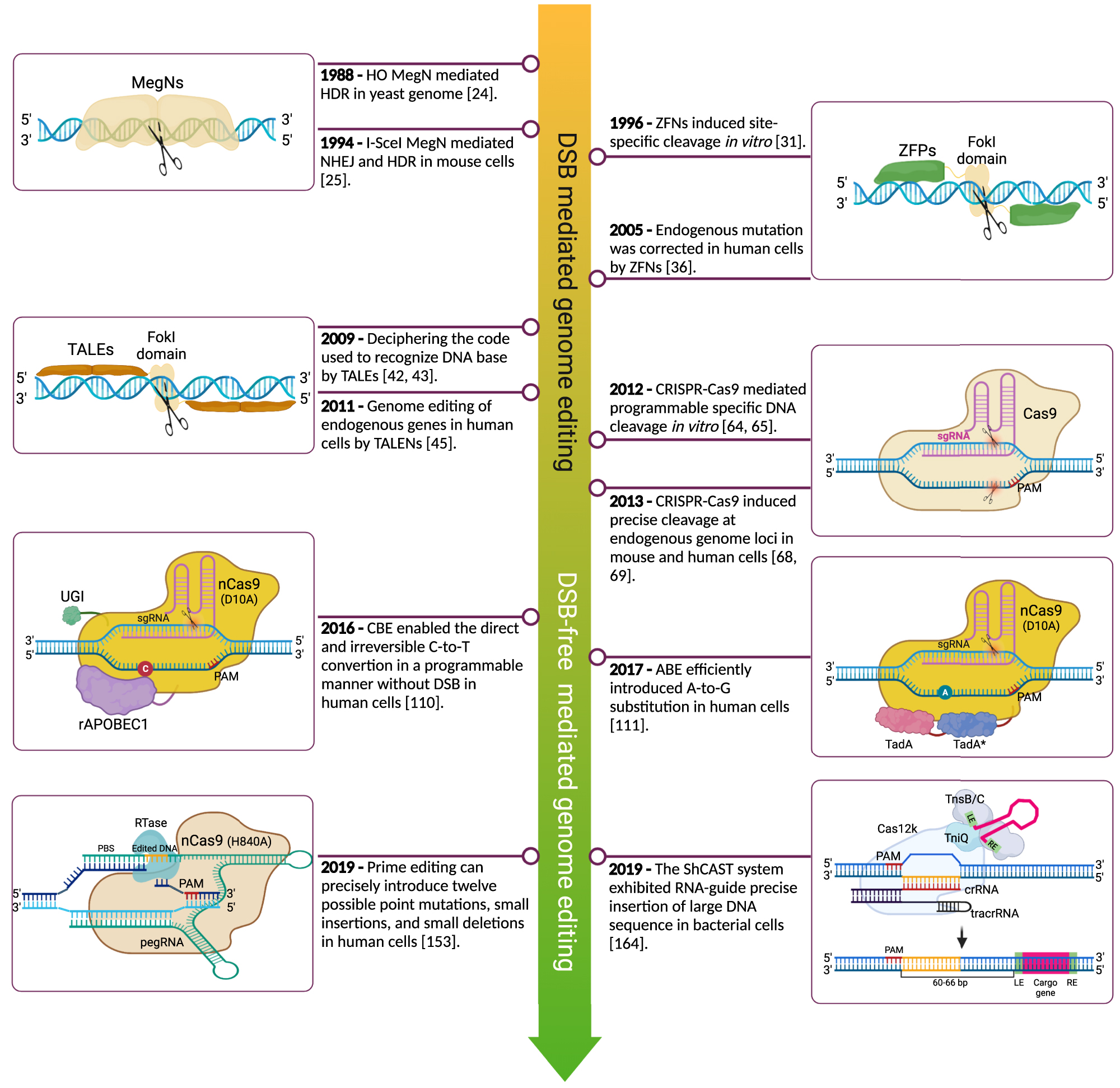
Timeline of the genome editing methods. The significant genome editing events from meganucleases mediated DNA double-strand breaks to transposases induce targeted insertions. After the emergence of CRISPR-Cas9 technology in 2012, it opened a new era of genome editing and derived a series of new agents that can efficiently rewrite the genome DNA. CRISPR-Cas9, clustered regularly interspaced short palindromic repeats-CRISPR-associated protein 9.
MegNs
MegNs, also termed homing endonucleases, are highly specific DNA cleaving endo-deoxyribonucleases. They found in mitochondria and chloroplasts of microbial life, which can be divided into six families based on amino acid sequence and structure motifs: LAGLIDADG, GIY-YIG, HNH, His-Cys box, PD-(D/E) XK, and EDxHD [17, 18]. These enzymes are small in size, and specially recognize and cleave double-strand DNA at specific sites compromising 14–40 bp, which can lead to site-specific recombination events resulting in insertion, deletion, and mutation correction [19]. Unlike restriction enzymes, MegNs generate at individual loci of their genome and facilitate site-specific gene/element conversion from the host to the recipient, thus referred to as homing [19]. One of the most well-studied MegNs families is the LAGLIDADG family, which is generally encoded within introns or inteins, such as I-CreI, I-SceI, and HO [17]. I-CreI is a homo-dimeric MegNs encoded by an ORF contained within a group I intron in the 23S rRNA gene of Chlamydomonas reinhardtii chloroplast, which recognizes and cleaves a 22-bp pseudo-palindromic recognition sequence [20]. I-SceI is a rare-cutting MegNs encoded by a mitochondrial group I intron of Saccharomyces cerevisiae with an 18 bp nonpalindromic target [21]. The HO protein makes a DSB at the yeast MTA locus, contains two LAGLIDADG motifs, and has a C-terminal zinc finger domain [22, 23].
MegNs enable inserting intended external DNA into the specific sites by HDR after DSB, which had been proved in model organisms by HO and I-SceI endonucleases [24], [25], [26]. In 1988, Rudin and Haber first reported that HO endonuclease stimulates efficient HDR between the homologous flanking sequences after site-specific DSB in yeast [24]. Furthermore, expression of I-SceI in mouse cells carrying cleavage sites in external DNA or endogenous genome stimulated HR using donor DNA as a template [25, 26], which sheds light on treating genetic diseases through HDR. However, while MegNs show promising applications for genome editing owing to their long DNA recognition sequence, the lack of target site in mammalian cells and the difficulty of engineering DNA-binding and cleave motifs ultimately make them hard for routine applications.
ZFNs
In principle, genome editing originated from the application of ZFNs technology. ZFNs are artificially engineered architectures by fusing the DNA-binding Zinc finger proteins (ZFPs) and the nuclease domain of the IIS restriction enzyme FokI. They are designed to target specific DNA sites [27]. ZFPs enable ZFNs to bind DNA sequences with specificity and contain a tandem array of Cys2His2 zinc fingers, one of which consists of approximately 30 amino acids and especially recognizes 3 bp DNA sequences [27, 28]. The FokI nuclease needs to be dimerized to cut DNA sequences, so a pair of ZFPs targeted upstream and downstream of the genomic site of interest need to be designed to induce DSB [29]. One ZFN can especially recognize DNA sequences up to 18 bp, typically 9 bp, depending on the number of fingers and the nature of critical amino acid residues of the DNA binding motif [27, 30].
Generally, DSBs generated by ZFNs can be repaired by NHEJ or HDR pathway, eventually resulting in random indels or exact replacement for genome editing (Figure 2). In 1996, Chandrasegaran’s group first constructed hybrid ZFNs to target intended DNA sequences in vitro, which opened a new way for genome editing [31]. Subsequently, ZFNs were applied to mutate genes in organisms, such as Drosophila [32], Caenorhabditis elegans [33], and zebrafish [34, 35]. A breakthrough came in June 2005 when Holmes’s group designed ZFNs to repair severe combined immune deficiency mutation of the IL2Rγ gene in human cells through HDR [36]. The authors proved that ZFNs permanently corrected the DNA sequences at a specific site, encouraging gene therapy in the clinic [36]. However, despite many successful and inspiring achievements at the cellular and organismal levels, some drawbacks limit the wide applications, such as customized protein for every DNA sequence [37], over toxicity to cells [38, 39], and high efficiency of the off-targets [40, 41]. Many efforts have been made to address these issues to improve the specificity and safety of ZFNs technology [27].
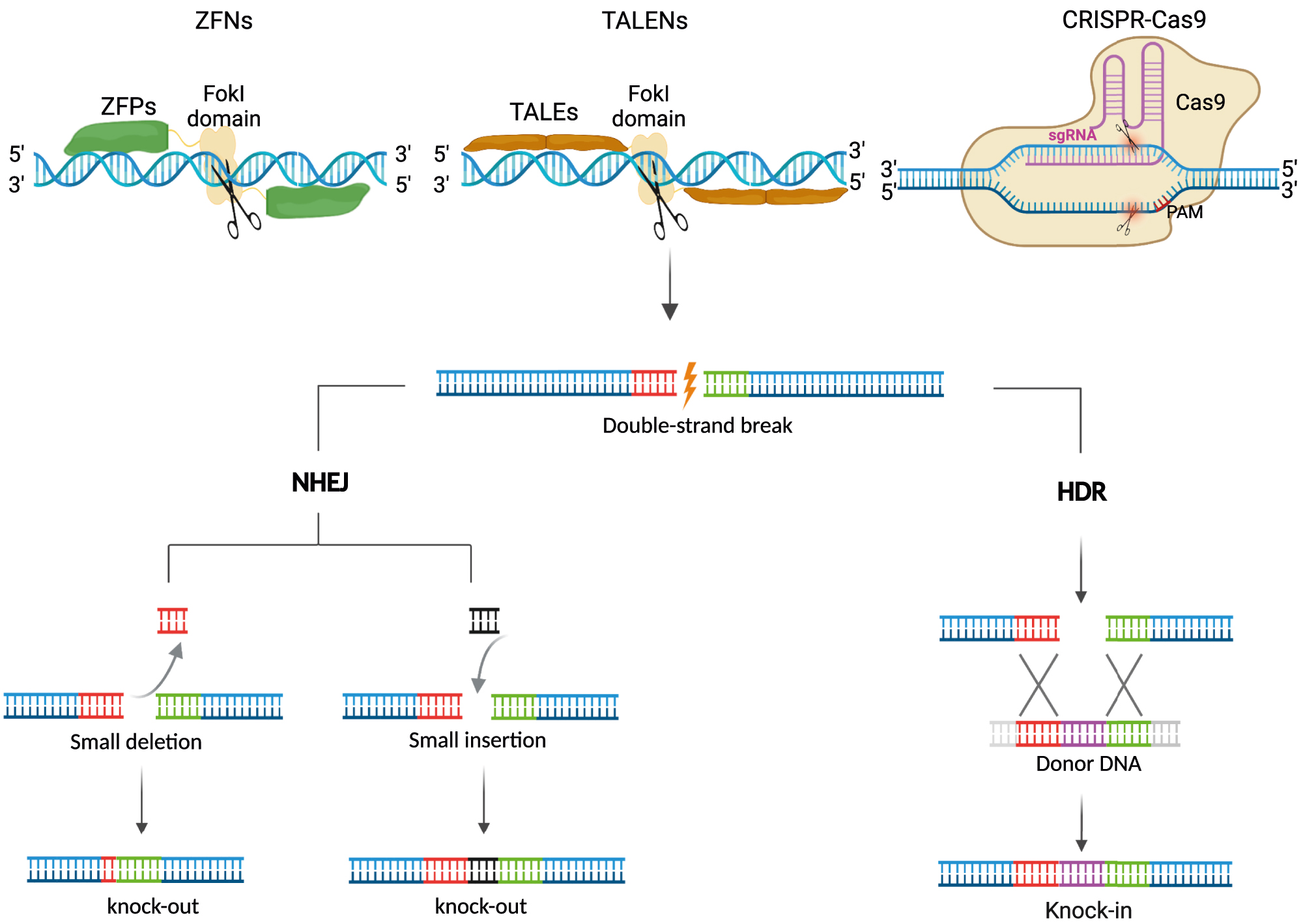
DSBs mediated genome editing. DNA DSBs at the targeted sites induced by nucleases (ZFNs, TALENs, and CRISPR-Cas9), which can be repaired by NHEJ without a donor or HDR in the presence of a donor DNA template. NHEJ can disrupt gene expression by forming indels (deletion and insertion), producing stop codons by frameshift mutations. HDR can precisely restore genetic mutation or insert gene fragments at a specific site. DSBs, double-strand breaks; ZFNs, zinc finger nucleases; TALENs, transcription activator-like effector nucleases; NHEJ, nonhomologous end-joining; HDR, homology-directed repair.
TALENs
Surprisingly, TALENs followed in the footsteps of ZFNs and ignited the genome editing revolution. In 2009, two groups deciphered the code used by transcription-activator-like effectors (TALEs) from plant pathogenic Xanthomonas to recognize the DNA sequences [42, 43]. The TALE protein contains three functional domains: a central DNA binding domain of 34 amino acids tandem repeats, two nuclear locational signals (NLSs), and an acidic transcriptional activation domain (AD) [43]. Unlike zinc fingers, which recognize DNA triplets, each repeat domain in the TALE proteins especially recognizes a single base, the specificity of which is determined by the dual residues at positions 12 and 13 of 34 amino acids repeat [42, 43]. These two amino acid pairs are the repeat variable di-residues (RVDs), including NI, NG, HD, and NN. Their TALEs preferentially recognize adenine, thymine, cytosine, and guanine/adenine [43, 44]. Based on ZFNs, TALE truncation variants fused with FokI nuclease (TALENs) enable efficiently cleaving of endogenous human genes at specific sites [45, 46]. Compared to ZFNs, TALENs showed easy operation, improved specificity, and reduced toxicity, improving their applications to fundamental research and gene therapy [47], [48], [49], [50]. Notably, many works have further optimized the TALENs for genome editing, such as the assembly of custom TALE arrays [51, 52], the DNA-binding specificity of the TALEs [53], and the delivered contents (plasmids, mRNA, or protein) into the cells [54]. Unfortunately, the discovery and rapid development of CRISPR–Cas technology made the reign of TALENs short-lived (Figures 1 and 2).
CRISPR-Cas
CRISPR-Cas technology originated from the discovery of a series of short, direct repeats (29 nt) interspaced by five intervening nonrepetitive sequences (32 nt) downstream of Escherichia coli iap gene in 1987 [55], which were named the clustered regularly interspaced short palindromic repeats (CRISPRs) in 2002 [56]. CRISPRs were later widely detected originating from extra-chromosome and phage-associated sequences in bacteria and archaea [57, 58]. The CRISPR-associated (Cas) genes that locate around CRISPR sequences encode proteins with putative nuclease and helicase activity [59]. In 2007, the CRISPR-Cas system was applied to resist phage infection in bacteria using CRISPR and enzymatic machinery of Cas protein-first experimental evidence for CRISPR-Cas as adaptive immunity [60].
The CRISPR-Cas systems can be classified as 1 and 2 based on multiple or single Cas effector modules for CRISPR RNAs (crRNAs) processing and target binding [61]. Furthermore, each class is composed of at least three types and multiple subtypes, including types I (such as Cas3), III, and IV in class 1 and types II (such as Cas9), V (such as Cas12 and Cas14), and VI in class 2 [61]. Besides, the protospacer adjacent motif (PAM), a short 2–5 bp sequences adjacent (e.g., NGG for Cas9; TTTV for Cas12a or Cpf1) to one end of the crRNA-targeted sequence, plays an essential role in recognition of targeted DNA sequences, especially for Type I and II [62, 63].
In 2012, Charpentier’s, Doudna’s, and Siksnys’s groups demonstrated that Cas9-crRNA ribonucleoprotein cleaved double-strand DNA guided by crRNA in vitro, which paved the way to use the CRISPR-Cas system for genome editing [64, 65]. Cas9 protein was derived from the pathogen Streptococcus pyogeneshas (named SpCas9). It contained two distinct nuclease domains, HNH and RuvC, which cleaved the complementary strand of crRNA and the opposite strand with NGG PAM, respectively [64, 66]. Inactivation of either the HNH (H840A) or the RuvC domain (D10A) in Cas9 creates a variant protein that cleaves only one DNA strand (named nickase Cas9, nCas9), whereas mutating both domains generated dead Cas9 (dCas9) with RNA-guided DNA binding ability [64, 67]. Based on these findings, the CRISPR-Cas9 system was quickly applied to engineer human and mouse genomes at multiple loci, opening a new age of genome editing [68], [69], [70]. Compared to ZFNs and TALENs, the customized RNA-guided CRISPR-Cas9 system is a facile, robust, and low-cost approach for fundamental research and gene therapy (Figure 2) [63].
Although wild-type SpCas9 was widely used in eukaryotic cells and animals, its specific NGG PAM requirement, high off-target, and large protein size (about 1,368 aa) limits its application for gene therapy. To expand the targeting scope of CRISPR-Cas9, the researchers generated various Cas9 variants with altered PAM profiles through structure-guided engineering or protein-directed evolution [63, 71]. Kleinstiver et al. identified SpCas9-VQR (NGA PAM), SpCas9-EQR (NGAG PAM), and SpCas9-VRER (NGCG PAM) through bacterial selection-based directed evolution combined with structural information [72]. Hu et al. used phage-assisted continuous evolution to evolve an xCas9 that can recognize NG, GAA, and GAT PAM sequences [73]. Nishimasu et al. reported a SpCas9-NG harboured relaxed NG PAM guided by structural information [74]. Miller et al. evolved three variants through phage-assisted evolution, including SpCas9-NRRH, SpCas9-NRTH, and SpCas9-NRCH, collectively recognising NRNH PAMs (where R is A or G and H is A, C, or T) [75]. Recently, Walton et al. engineered SpG-Cas9 with high activity to NG PAM and SpRY-Cas9 (where Y is C or T) with NRN and NYN PAMs, which offer capabilities to target almost all genome sites [76]. These PAM flexible Cas9 variants significantly facilitate genome editing. Besides, other CRISPR-Cas enzymes improved the targeting abilities of the genome, such as Cas12a (formerly known as Cpf1) with TTTV PAM [77], Cas12j (formerly CasФ) with TTN PAM [78], and Cas12f (formerly Cas14) with TTTR PAM [79]. Collectively, these Cas proteins will expand the future applications of genetic disease treatment.
One major concern associated with using the CRISPR-Cas system for gene therapy was the high efficiency of off-target mutations [80, 81]. Currently, there are many attempts to overcome this concern and improve the specificity of this system, including reusing and engineering Cas proteins and modifying single-guided RNA (sgRNA). First, nCas9 with appropriately paired sgRNAs was applied to introduce two different nicks at the single-strand DNA, resulting in a targeted double-strand DNA break, which showed high fidelity owing to individual nicks in the genome being repaired [82, 83]. Two hybrid nucleases, a programmable DNA binding domain (Zinc fingers or TALEs) fused with SpCas9 variant that disabled DNA and dCas9 fused with FokI nucleases binding, also increased the specificity of the Cas9 system [84, 85]. Secondly, several studies have evolved showing Cas9 variants reduced off-targets while maintaining editing efficacy, including SpCas9-HF1 [86], eSpCas9 [87], HypaCas9 [88], HeFSpCas9 [89], evoCas9 [90], HiFiCas9 [91], and Sniper-Cas9 [92]. Finally, by computerized analysis, engineering sgRNA structure and optimizing the sgRNA sequence decreased the off-target mutagenesis and improved the editing specificities [93], [94], [95].
For gene therapy, adeno-associated viruses (AAVs) are a popular way to package and deliver genes, but the cargo capacity is only about 4.7 kb [96, 97]. An all-in-one AAV delivery system would boost the therapeutic effect. CRISPR-Cas9 system, though, including the promoters, sgRNA, Cas9, and cis-regulatory elements, exceeds the cargo size of a single AAV vector for efficient delivery in vivo [98]. The split AAVs with separate SpCas9 and sgRNA, or split-Cas9 architecture, have been developed to overcome this limitation but with reduced fusion protein expression [99, 100]. An all-in-one AAV delivery system would boost the therapeutic effect, and the smaller Cas proteins mediated in vivo genome editing raised the potential of efficient gene therapy, including SaCas9 (1,053 aa) [98], CjCas9 (984 aa) [101], Cas12j (700–800 aa) [78], Cas12f (529 aa) [102], etc.
In summary, the multiple CRISPR-Cas systems are a versatile and robust tool for treating genetic diseases. Still, because they require DSBs, they lead to the generation of a minimum of unwanted structural variants [103], chromosomal deletions and rearrangement [104], [105], [106], and p53 activation [107, 108].
BEs
In recent years, CRISPR-Cas-derived BEs efficiently converse one base of genome DNA into another in a programmable manner without requiring DSBs or a donor template [15, 109]. DNA base-editors are mainly composed of the nCas or dCas systems for recognizing the specific DNA site and cytosine or adenine deaminase to catalyze nucleotide conversion in a single-strand DNA [109]. Currently, three types of BEs have been developed, including cytidine base editor (CBE, C to T) [110], adenosine base editor (ABE, A to G) [111], and glycosylase base editors (GBE, C to A or C to G) [112, 113]. In addition, recent studies reported a dual adenine and cytosine base editor by fusing both deaminases with nCas9 to simultaneously achieve C-to-T and A-to-G conversions at the same target site in human cells, in which RNA off-target activity is substantially decreased [114], [115], [116]. As approximately half of all pathogenic genetic variants are due to single-nucleotide variants (SNVs) [117], these new BEs offer great potential for treating numerous genetic diseases through permanent DNA transversion.
The first powerful CBE system (BE3), consisting of the nCas9, the rat cytidine deaminase rAPOBEC1, and uracil glycosylase inhibitor (UGI), was developed by Liu’s group in 2016 [110], which was used to correct disease mutation and genetic screening by inducing C•G to T•A conversion [118, 119]. There are three steps to convert base C to T for the BE3 system (Figure 3A) [120]: (i) The BE3 complex is targeted to the specific site of genome-guided by the sgRNA; (ii) The rAPOBEC1 changes base C to U within a window of 4–8 bases away from the PAM of the sgRNA, and the nCas9 simultaneously creates a nick in the non-deaminated strand; (iii) Owing to the inhibition of UGI to the uracil N-glycosidase (UNG), which can remove the base U and perform error-free or error-prone repair and generate different substitutions, the intact deaminated strand is preferred template for endogenous mismatch repair, generating a U•A base pair that is then resolved as a T•A base pair after DNA replication or repair. Subsequently, other cytidine deaminases were also shown to efficiently convert specific base C to T by coupling with nCas9, such as activation-induced cytidine deaminase (AID) [121, 122], human APOBEC3A [123, 124], human APOBEC3G [125, 126], and CDA1 [125] (Figure 3B).
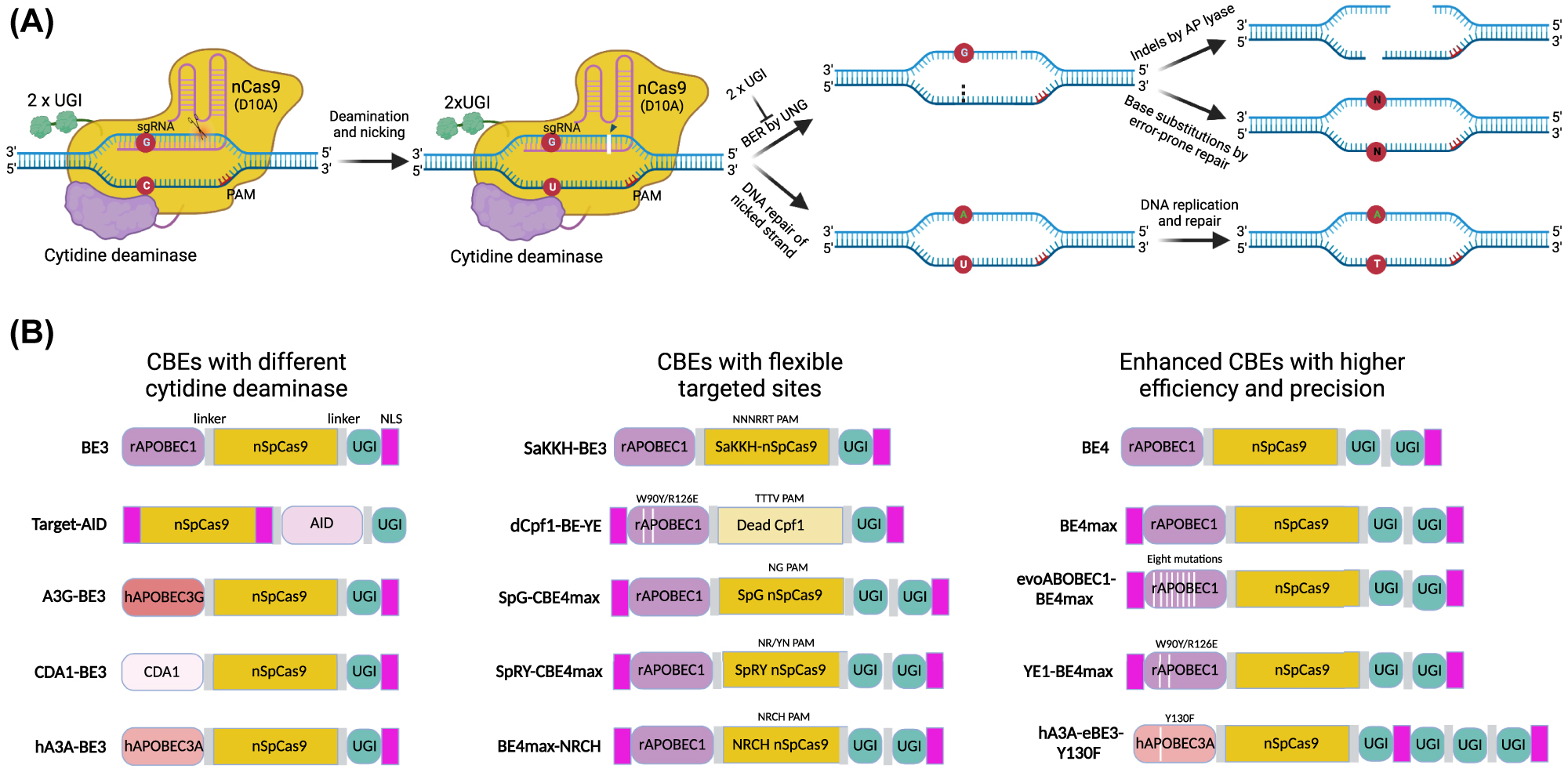
Cytosine base editing. (A) Schematic diagram of a CBE system using nCas9 (D10A) linked with cytosine deaminase and double UGI as an example. The nCas9-sgRNA complex binds to the targeted DNA, forms a single-stranded R-loop, and nicks the non-deaminated strand. The linked cytidine deaminase catalyzes the hydrolytic deamination of cytosine (C) to uridine (U). An additional linked protein, UGI, inhibits the activity of UNG, which excises the U from DNA in cells and initiates BER, with the reversion of the U•G pair to an n•n pair (C•G as the most common outcome). The nick induced by apyrimidinic site lyase (AP lyase) on the deaminated strand combined with the nick on the non-deaminated strand by nCas9 results in the DSB and indels. The intact deaminated strand is preferred for templated repair, and DNA polymerase reads U as thymine (T), generating the U•A base pair to T•A base pair. (B) Schematic diagrams showed different versions of cytosine base editors, including deamination by different enzymes, targeting DNA by various Cas proteins, and evolved CBEs with higher activity and precision. CBE, cytidine base editor; nCas9, nickase Cas9; UGI, uracil glycosylase inhibitor; UNG, uracil N-glycosylase; BER, base-excision repair.
In addition, CBE systems still face some challenges for gene therapy, including low editing efficiency and bystander mutations [118, 119], PAM and editing window limitations [127], DNA and RNA off-target effects [128], [129], [130], [131], and packaging constraints [132]. Recently, we have witnessed an improvement in their product purities and efficiencies, expanded the scope of targeting, minimized the undesirable DNA and RNA off-targets and engineered more compact CBE systems (Figure 3B) [133]. To improve the intended editing efficiency (C-to-T mutation) of BE3, additional nuclear-locational sequences [118, 134] and more UGIs [125, 135] both dramatically improved the editing efficiency with fewer indels or by-products. Different CRISPR-associated nucleases fused with evolved cytidine deaminases can increase the target scope and efficiencies of CBEs, such as dCpf1-BE (TTTV PAM) [136], SaBE3 (NNGRRT PAM) [127], SaKKH-BE3 (NNNRRT PAM) [127], SpG-or SpRY-CBE4max [76], evoAPOBEC1-BE4max [137], and CBE-NG [75]. Protein engineering showed that W90Y and R126E mutations in the rAPOBEC1-derived YE1 CBE system showed minimal Cas9-independent DNA and RNA off-targets but retained high on-target activity [138, 139]. Besides, other cytidine deaminases, hAPOBEC3A, hAPOBEC3G, AmAPOBEC1, SsAPOBEC3B, or PpAPOBEC1, derived CBE systems exhibited minimum off-target effects [123, 126, 140]. Due to the limitation of molecular size for AAV delivery, CBE has been split into two AAV vectors for delivery in vivo, demonstrating the potential for therapeutic application [132], thus guarantying the single-AAV system as a promising approach for delivery [141]. Thus, CBE is a popular and promising genome editing tool and could potentially reverse about 5,000 pathogenic point mutations associated with human genetic diseases [109].
Drawing inspiration from the CBEs, Liu’s group quickly performed seven rounds of evolution for E. coli tRNA-specific adenine deaminase TadA and identified a mutant deaminase TadA* that deaminated ssDNA adenosine to inosine (A-to-I editing) in human cells, which was read or replicate as guanine (G) (Figure 4A) [111]. The original ABE version, ABE7.10, consists of wild-type and mutant TadA, nCas9, and sgRNA, which can efficiently convert targeted A•T base pairs to G•C [111]. Recently, several strategies have been adopted to optimize the ABE systems (Figure 4B): (i) ABEmax, an efficient editor obtained by codon optimization of ABE7.10 with bis-bpNLS [142], fused with SpCas9-NG to construct NG-ABEmax with expanded targeting scope [143], the efficiency of which also been improved by NG-ABEmax-KR obtained through human cell-based direct evolution [144]. ABEmax, combined with SpG or SpRY Cas9, targeted almost all PAMs with vigorous activities in the human genome [76]. (ii) Protein evolution of ABE7.10 resulted in ABE8e [145] and ABE8s [146] with only a TadA variant monomer, showing greatly editing efficiency and lower RNA off-target [147]. (iii) V106W or V82G mutation in the TadA* domain [145, 147, 148] or F148A mutation in both TadA and TadA* [131] derived ABEs possess significantly lower RNA off-target effects. (iv) ABEs also catalyzed bystander cytosine deamination at the target site [149], which was significantly reduced by introducing the D108Q mutation in the TadA* domain [150]. (v) Compact Cas9 ortholog combined with ABE8e was efficiently delivered by a single AAV vector with high efficiency of in vivo base editing [151, 152]. These evolved ABEs are functional classes of editing agents because approximately half of the pathogenic single-nucleotide polymorphisms (SNVs) can be corrected by installing A•T→G•C conversions [109].
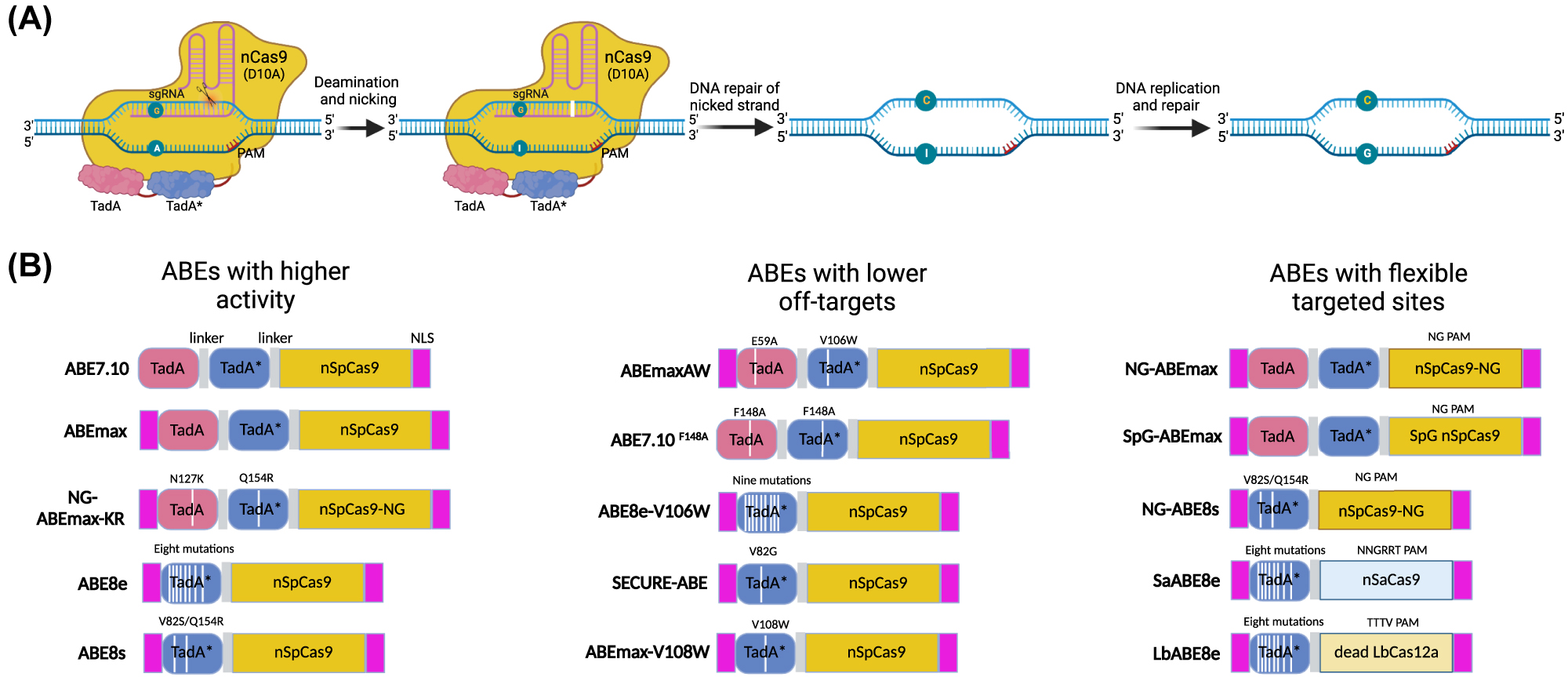
Adenine base editing. (A) Schematic diagram of an ABE system using ABE7.10 as an example. The nCas9-sgRNA complex binds to the non-deaminated strand and forms a nick on it. The fused evolved TadA* catalyzes the deamination of adenosine (A) into inosine (I), which is read as guanosine (G) by DNA polymerase during DNA replication. In the example shown, two deoxyadenosine domains exist, including one wild-type TadA domain and an evolved TadA* domain. Subsequent research found that a single TadA* domain can achieve A•T to G•C base editing. As with uracil, inosine can also be excised by endogenous glycosylases, generating an abasic site that leads to base substitutions or indels, but this process is inefficient. (B) Schematic diagrams showed different versions of ABEs. The evolved ABEs showed higher editing efficiency and lower off-target effects. The Cas variants fused with deoxyadenosine deaminase can target broader genome loci, which are suitable for more genetic mutation repair. ABE, adenosine base editor.
Currently, the CBEs and ABEs catalyzed DNA base transitions, pyrimidine-to-pyrimidine or purine-to-purine. However, base transversions (pyrimidine-to-purine) are challenging to achieve through base editing [112, 113], which also plays an essential role in repairing pathogenic SNVs [109]. Therefore, the researchers have developed GBE that can efficiently transverse C-to-G mutation in human cells and C-to-A in E. coli by replacing the UGI on the BE3 with UNG or directly deleting UNG [112, 113]. Although the optimizations of these BEs have shown explosive growth, there is still a lot of room to maximize their efficiency, specificity, safety, and ability to be delivered in vivo.
PEs
PE system is another newly developed genome editing tool that can achieve other functions besides BEs, such as small DNA insertions and deletions, larger deletions, and A-to-C or A-to-T base substitution [153, 154]. The original PE1, a versatile and precise gene-editing method, is composed of a Cas9 nickase (H840A) fused to engineered reverse transcriptase (M-MLV RTase) and a modified sgRNA known as prime editing guide RNA (pegRNA), which can induce all twelve types of base substitutions and small indels without DSBs [153]. In addition, the engineered M-MLV RTase improved the editing efficiency of PE1 (hereafter named PE2). Furthermore, an additional sgRNA was used to introduce nicks on the non-edited strand by nCas9, further boosting the activity of PE2 (designated as PE3 and PE3b) (Figure 5) [153], which quickly applied to genome editing in animals and plants [155], [156], [157].
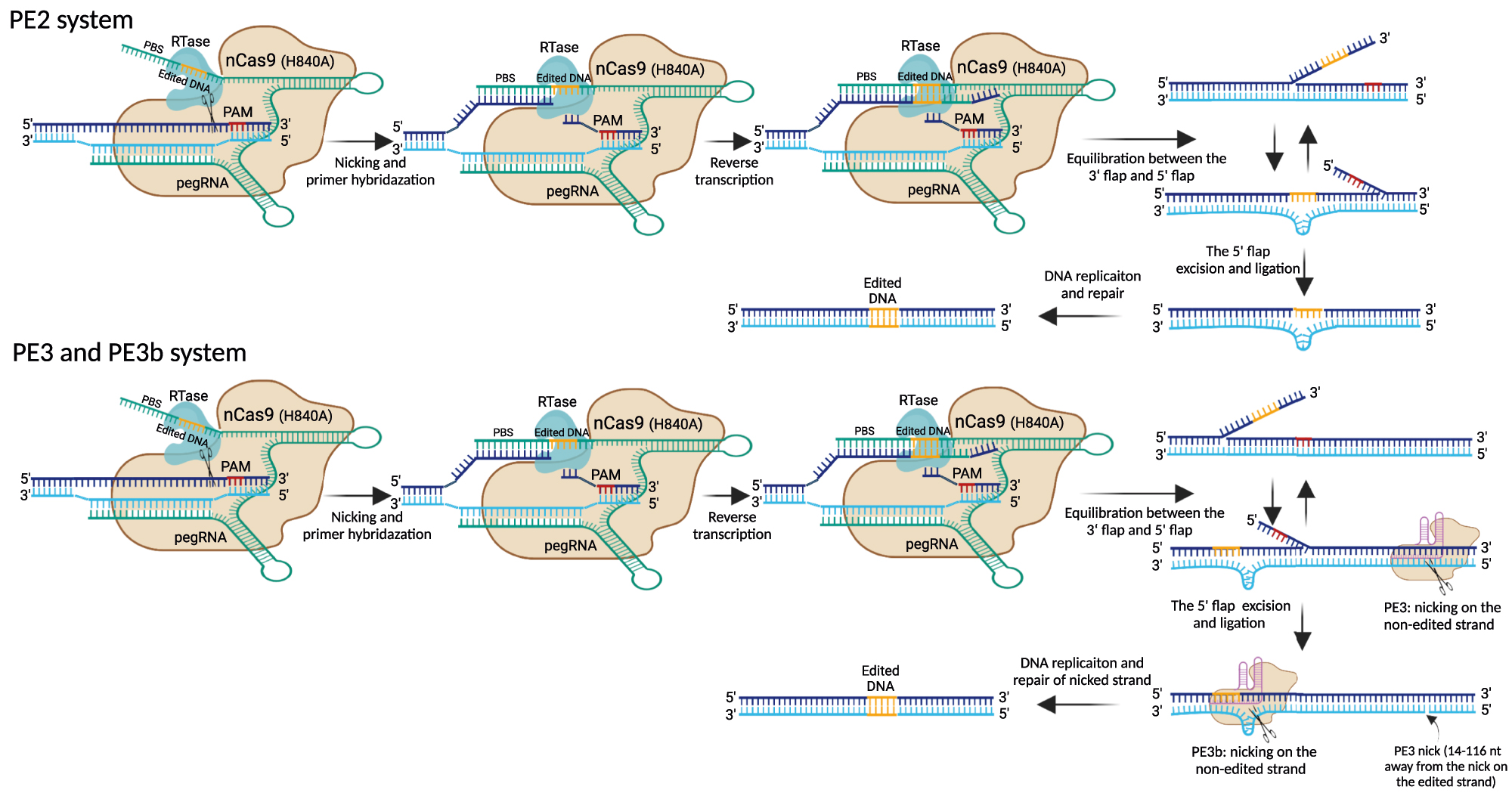
Overview of prime editing by PE2, PE3, and PE3b. The prime editors consist of nCas9 (H840A) fused to RTase via a linker and pegRNA, which consists of gRNA sequence for targeting DNA, PBS for initiating reverse transcription, and RT template for introducing edited DNA. After nicking the PAM-containing strand by the nCas9, the newly released DNA 3′ end hybridizes to the PBS sequence of pegRNA to form a primer-template complex. The RTase domain then initiates reverse transcription and adds the edited DNA from the pegRNA into the genomic DNA, resulting in a heteroduplex DNA. After reverse transcription, the PE2 system involves flap equilibrium of edited 3′ and unedited 5′ flap. Endonucleases excise the unedited 5′ flaps, and then ligation and DNA replication/repair lead to stable editing. PE3 and PE3b involve a second nick on the non-edited strand to ensure that the edited strand is used for DNA replication and repair. In the PE3 approach, the second nick is introduced by the nCas9-sgRNA complex in the unedited strand 14–116 nt away from the initial pegRNA nick site. In the PE3b approach, the nCas9-sgRNA complex can only match the edited strand after the edited DNA was incorporated and induces a nick on the non-edited strand, which can prevent the presence of concurrent nicks. PE, prime editor; RTase, reverse transcriptase; pegRNA, prime guide RNA; PBS, primer binding site.
However, a significant determinant of excellent editing efficiency was the selection of the pegRNA, which contains 3′ extensions with a primer binding sequence (PBS) that anneals to the target genomic site and an RT template that encodes the intended edit [153]. Recently, computer models from deep learning enabled the quick prediction of the editing efficiency of pegRNA for PE2 in human cells [158]. Furthermore, a web-based pegFinder, for the rapid design and score of pegRNAs from reference was developed [159], which would facilitate the widespread applications of prime editing. It has been shown that engineered pegRNA with stabilized 3′ terminus (epegRNA) or same-sense mutations at proper positions significantly improved the editing efficiency [160, 161]. In addition, CRISPRi screens found that DNA mismatch repair (MMR) inhibited prime editing efficiency and promoted undesired indel byproducts, which could be blocked by an engineered MLH1 protein [162]. Thus, the PE4 and PE5 obtained by co-expression of PE2 and PE3 with MLH1 significantly enhanced editing efficiency and reduced indels [162].
While prime editing demonstrates a more versatile new approach to rewriting the genome compared to base editing and nucleases mediated HDR, base editors (CBEs and ABEs) also show higher efficiency and precision when a single target nucleotide is present within the base editing window [15, 153]. Thus, prime editing and base editing offer complementary strengths and weaknesses to make targeted gene mutations through DSB-free genome editing [163]. However, to achieve the therapeutic potential, efficiency, safety, and delivery strategies remain to be stressed for prime editing.
CRISPR-associated transposases (CASTs)
CAST system is emerging as a powerful tool for targeted genomic knock-in and knock-out in bacterial cells. This prevents the need for DSBs in the target DNA and host-cell repair machinery [164], [165], [166], [167]. In 2019, two groups demonstrated that Tn7-like transposons from cyanobacteria combined with the CRISPR-Cas system to efficiently perform sgRNA-guided targeted insertions [164, 165]. Strecker et al. reported a CAST system from cyanobacteria Scytonema hofmanni (ShCAST) that can insert cargo DNA into a specific site [164]. The ShCAST consists of three main parts: (i) the inactive type V-K (Cas12 k) Cas with guide RNA array; (ii) the Tn7-like transposase subunits (TnsB-TnsC-TniQ); (iii) a cargo DNA flanked by the transposon left end (LE) and right end (RE) elements [164]. CRISPR-Cas12k was solely a DNA binding function and efficiently targeted a 24 bp spacer sequence with requisite 5′-NGTN-3′ PAM. Among the transposition machinery, TnsB and TnsC specifically recognize the LE and RE sequence and catalyze the cleavage of the cargo DNA. At the same time, TniQ is required for RNA-guided insertions in E. coli [164]. ShCAST catalyzes RNA-guided DNA insertion predominantly at 60–66 bp downstream of the PAM sequences in the E. coli genome, which displays a T or A preference (about 3 nt) upstream of the insertion site. The insertion only occurred in the forward 5′-LE-cargo-RE-3′ orientation, which was highly efficient and can insert up to 10 kb [164].
Meanwhile, Klompe et al. established the VchCAST (Tn6677) system from Vibrio cholera. They proved that it had the potential to integrate the donor DNA in two possible orientations at 79 to 83 bp downstream of the PAM sequence (5′-CC-3′) [165]. The Tn6677 system contains three plasmids: pQCascade, pTnsABC, and LE-cargo-RE transposon. pQCascade encodes the CRISPR–Cas associated machinery (crRNAs, Cas6, Cas7, and Cas8) and the transposition protein TniQ. TniQ binds to the Cascade complex as a dimer in a head-to-tail configuration and links the Cas machinery and the transpososome (transposase complex with DNA transposon) [168, 169]. Because the Tn6677 system consists of the type I-F CRISPR system, it is more complicated than the ShCAST system. Still, since no crRNA is required, it is more suitable for directing multiple cargos into the same or multiple genomic targets [170, 171]. Together, the CAST systems stir exciting opportunities in genome editing for specific rearrangements of large DNA sequences or chromosomes. Still, continued efforts need to improve these systems for genome editing in mammalian cells [170].
Progress of genome editing in the treatment of inherited diseases
Therapeutic genome editing aims to target the human genes “hiding” in the cells. In the pathogenesis of inherited diseases, gene mutations often lead to the dysfunction of different systems of the patients. Considering that, it is essential to design and implement a suitable genome editing strategy involving target cell types, the tissues (organs or systems) and their corresponding delivery approaches. In our bodies, somatic cells mainly contribute to the function of various tissues. In contrast, germ cells are responsible for giving rise to gametes and embryos, a process that could pass the disease-causing mutation to the offspring. Notably, genome-editing therapy in somatic and germ cells is quite different. Therapeutic somatic cell editing only affects the patient himself. However, germ cell editing would potentially introduce genetic changes into the offspring. This will inevitably raise enormous ethical challenges beyond safety issues. In this section, we review the progress of therapeutic somatic cell editing and discuss the possible attempts for therapeutic germ cell editing in research and clinical use.
Ex vivo editing in somatic cells
All therapeutic genome editing in development is based on somatic cell editing (Tables 1 and 2). As mentioned above, editing the somatic cells of the different tissues requires a feasible approach with the considerations of target cells and ways of delivering and editing. Somatic cell editing can be divided into ex vivo and in vivo (Figure 6).
Summary of therapeutic genome editing ex vivo for inherited diseases in clinical trials.
| Editing tools | Diseases | Target cells | Target gene | Delivery | Mechanisms | Drug name | Year | Current status | Sponsor | Trial numbera |
|---|---|---|---|---|---|---|---|---|---|---|
| CRISPR-Cas9 | Sickle cell disease | HSPCs | BCL11A ESE | NA | NHEJ | OTQ923/HIX763 | 2020 | I/II, Recruiting | Novartis pharmaceuticals | NCT04443907 |
| Sickle cell disease | HSCs | HBB | Electroporation | HDR? | CRISPR_SCD001 | 2021 | I/II, not yet recruiting | University of California, san francisco | NCT04774536 | |
| Sickle cell disease | HSCs | HBB | RNP electroporation, AAV6 for teample | HDR | GPH101 | 2021 | I/II, Recruiting | Graphite bio | NCT04819841 | |
| Sickle cell disease | HSPCs | BCL11A ESE | Electroporation of RNP | NHEJ | CTX001 | 2018 | II/III, active, not recruiting | Vertex pharmaceuticals | NCT03745287 | |
| Sickle cell disease (pediatric) | HSPCs | BCL11A ESE | Electroporation of RNP | NHEJ | CTX001 | 2022 | III, Recruiting | Vertex pharmaceuticals | NCT05329649 | |
| Sickle cell disease, β-Thalassemia | HSPCs | BCL11A ESE | Electroporation of RNP | NHEJ | CTX001 | 2019 | n/A | Vertex pharmaceuticals | NCT04208529 | |
| β-Thalassemia | HSPCs | BCL11A ESE | Electroporation of RNP | NHEJ | BRL-101 | 2020 | I/II, Recruiting | Biorary laboratories | NCT04211480 | |
| β-Thalassemia | HSPCs | BCL11A ESE | Electroporation of RNP | NHEJ | CTX001 | 2018 | II/III,Active, not recruiting | Vertex pharmaceuticals | NCT03655678 | |
| β-Thalassemia | HSPCs | BCL11A ESE | NA | NHEJ | ET-01 | 2021 | I, active, not recruiting | EdiGene | NCT04925206 | |
| β-Thalassemia with CVS-654 mutation | HSCs | HBB | Electroporation | NHEJ? | NA | 2021 | I/II, enrolling by invitation | Biorary laboratories | NCT04205435 | |
| CRISPR-Cas12a | Sickle cell disease | HSCs | HBG1andHBG2 promotersb | Electroporation of RNP | NHEJ | EDIT-301 | 2021 | I/II, recruiting | Editas medicine | NCT04853576 |
| ABEs | Sickle cell disease | HSPCs | HBB | Electroporation | T to C | BEAM-101 | 2022 | I/II, not yet recruiting | Beam therapeutics | NCT05456880 |
| ZFNs | Sickle cell disease | HSPCs | BCL11A ESE | mRNA transfection | NHEJ | BIVV003 | 2019 | I/II,Recruiting | Sangamo therapeutics | NCT03653247 |
| Sickle cell disease | HSPCs | BCL11A ESE | mRNA transfection | NHEJ | BIVV003 | 2021 | n/A, recruiting | Sangamo therapeutics | NCT05145062 | |
| β-Thalassemia | HSCs | BCL11A ESE | Electroporation and AAV | NHEJ | ST-400 | 2018 | I/II, active, not recruiting | Sangamo therapeutics | NCT03432364 | |
| β-Thalassemia | HSCs | BCL11A ESE | Electroporation and AAV | NHEJ | ST-400 | 2021 | n/A, recruiting | Sangamo therapeutics | NCT05145062 |
-
a www.clinicaltrials.gov; bThe regions are BCL11A binding sites.
Summary of therapeutic genome editing in vivo for inherited diseases in clinical trials.
| Editing tools | Diseases | Target cells | Target gene | Delivery | Mechanisms | Drug name | Year | Current status | Sponsor | Trial number |
|---|---|---|---|---|---|---|---|---|---|---|
| ZFNs | Hemophilia B | Hepatocytes | Albumin intron 1 | rAAVs | HDR | SB-FIX | 2016 | I, terminated | Sangamo therapeutics | NCT02695160; NCT04628871 |
| Mucopolysaccharidosis I | Hepatocytes | Albumin intron 1 | rAAV2/6 | HDR | SB-318 | 2016 | I/II, terminated | Sangamo therapeutics | NCT02702115; NCT04628871 | |
| Mucopolysaccharidosis II | Hepatocytes | Albumin intron 1 | rAAV2/6 | HDR | SB-913 | 2017 | I/II, terminated | Sangamo therapeutics | NCT03041324; NCT04628871 | |
| CRISPR-Cas9 | Hereditary transthyretin amyloidosis | Hepatocytes | TTRs | LNP | NHEJ | NTLA-2001 | 2020 | I, recruiting | Intellia therapeutics | NCT04601051 |
| Hereditary angioedema | Hepatocytes | KLKB1 | LNP | NHEJ | NTLA-2002 | 2021 | I/II, recruiting | Intellia therapeutics | NCT05120830 | |
| Leber congenital amaurosis | Photoreceptor cells | CEP290 | rAAV5 | NHEJ | EDIT-101 | 2019 | I/II, recruiting | Editas medicine | NCT03872479 | |
| ABEs | Heterozygous familial hypercholesterolemia | Hepatocytes | PCSK9 | LNP | A to G | VERVE-101 | 2022 | I, recruiting | Verve therapeutics | NCT05398029 |
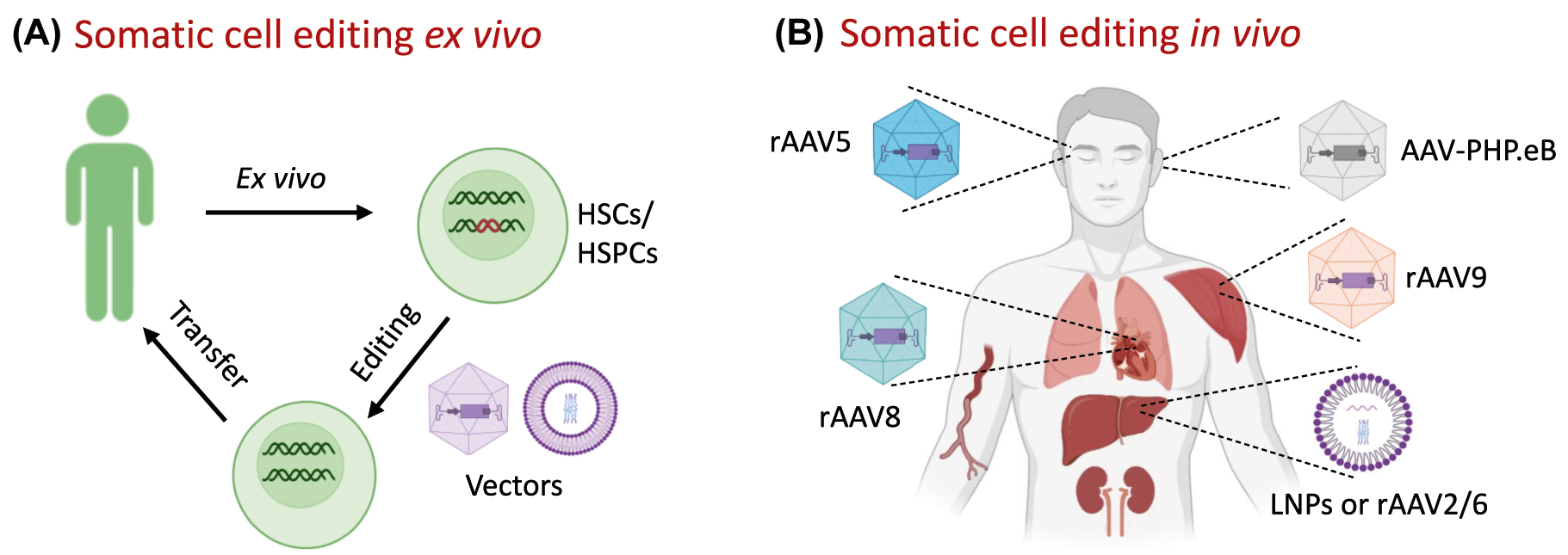
Strategies for therapeutic genome-editing. (A) Schematic diagram for somatic cell editing ex vivo. Bone marrow cells of patients are mobilized and HSCs or HSPCs are isolated and cultured ex vivo. The genome editing components are packed and delivered into cells, usually by electroporation or AAVs. After quality control, the edited HSCs/HSPCs are infused into the patients for treatments. (B) Schematic diagram for somatic cell editing in vivo. The genome editing components are packed into the delivery vehicles with organ tropism, such as AAVs and LNPs, then injected directly into patients. HSCs, hematopoietic stem cells; HSPCs, hematopoietic stem and progenitor cells; AAVs, adeno-associated viruses; LNPs, lipid nanoparticles.
Ex vivo editing followed by adoptive transfer
Ex vivo somatic editing is suitable for the inherited disorders of the hematopoietic system, such as immune deficiencies, erythrocyte diseases and platelet disorders. The most prevalent attempts aim to cure SCD and β-thalassmia, both caused by a single nucleotide mutation in the gene that encodes the β-globin (HBB). In erythrocytes, two β-globin subunits assemble into a tetramer with two α-globin subunits, called adult hemoglobin (HbA), which binds oxygen with high affinity. In SCD erythrocytes, the sickle hemoglobin (HbS), resulting from A-to-T mutant at position 20, polymerizes into fibers under hypoxic or acidic conditions, distorting the erythrocytes to the “sickle” shape with shortened lifespan and reduced function, causing hemolytic anemia and multi-organ damage [172]. Various single nucleotide substitutions, small deletions or insertions within the HBB gene cause β-thalassemia, resulting in reduced (β+) or absent (β0) β-globin synthesis and anemia [173]. Less than 15%–20% of the SCD and β-thalassemia patients have histocompatible (human leukocyte antigen–matched) healthy donors and are curative by allogenic HSC transplantation [174], [175], [176]. HSC gene therapy provides a promising way to treat SCD, which relay on gene correction of autologous HSC followed by transplantation (Figure 6A). Several gene targets involved in regulating the β-globin gene or γ-globin (forming fetal Hb, abbreviated as HbF), which has anti-sickling effects, have been proposed and proved useful [177, 178]. One of the employed strategies is gene addition by lentivirus in patient CD34+ HSCs that are widely tested in clinical trials and reviewed elsewhere [179].
CRISPR-Cas9 in erythrocyte diseases
Currently, mutant HBB and BCL11A erythroid-specific enhancers (ESE) are the two most popular targeting sites in gene-editing trials. Two clinical trials regard to HBB editing by CRISPR-Cas9 for the treatment of serve SCD are in progress at phase 1/2 in the USA [180, 181]. One of them, GPH101 from Graphite Bio, is the first CRISPR-based genome correction therapy that harnesses CRISPR-Cas9 and HDR mechanisms to cut out the mutation and repair with wild-type (WT) DNA sequence [182] (Figure 7A). In this platform, Cas9-sgRNA, is delivered through ribonucleoprotein (RNP) complexes. In contrast, the DNA template is delivered through an AAV6 vector ex vivo, and the preclinical data demonstrated this strategy’s safety, efficacy, and reproducibility [183]. Furthermore, in 2021, a clinical study of β-globin restored autologous HSCs using CRISPR/Cas9 for β-thalassemia major patients with CVS-654 mutation started (Shanghai Bioray Laboratories and PLA 923 Hospital) in China [184]. This disease subtype is also known as transfusion-dependent β-thalassemia (TDT), the most severe form of β-thalassemia.
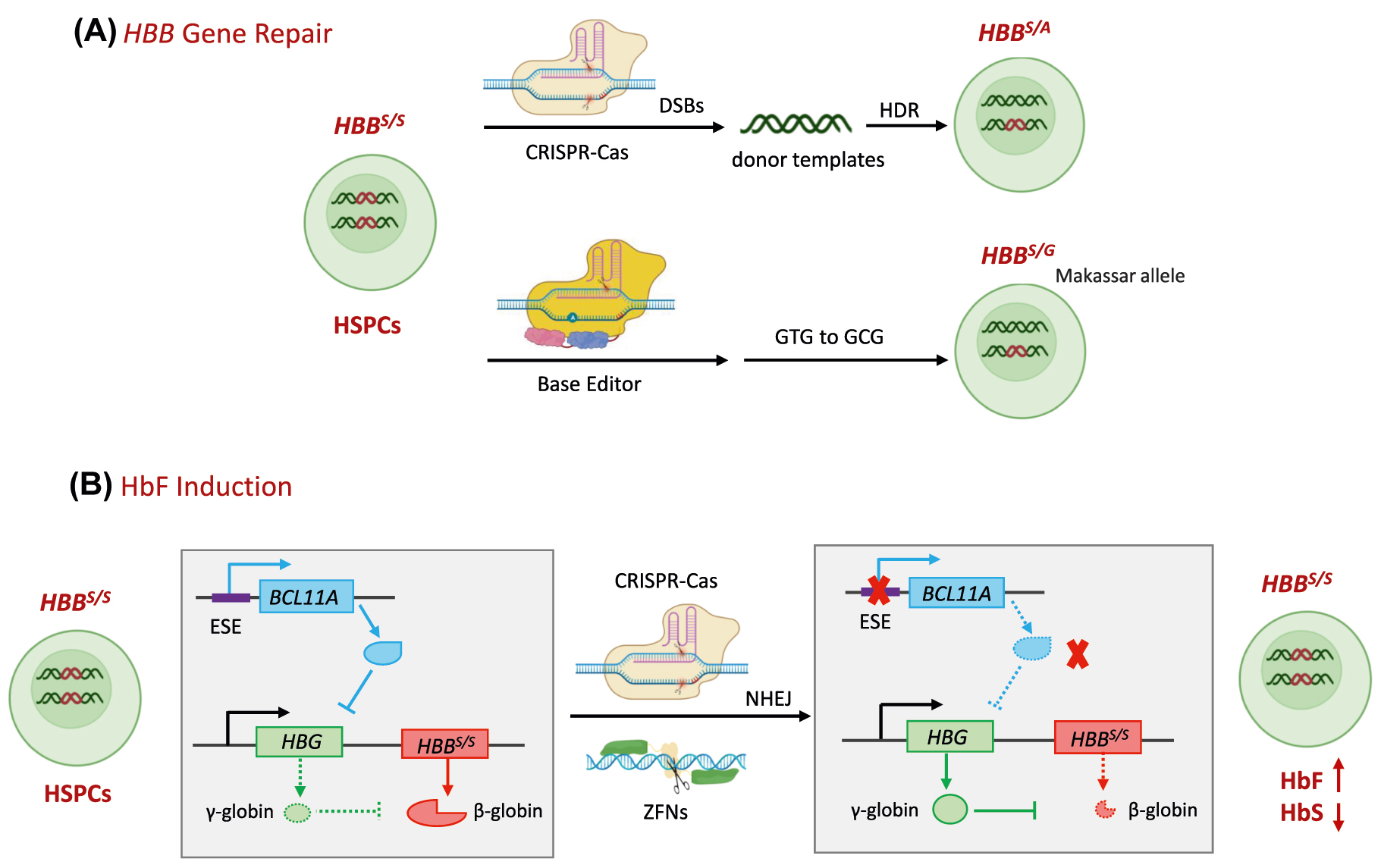
Molecular mechanisms of therapeutic genome editing ex vivo for SCD. (A) HBB gene repair. Upper: The CRISPR-Cas9 recognizes and cuts the locus of HBB S/S in HSPCs and DSBs are induced, triggering DNA repairing. At the presence of DNA donor as a template, DSBs are repaired by HDR and the HBB S/S is converted to HBB S/A or HBB A/A. Below: the pathogenic HBB S allele (codon GTG) to a non-pathogenic, naturally occurring variant HBB G (GCG) in HSPCs by ABEs. (B) HbF induction: After birth, BCL11A binds to the γ-globin promoter directly, repressing γ-globin expression, a component of HbF that has anti-sickling effects. HBB S/S in patients produces sickle β-globin. CRISPR-Cas9 or ZFNs targeting BCL11A ESE, will induce DSBs and NHEJ. As a consequence, BCL11A expression and the inhibition to γ-globin is disrupted, leading to an increase in γ-globin. SCD, sickle cell disease; HbF, fetal hemoglobin; ESE, erythroid-specific enhancers; HbS, sickle hemoglobin.
BCL11A ESE attracts more attention and go further in clinical trials. BCL11A plays a critical role in HbF-HbA switching via binding to the γ-globin promoter directly, repressing γ-globin expression [185, 186]. Disrupted BCL11A expression showed therapeutic potential but negatively affected B-cell differentiation and HSC engraftment [177, 187]. Further studies found that deletion of the BCL11A ESE region (especially the site of +58 bp from the transcriptional start site) led to an increase in γ-globin and resulted in survival advantage of the editing cells without affecting their proliferation and differentiation [188], [189], [190] (Figure 7B). CTX001 from Vertex Pharmaceuticals, which is autologous CD34+ hHSPCs (hematopoietic stem and progenitor cells) modified with CRISPR-Cas9 at BCL11A ESE, was used in one SCD patient and one TDT patient and showed a striking curative effect in more than one year follow-up [191]. Of note, no evidence of off-target editing was observed. The multi-site, observational study to evaluate the long-term safety and efficacy of CTX001 in the TDT and serve SCD patients who received CTX001 is ongoing [192]. A phase 3 clinical trial of CTX001 in pediatric participants with severe SCD and hydroxyurea failure or intolerance was started in 2022 [193]. OTQ923 and HIX763 from Novartis Pharmaceuticals are similar autologous products edited with CRISPR-Cas9, which are also in phase 1/2 for treating adult/children SCD [194]. More recently, a study from Xiangya Hospital, Central South University, reported the preliminary data of an ongoing phase 1/2 trial (NCT04211480) evaluating the safety and efficacy of BCL11A ESE editing, autologous, HSPCs (BRL-101 from Shanghai Bioray Laboratories) in two children with TDT. Both patients were clinically well with multilineage engraftment and achieved transfusion independence for more than 18 months without incidence of adverse events [195]. Longer follow-ups of a larger cohort of patients are wanted to confirm whether this treatment can cure pediatric TDT patients. In 2021, EdiGene (GuangZhou) Inc. from China also started a phase 1 study in subjects with TDT, in which HSPCs edited by CRISPR-Cas9 for BCL11A ESE will be evaluated (NCT04925206) [196].
CRISPR-Cas12a, ZFNs and BEs in erythrocyte diseases
Except for CRISPR-Cas9, CRISPR-Cas12a and ZFNs were also used for therapeutic ex vivo. EDIT-301 from Editas Medicine using Cas12a, targeting a BCL11A binding region in HBG1 and HBG2 promoters for HbF induction, was tested in phase 1/2 for the efficacy, safety and tolerability in adult subjects with severe SCD in 2021 [197, 198]. Using ZFNs, BIVV003 and ST-400 from Sangamo Therapeutics for treating severe SCD and TDT, respectively, are in phase 1/2. The data in the five TDT patients treated with ST-400 showed that the treatment was generally well tolerated and increased the levels of HbF transiently after reconstitution, which might need resumption of transfusions [199]. In another recent study, four SCD patients were successfully infused with a single dose of BIVV003. They had no SCD-related events or infusion-related reactions with 26 weeks of follow-up, with increased total Hb, HbF, and HbF-containing cells, and clinical improvements [200] (Figure 7B).
Using a custom adenine base editor (ABE8e-NRCH), Liu et al. edited the pathogenic HBB S allele (codon GTG) to a non-pathogenic, naturally occurring variant HBB G (GCG), in HSPCs from SCD patient [201] (Figure 7A). In the 16 weeks of follow-up, edited HSPCs were durable after engraftment in immunodeficient mice with 68% HBBG and reduced propensity for hypoxic sickling. Furthermore, HBB S -to-HBB G base editing alleviates pathology in a mouse model of SCD, identifying no off-target mutations with anticipated clinical relevance [201]. These data suggest that this platform provides a promising treatment for SCD. The corresponding manufactured product BEAM-101 was initiated in a clinical trial this year (NCT05456880) [202].
Potential candidates
The above studies give us great hints that many other hereditary diseases of the hematopoietic system also could be cured by transplantation of genome edited autologous HSPCs (Table 1). Fanconi anemia (FA) is an autosomal recessive disorder that leads to bone marrow failure and a predisposition to malignancies. More than 22 gene mutations are involved [203]. CRISPR-Cas9, ZFNs mediated genome editing in FA patient-derived HSCs followed by transplantation in mouse is feasible for correcting disease related phenotypes [204, 205]. Particularly, Paula Rı´o et al. showed highly efficient NHEJ-mediated repair of mutated FA genes to generate compensatory mutations that correct the phenotype of FA patient-derived HSCs, where the efficacy of NHEJ is enhanced in FA-deficient cells and secondary mutations in FA genes restored the function of mutated alleles [205]. These studies suggest the feasibility of NHEJ-mediated gene editing in treating FA and other monogenic diseases of the hematopoietic system.
In humans, primary immunodeficiency syndromes (PIDs), which cause dysfunction or nonfunctioning of one or more components of the human immune system, represent a group of monogenic disorders of leukocytes. Over 300 gene mutations cause PIDs, such as IL2Rγ mutation in X-linked severe combined immunodeficiency (SCID-X1), ADA mutation in adenosine deaminase deficiency (ADA-SCID), WAS mutation in Wiskott-Aldrich-Syndrome (WAS) etc. [206]. Autologous HSCs transplantation combined with gene addition by lentivirus or retrovirus vectors has attracted much attention in clinical trials for PIDs. The commercial product Strimvelis was approved by the European Medicines Agency (EMA) for the treatment of ADA-SCID in 2016 (reviewed elsewhere [206, 207]), even though long-term efficiency and safety in patients remain challenged. In contrast to erythrocyte diseases, there is a limited NHEJ-based genome editing strategy for PIDs. HDR-based editing is more challenging since the low editing efficiency in primitive HSPCs. For example, only about 5% targeting efficiency of HDR in ZFN-mediated editing of SCID-X1 HSPCs was observed in the previous study [208], which did not reach the threshold (10%) for functional HSPCs required for immune reconstitution [209]. Optimizing editing reagents and treatment protocols such as delivery method and culture conditions might help its application of genome editing in PIDs.
In vivo genome editing in somatic cells
Delivery systems
In vivo genome editing involves introducing therapeutic components to the cells in the patient’s body directly (Figure 6B). There are some advantages over ex vivo therapy: (i) it bypasses the complicated steps of cell culture, quality control and cell transfer; (ii) it is transplantation-independent, making it possible to edit cells that are difficult for manipulation ex vivo or poor engraftment; (iii) potentially corrects the gene mutations of multiple tissue types when the delivery vehicles are well designed. Sometimes, this might also be a disadvantage of in vivo editing. Indeed, the delivery vehicles for in vivo are much more challenging than the counterpart used ex vivo. The most challenging part is delivering the gene-editing components to the target cells precisely with high efficacy [210]. Ex vivo editing strategy often employs non-virus deliver systems such as electro-transfection or microinjection of the RNP (protein–RNA complex), which might be more controllable and suitable for stem cells, mainly because of no genomic integration and no long-term expression and fewer off-target [211]. Instead, viral vectors or nanoparticles are favored for in vivo delivery. Viral vehicles, including lentivirus, adenoviruses (AVs), and AAVs, have high efficacy and apply to various tissues. Especially, AAVs are the most popular vehicles used in clinical gene therapy. AAVs can infect the dividing and non-dividing human cells efficiently, making them a promising tool for therapeutic components to deliver and cure diseases of various tissues. Moreover, a variety of serotypes of AAVs show tissue-specific tropism, increasing the specificity of the treatment. The non-integrative property of AAVs into the host genome is another advantage compared to integrating lentiviral vehicles. The integrating feature is a hidden danger both in vivo and ex vivo. In the phase 1/2 study about transplantation of autologous HSCs transduced ex vivo with the LentiGlobin BB305 lentiviral vector in SCD patients, participants developed acute myeloid leukaemia/myelodysplastic syndrome where the transgene was present in the patient’s malignant cells [212]. However, AAVs have limited cargo size, with a maximum capacity of 4.7 kb. As a result, when using large nucleases such as SpCas9 (encoding by about 4.2 kb gene), a second AAV for sgRNA and repair template is needed, leading to reduced efficiency as two AAVs need to be introduced into one cell for function [213]. Moreover, sustained expression of genome-editing molecules probably leads to undesired off-target editing or immune reactions in patients [211, 214], which are common problems in applications in vivo and ex vivo when using viral vectors. Cationic lipid-based nanoparticles and inorganic nanoparticles are emerging delivery systems that could be used in clinical applications of in vivo genome editing-based therapy [215], [216], [217]. In size, nanoparticles are much bigger than viruses and RNPs, ranging from 50 to 500 nm [211]. They show low cost, low immunogenicity and no genomic integration, but limited tissue targeting spectrum and low systemic delivery efficiency [218].
In summary, several delivery vehicles are available for in vivo genome editing, with distinctive drawbacks and advantages. Nonetheless, with efforts in optimization of the types of delivery vehicles, the dosages, the sites of administration and editing strategies, we are delighted to see real progress in the in vivo genome editing in treatments of genetic diseases from laboratory research to clinical practices (Table 2).
In vivo genome editing of CRISPR for inherited diseases in clinical trials
NTLA-2001 is the first-ever CRISPR-Cas9-based genome-editing therapy in vivo. NTLA-2001 was designed by Intellia Therapeutics to treat hereditary transthyretin amyloidosis (ATTR), including ATTR with polyneuropathy (ATTR-PN) and ATTR-related cardiomyopathy (ATTR-CM) [219]. These diseases are autosomal-dominant disorders and associated with over 100 different missense mutations in the TTR gene encoding transthyretin, resulting in misfolds of TTR tetramer, which further develops into TTR protein aggregates, generating amyloid fibrils, protein deposits in multiple tissues [220]. Given that TTRs are exclusively produced by hepatocytes of the liver, NTLA-2001 are designed as a TTR-specific guide RNA and human-optimized SpCas9 mRNA packing in lipid nanoparticles (LNP) with liver tropism. After intravenous injection of NTLA-2001, the circulatory LNPs are opsonized by apolipoprotein E (ApoE) and bind to hepatocytes via interaction of ApoE and the low-density lipoprotein receptor (LDLR) on hepatocytes, followed by endocytosis and endosome formation (Figure 8A and B). The mutant TTRs could be abrogated upon missense or nonsense mutations induced by Cas9 cut and NHEJ-mediated repair. In an ongoing phase 1 clinical study (NCT04601051), NTLA-2001 led to a reduction in the serum TTR protein in a dose-dependent effect, with a 52% decrease in patients who received 0.1 mg per kilogram and an 87% decrease for 0.3 mg per kilogram at 28 dpi, even though mild adverse effects were observed [219].
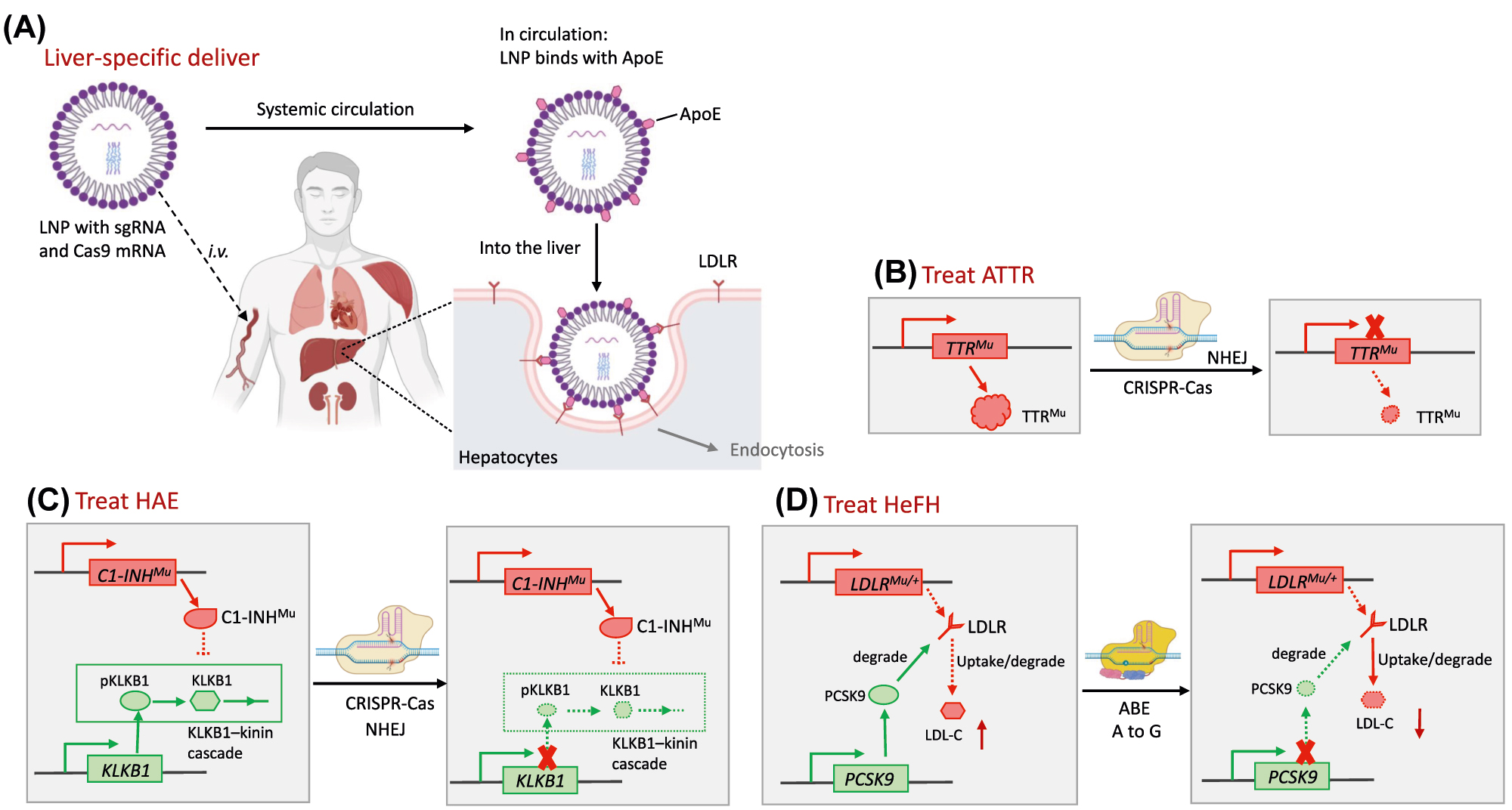
Molecular mechanisms of therapeutic genome editing in vivo for hereditary metabolic diseases. (A) Liver-specific deliver of genome editing agents: LNPs with sgRNA and Cas9 are i.v. injected and coated by ApoE in circulation. When circulating into the liver, interaction between ApoE and LDLR take the LNPs into the hepatocytes by endocytosis specifically. (B) In patients with ATTR, missense mutations in the TTR gene encoding transthyretin result in the misfolds of TTR tetramer, which further develop into TTR protein aggregates. TTR-specific gRNA and Cas9 delivered into hepatocytes via LNPs, abrogate mutant TTRs upon missense or nonsense mutations induced by NHEJ-mediated repair. (C) In patients with HAE, mutant C1 esterase inhibitor (encoding by C1-INH) lose its function of regulating the complement system and kallikrein–kinin cascades. CRISPR mediated knocking out of KLKB1 that encoding a precursor of plasma kallikrein, achieved therapeutic outcomes. (D) Loss-of-function mutation in the LDLR gene down-regulates LDLR expression, leading to high LDL-C and HeFH. ABEs make a A-to-G conversion at the splice site in the PCSK9 gene (causing aberrant splicing) for disrupting its expression and degrading of LDLR, subsequently lowers the LDL-C. ApoE, apolipoprotein E; ATTR, hereditary transthyretin amyloidosis; HAE, hereditary angioedema; LDLR, low-density lipoprotein receptor; LDL-C, low-density lipoprotein cholesterol; HeFH, heterozygous familial hypercholesterolemia.
With a similar delivery system, NTLA-2002 is an in vivo CRISPR-Cas9 genome editing drug that is designed to knock out the gene kallikrein B1 (KLKB1) in hepatocytes for the treatment of hereditary angioedema (HAE) (Figure 8C). HAE is a rare, autosomal dominant disorder characterized by severe, recurring inflammatory attacks with swelling in various organs [221]. HAE is caused by a deficiency or dysfunction of the C1 esterase inhibitor, a vital regulator of the complement system and kallikrein-kinin cascades. When the C1 inhibitor loses function, activation of the kallikrein-kinin cascade leads to the uncontrolled generation of plasma kallikrein and excessive production of bradykinin, which causes vascular leakage and subsequent angioedema [222]. Pre-clinical studies proved that knocking out KLKB1, encoding a precursor of plasma kallikrein, achieved sustained therapeutically reduction in plasma kallikrein activity in non-human primates (NHPs) [223]. Inspired by the results, Intellia Therapeutics started the Phase1/2 clinical trial of NTLA-2002 in adults with HAE in 2021 [224].
Another CRISPR-Cas9-based genome editing trial in the clinical is EDIT-101, using DSBs and NHEJ to remove the IVS26 mutation in the CEP290 gene (NCT03872479) [225]. IVS26 is a well-known Leber congenital amaurosis (LCA)-associated mutation in the CEP290, which is an A-to-G point mutation located within intron 26 (referred to as c.2991+1655A>G), resulting in aberrant splice donor. It accounts for almost one-third of all LCA cases [226, 227]. Jiang et al. subtly developed a pair of sgRNAs that flanked the IVS26 mutation, induced deletion or inversion of the intervening sequence, and achieved productive editing that met the therapeutic threshold [228] (Figure 9). In humans, EDIT-101 is delivered directly to the photoreceptor cells via the AAV5 vector after subretinal injection. It is worth noting that this is the first in vivo administration of CRISPR therapy to a child.
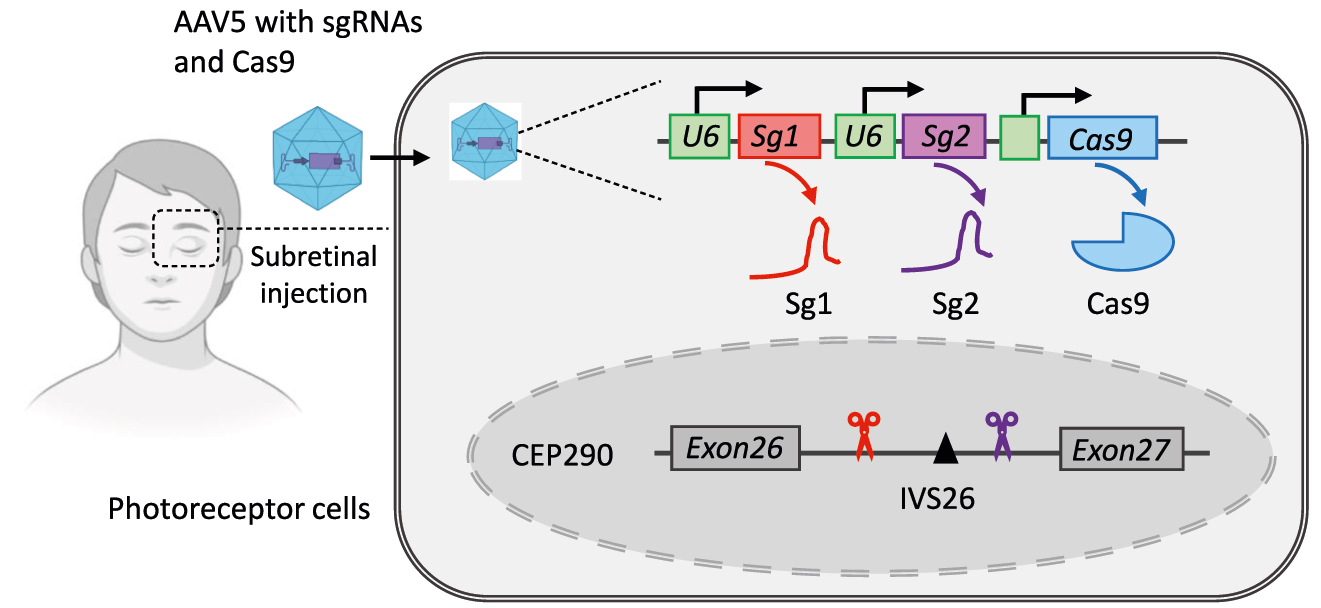
Strategy of therapeutic genome editing in vivo for LCA. A pair of sgRNAs and Cas9 are delivered directly to the photoreceptor cells via AAV5 vector after subretinal injection. Cas9 induces deletion of IVS26 mutation which is LCA-associated mutation in the CEP290. LCA, Leber congenital amaurosis.
In vivo genome editing of BEs for inherited diseases in clinical trials
The world’s first in vivo genome editing using BEs in humans, the VERVE-101, was also initiated (NCT05398029) [229]. It was designed by Verve Therapeutics and also utilized a LNP-mediated vector to target the liver and ABEs to make an A-to-G conversion at the splice site in the PCSK9 gene (causing aberrant splicing) for disrupting its expression and subsequently lowering the LDL cholesterol (LDL-C) levels in patients with heterozygous familial hypercholesterolemia (HeFH) [230] (Figure 8D). In patients with HeFH, a heterozygous loss-of-function mutation in the LDLR gene down-regulates LDLR expression, leading to high LDL-C in the blood and ultimately resulting in a heart attack or atherosclerotic cardiovascular disease [231]. In a preclinical study, a single intravenous injection achieved near-complete knockdown of PCSK9 in the liver. It resulted in an approximately 90% reduction in blood levels of PCSK9 protein and about a 60% reduction in blood levels of LDL-C in NHPs compared to the baseline [232]. Verve Therapeutics plans to develop a second gene editing program that targets the ANGPTL3 gene, a regulator of cholesterol and triglyceride metabolism, for patients with FH [233]. Notably, in the application of VERVE-101 in patients with HeFH, an issue arose that a low level of LDLR in patients might influence the endocytosis of the LNPs by hepatocytes in the liver as LDLR plays an essential role in LNP uptake.
In vivo genome editing of ZFNs for inherited diseases in clinical trials
Sangamo Therapeutics developed ZFN-mediated in vivo genome editing products-SB-318, SB-913, and SB-FIX to treat Mucopolysaccharidosis Type I (MPS I), MPS II, and severe Haemophilia B, respectively [13]. The three drugs were ZFN-mediated genome editing for knocking in therapeutic cDNA to the endogenous albumin locus of hepatocytes precisely, which are delivered by AAV-derived vectors in vivo and enable the patient’s liver to produce stable circulating therapeutic levels of a corrective protein for the lifetime of the patient [234]. As the first therapy based on ZFN-mediated genome editing, SB-FIX is designed to treat Hemophilia B, caused by the FIX (factor IX)-mutation. The editing machinery comprises three liver-tropic rAAVs, two of which carry left and right ZFNs, and one carries the WT FIX gene [235]. Under this setup, rAAVs delivered the components into the liver. In addition, ZFNs were responsible for recognizing and cutting albumin intron 1 precisely, followed by HDR with AAV-delivered donor DNA as templates. This clinical trial (NCT02695160) was started in 2016 and terminated in 2021 with only 1 participant enrollment. To date, no study results are available [236].
SB-913 was delivered by rAAV2/6 to insert the copy of the IDS gene into the albumin locus for treatment of MPS II (also known as Hunter syndrome), which is an X-linked disorder caused by mutations of the IDS gene encoding the iduronate-2-sulfatase enzyme, characterized by accumulation of glycosaminoglycans (GAG), dermatan sulfate, and heparan sulfate in the body, and widespread tissue damage. However, according to the latest results from the 9 subjects in phase 1/2 clinical trials (NCT03041324), Sangamo Therapeutics has terminated the study due to the lack of observed clinical benefit and serious adverse events including pyrexia, intestinal obstruction, bronchitis, spinal stenosis [237].
SB-318 is a similar drug (also rAAV2/6) to insert the IDUA gene (encoding for α-L-iduronidase) for the treatment of IDUA mutation-induced MPS I (also known as Hurler syndrome) [238]. The corresponding clinical study (NCT02702115) was also terminated, possibly due to low transgene expression [239].
To solve this problem, the same group designed a proprietary system with CRISPR-Cas9 to insert IDUA to the albumin locus through HDR with IDUA cDNA or an alternative pathway of NHEJ-mediated whole AAV sequence (with IDUA cDNA) integration, which was able to work in both dividing and non-dividing cells [239]. AAV8 instead of AAV2/6 was used and showed efficacy and safety in mice, indicating a potential treatment for a clinical study.
Potential candidates
With efforts in exploring the potential gene targets, genome editing tools and delivery systems, researchers have achieved massive progress in treating monogenic diseases using in vivo genome editing therapy (Table 2). These results prompt us to try this therapy further in hereditary diseases of other systems, such as DMD and cystic fibrosis. DMD has attracted lots of investigations regarding genome editing therapy from the beginning. DMD is a progressive and fatal muscle-wasting monogenic disease caused by loss-of-function mutations of the X-linked DMD gene encoding the dystrophin protein. This essential scaffolding protein stabilized striated muscles and protected the muscles from contractile-induced damage [8]. Due to the lack of normal function of DMD in the muscle, DMD patients gradually lose the ability to walk at an early age and eventually develop into heart and respiratory failure. DMD is one of the largest human genes spanning 2.3 Mb with 79 exons, about 11 kb complete coding sequence, encoding for the 427 kDa protein, and over 7,000 pathogenic mutations are reported. Among them are large deletions spanning one or more exons (about 66%), exon duplications (about 11%), small deletions/insertions (10%–15%), nonsense mutations (10%–15%) [8, 240]. Most mutations lead to a shift in the reading frame that produces a truncated and dysfunctional protein without the essential C-terminal domain. Therefore, reframing or skipping one or two exons would achieve therapeutic benefit in most DMD patients, accounting for 83% as estimated [241]. Numerous in vivo experiments in mouse DMD models and other large animal models such as dogs and pigs have shown that NHEJ-mediated exon skipping, deletion or reframing induced by CRISPR or base editing of the splice site could restore functional dystrophin expression and achieve the therapeutic effect (reviewed in elsewhere [218, 240, 242]). However, differences in the sequences and mutations between humans and animals exist. Humanized DMD animal models are needed, providing a powerful platform for developing and verifying sequence-dependent therapies in vivo and helping the preclinical studies translated to the clinic. Furthermore, an efficient, safe, and muscle-specific delivery system is lacking and highly desirable. By addressing these challenges, we will see the translation of genome editing therapies for DMD.
Germline genome editing
Germline genome editing includes editing the genome of preimplantation embryos, sperm, eggs and their precursors-germ cells, including spermatogonia, spermatocytes, spermatids from males and oocytes from females (Figure 10). Unlike somatic cell editing, germline editing introduces genetic changes to the next generations, which are heritable. The last raise societal and ethical issues in human application and must be rigorously regulated. No country explicitly permits heritable human genome editing [243]. However, germline genome editing provides potential prevention of the transmission of pathogenic mutations from parent to child and treatments of infertility due to mutations that affect gamete or embryonic development [244].
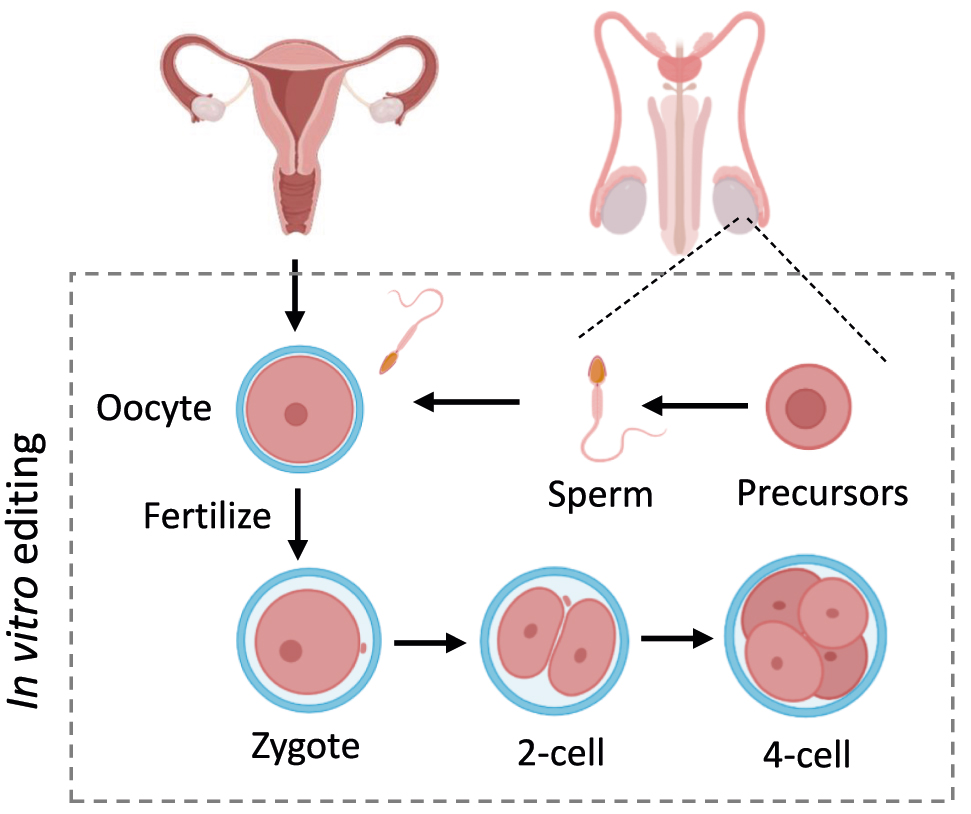
Germline genome editing. Editing the genome of preimplantation embryos, sperm, eggs and their precursors, including spermatogonia, spermatocytes, spermatids from males and oocytes from females.
Heritable genome editing is not a new concept. In many fields, heritable genome editing in embryos (zygotes) is now widely used for producing genetically modified animals, such as mice, rats, pigs, goats, rabbits, dogs, and monkeys, primarily by the CRISPR-Cas9 system as it is easy and efficient [245]. In addition, the editing of spermatogonial stem cells (SSCs) can also be applied to germline-modified animals [246]. In these strategies, mosaic Founder 0 animals require genetic screening and crossbreeding to produce stable and heritable genetically modified animals. In addition, our group and others developed novel haploid embryonic stem cells (haESC) from mice, rats, monkeys, and humans, which could be used for generating germline editing animals [245].
Regarding treatments for hereditary diseases, proof-of-concept studies in mouse models have proved the potential of germline genome editing to cure genetic diseases. Previously, our group co-injected the Cas9 mRNA and sgRNA targeting dominant mutation in the Crygc gene that caused cataracts into zygotes and corrected the pathogenic mutation via HDR based on exogenous oligo or endogenous WT allele [247]. With a similar strategy, gene correction of Crygc mutation in SSCs was also achieved [248]. Importantly, all the corrected alleles were stably transmitted to the offspring. Olson et al. used CRISPR-Cas9 to correct the DMD mutation in the germline of the DMD mice model and generated mosaic mice with 2%–100% correction of DMD mutations, in which the muscle phenotype was rescued [249]. Recently, Lu et al. used CRISPR-Cas9 to edit the germline of mouse embryos containing a point mutation in Hbb that leads to β-thalassemia [250]. Microinjection of Cas9 mRNA and sgRNA into embryos generated 70% of the born mice with corrected genotypes and significantly improved symptoms. In a mouse model of azoospermia, Wang et al. isolated the Tex11 mutation SSCs and repaired the genetic defect with CRISPR-Cas9-mediated HDR ex vivo, restoring spermatogenesis after transplantation into testis [251].
So far, there have been limited studies on the germline editing of human embryos. Using CRISPR-Cas9, Liang et al. conducted editing of HBB in tripronuclear (3 PN) zygotes [252]. They found low efficiency of HDR and off-target cleavage in the mosaic embryos. In contrast to these results, Tang et al. demonstrated that CRISPR-Cas9-mediated HDR-based correction of point mutations in HBB and G6PD of human 2 PN zygotes was efficient [253]. They did not find evidence of off-targeting, but mosaicism did exist. To eliminate mosaicism, Mitalipov’s group co-injected a mixture of Cas9, sgRNA, DNA templates and sperm from the patient heterozygous for the MYBPC3 mutation into metaphase II (MII) oocytes [254]. This approach avoided mosaicism and achieved over 70% of HDR-mediated repaired embryos. Interestingly, the mutation alleles were repaired using the maternal WT alleles instead of exogenous DNA templates, a process termed gene conversion [244, 254]. Others challenged these results. The main argument was that gene conversion seemed impossible because paternal and maternal pronuclei were separated in early zygotes [255]. Deletion of large fragments by CRISPR-Cas9 might remove the primer-binding site and lead to amplification of only the maternal allele [255, 256]. Mitalipov’s group gave additional evidence to support the mechanism of gene conversion, and their further work demonstrated that gene conversion and NHEJ were two major repair mechanisms induced by DSB in preimplantation human embryos [257, 258]. However, Zuccaro et al. showed that the significant repair was mediated by micro homology-mediated end joining (MMEJ) but not NHEJ, resulting in frequent chromosome loss in the context of Cas9-induced DSB [105]. This fundamental question is still under debate, which challenges human germline editing.
In contrast to CRISPR-Cas9, BEs can circumvent the problem induced by DSB in human germline editing. Instead, BEs might lead the trouble with unintended mutations in the target site and base conversion close to the target site [109]. Indeed, CBE and its variants were tested in 3 PN zygotes, 2 PN zygotes, MII and 2-cell embryos in previous studies, involving genes such as RNF2 [259], HBB [260, 261], FANCF [260], FBN1 [262], OCT4 [263]. Overall, CBEs achieved higher precise editing efficiencies compared to CRISPR-Cas9. Only one study reported ABE editing in 3 PN zygotes targeting multiple gene sites with high efficiency [264]. However, mosaicism and unwanted base conversion proximity to the target sites existed in almost all BE applications in human embryos.
Altogether, human germline genome editing faces many obstacles in theory and technology. The ethical issues also hinder their further applications. Besides safety concerns, informed consent is impossible for the edited generations. The edited generations might also suffer from discrimination. Gene enhancement by germline editing would lead to serious social problems. More seriously, germline editing would introduce unintended mutations, leading to perturbation of the human gene pool [265, 266]. Once the mutations were introduced, dealing with them would be quite complicated. To be clear, it is a consensus that any germline genome editing for clinical application is forbidden and illegal for a long time in the future. Any research use must be under rigorous regulation according to national law.
Challenges and perspectives
Approximately twenty therapeutic genome editing drugs for inherited disorders are in ongoing clinical trials, as described above. Most studies’ preliminary data look promising except for the in vivo therapy using ZFNs (Tables 1 and 2). In view of the target cells, all are aiming to edit the somatic cells, including HSCs/HSPSs ex vivo and hepatocytes or photoreceptor cells in vivo. In ex vivo studies, therapeutic editing for SCD and TDT are pioneers. The CRISPR-Cas9-based CXT001 is the first to enter phase 3, even though the clinical applications in inherited diseases are generally behind their applications in cancer immunotherapy and treatment of HIV infection. CRISPR-Cas9 is the most used in ex vivo genome editing, followed by ZFNs. Emerging tools, such as CRISPR-Cas12a and ABEs, are also used. With the development of tissue-specific delivery methods, genome editing in vivo has also come to the clinic. Two CRISPR-Cas9 mediated editing drugs and one ABE meditated drug are reported, all targeting hepatocytes delivered by LNP for treatment of metabolic diseases. Another three ZFN-mediated editing drugs are terminated due to the lack of observed clinical benefit and serious adverse events. Human germline editing is now in the initial stage but full of difficulties for several reasons. First, our understanding of the biology of human development is limited, and the consequence of human germline editing is unforeseeable. Pioneering research has raised the fundamental theory controversy and technological problems. One of the most challenging issues is the ethical problem, such as discrimination, inequality, genetic enhancement and perturbation of the human gene pool. So human germline editing for clinical uses is prohibited and out of reach.
Our ultimate purpose for every drug or treatment is to cure disease with high safety, efficiency, and durability. The era of therapeutic genome editing in humans has already arrived and provided potential durable and curable therapy for inherited disorders, an advantage that traditional therapy is hard to achieve. However, safety and efficiency still limit their further applications. These issues exist in every process of genome editing therapy, including (i) delivery, (ii) recognition and cut, and (iii) repair. Nevertheless, we suppose that, as a rapidly evolving technology, efforts addressing the following challenges might help their therapeutic outcomes in patients with inherited diseases.
Highly accurate, specific, efficient delivery vehicles are needed
It is more urgent for in vivo deliver than ex vivo since electroporation is very efficient, simple, and safe for ex vivo deliver, which is widely used in the clinic. In contrast, for in vivo use, viral delivery systems, especially AAVs, have high expectations because of their efficiency and tissue tropism. As mentioned above, the packaging capacity limits carrying all the editing components into a single AAV and further limits the scope of AAVs. The split-intein dual-AAV strategy has been reported to be useful, but an increasing dose of AAV is required. Another strategy is developing smaller genome editing tools, such as Cas9 ortholog from SaCas9, CjCas9, and Nme2Cas9, SauriCas9. Indeed, the ongoing clinical trial regarding treating LCA used SaCas9 and two sgRNAs packing into a single AAV [228]. AAVs have a prolonged expression of the editing components that might increase the risks of being off-target or trigger an immune response. Spatially or temporally regulating the expression of AAV-delivered editor offers helpful strategies [267]. As a non-viral delivery system, LNPs show advantages for liver delivery with lower immunogenicity, off-target, and toxicity. But LNP-mediated delivery for other organs remains challenging. The physical (changing the charge), chemical (changing the compositions), or biological (conjugating with antibody) modifications of LNPs provide help for targeting non-liver organs. Virus-like particles (VLPs) are emerging delivery vehicles usually assembled by non-infectious retroviral proteins and mRNA or RNP cargos, enabling transiently delivery of the mRNA or RNP into target cells [268]. Liu et al. anticipated that RNP-packaging VLPs would offer the shortest exposure to editors and, therefore, the lowest off-target and would be very popular in future applications [267]. The ability for delivery to different organs and feasibility in pre-clinical studies are also needed.
Highly precise, specific, efficient enzymes are obligatory
Even though various nucleases, nickases, and delivery vehicles are available, none are perfect. ZFNs and TALENs both tolerate a small number of positional mismatches, but CRISPR-Cas9 tolerates multiple consecutive mismatches, which largely compromise their specificities. Engineering Cas variants and sgRNAs are hopeful of generating editors with higher specificity. Three strategies are reported, including (i) paired Cas9 nickases with two sgRNAs or fused chimaeras with one inactivated Cas9 and one attenuated Cas9; (ii) alanine scanning for screening mutants with precise and efficient genome editing in human cells such as eSpCas9; (iii) engineered sgRNAs such as truncated sgRNAs or hp-sgRNAs [15]. BEs also have bystander editing, transversion mutations, indels at the target nucleotide, and undesired editing at the off-target DNA, impairing their accuracy and specificity. The off-targets are divided into Cas-dependent and Cas-independent [269, 270]. Anyway, unwanted editing at the target and off-target sites should be avoided and waiting for optimization in clinical use.
Repair: expand the scope of applications
In principle, genome editing can correct any mutation for therapy purposes. There are so many types incurable inherited diseases. However, clinical testing therapeutic editing for inherited diseases is no more than ten, accounting for a tiny percentage. In addition to the limitations of delivery vehicles and enzymes we have discussed above, the repairing mechanism is one of the most challenging issues. As we can see, most ongoing clinical trials use the NHEJ-mediated repairing mechanism, indicating that they employ it to induce another “mutation” to cure the pathogenic mutation. Indeed, the precise correction of the pathogenic mutation relies on HDR-mediated repairing, a process that is active in only dividing cells. Moreover, NHEJ is generally more active than HDR [271]. These features limit gene correction in most human cells, such as muscle cells, nerve cells, etc. To expand the therapeutic scope, improvement of donor templates, co-localization of templates with nick, synchronization of the cell cycle, and suppression of NHEJ activity were harnessed to promote the HDR but were not as effective as expected [15, 272]. BEs provided another repairing mechanism without DSBs. Expanding the scope of applications of BEs will be helpful, and developing Cas orthologs or variants recognizing alternative PAMs for targeting a broader region of the genome could achieve this goal [15]. The new finding CAST systems also provided a gene correction technology by a mechanism independent of HDR or base conversion. They potentially expanded the scope of therapy for inherited diseases after the system was fully developed.
In conclusion, site-specific genome editing systems showed up in clinical trials for several inherited disorders in the last five years. The performances of these systems are exciting, inspiring, and promising, especially the CRISPR-Cas and BEs. Certainly, each of the technologies has its limitations and drawbacks. However, as highly anticipated technologies, they are quick evolving and continuously optimized. With the continuous improvement of delivery vehicles, enzymes, repair pathways in their accuracy, specificity, efficiency, the scope of their applications in treatments of inherited diseases will expand. We anticipate that more clinical trials will be carried out in the next five years, and patients with inherited disorders will benefit from them in the near future.
Funding source: CAS Special Research Assistant Program
Funding source: Genome Tagging Project
Funding source: National Natural Science Foundation of China
Award Identifier / Grant number: 31730062
Award Identifier / Grant number: 31821004
Award Identifier / Grant number: 32030029
Award Identifier / Grant number: 3210060158
-
Research funding: This study was supported by the Genome Tagging Project and grants from the National Natural Science Foundation of China (31821004, 32030029, 31730062, and 3210060158), and CAS Special Research Assistant Program. The images in Figures were created with BioRender.com.
-
Author Contributions: C.H., Q.L. and J.L. conceived this study and wrote the manuscript. J.L. supervised this study.
-
Competing financial interests: The authors declare no competing financial interests.
-
Ethical approval: Not applicable.
References
1. Prasad, C, Galbraith, PA. Sir Archibald Garrod and alkaptonuria-‘story of metabolic genetics’. Clin Genet 2005;68:199–203. https://doi.org/10.1111/j.1399-0004.2005.00487.x.Suche in Google Scholar PubMed
2. Pauling, L, Itano, HA, Singer, SJ, Wells, IC. Sickle cell anemia a molecular disease. Science 1949;110:543–8. https://doi.org/10.1126/science.110.2865.543.Suche in Google Scholar PubMed
3. Jackson, M, Marks, L, May, GHW, Wilson, JB. The genetic basis of disease. Essays Biochem 2018;62:643–723. https://doi.org/10.1042/ebc20170053.Suche in Google Scholar
4. Hamosh, A. OMIM gene map statistics; 2022. Available from: https://www.omim.org/statistics/geneMap.Suche in Google Scholar
5. Bick, D, Bick, SL, Dimmock, DP, Fowler, TA, Caulfield, MJ, Scott, RH. An online compendium of treatable genetic disorders. Am J Med Genet C Semin Med Genet 2021;187:48–54. https://doi.org/10.1002/ajmg.c.31874.Suche in Google Scholar PubMed PubMed Central
6. Park, SH, Bao, G. CRISPR/Cas9 gene editing for curing sickle cell disease. Transfus Apher Sci 2021;60:103060. https://doi.org/10.1016/j.transci.2021.103060.Suche in Google Scholar PubMed PubMed Central
7. Ribeil, JA, Hacein-Bey-Abina, S, Payen, E, Magnani, A, Semeraro, M, Magrin, E, et al.. Gene therapy in a patient with sickle cell disease. N Engl J Med 2017;376:848–55. https://doi.org/10.1056/nejmoa1609677.Suche in Google Scholar PubMed
8. Duan, D, Goemans, N, Takeda, S, Mercuri, E, Aartsma-Rus, A. Duchenne muscular dystrophy. Nat Rev Dis Prim 2021;7:13. https://doi.org/10.1038/s41572-021-00248-3.Suche in Google Scholar PubMed
9. Landfeldt, E, Lindgren, P, Bell, CF, Schmitt, C, Guglieri, M, Straub, V, et al.. The burden of Duchenne muscular dystrophy: an international, cross-sectional study. Neurology 2014;83:529–36. https://doi.org/10.1212/wnl.0000000000000669.Suche in Google Scholar
10. Shirley, M. Casimersen: first approval. Drugs 2021;81:875–9. https://doi.org/10.1007/s40265-021-01512-2.Suche in Google Scholar PubMed
11. Heo, YA. Golodirsen: first approval. Drugs 2020;80:329–33. https://doi.org/10.1007/s40265-020-01267-2.Suche in Google Scholar PubMed
12. Syed, YY. Eteplirsen: first global approval. Drugs 2016;76:1699–704. https://doi.org/10.1007/s40265-016-0657-1.Suche in Google Scholar PubMed
13. Lu, Y, Mbakam, CH, Song, B, Bendavid, E, Tremblay, JP. Improvements of nuclease and nickase gene modification techniques for the treatment of genetic diseases. Front Genome Ed 2022;4:892769. https://doi.org/10.3389/fgeed.2022.892769.Suche in Google Scholar PubMed PubMed Central
14. Khalil, AM. The genome editing revolution: review. J Genet Eng Biotechnol 2020;18:68. https://doi.org/10.1186/s43141-020-00078-y.Suche in Google Scholar PubMed PubMed Central
15. Anzalone, AV, Koblan, LW, Liu, DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 2020;38:824–44. https://doi.org/10.1038/s41587-020-0561-9.Suche in Google Scholar PubMed
16. Li, H, Yang, Y, Hong, W, Huang, M, Wu, M, Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Targeted Ther 2020;5:1. https://doi.org/10.1038/s41392-019-0089-y.Suche in Google Scholar PubMed PubMed Central
17. Silva, G, Poirot, L, Galetto, R, Smith, J, Montoya, G, Duchateau, P, et al.. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 2011;11:11–27. https://doi.org/10.2174/156652311794520111.Suche in Google Scholar PubMed PubMed Central
18. Stoddard, BL. Homing endonucleases from mobile group I introns: discovery to genome engineering. Mobile DNA 2014;5:7. https://doi.org/10.1186/1759-8753-5-7.Suche in Google Scholar PubMed PubMed Central
19. Stoddard, BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 2011;19:7–15. https://doi.org/10.1016/j.str.2010.12.003.Suche in Google Scholar PubMed PubMed Central
20. Jurica, MS, Monnat, RJJr, Stoddard, BL. DNA recognition and cleavage by the LAGLIDADG homing endonuclease I-CreI. Mol Cell 1998;2:469–76. https://doi.org/10.1016/s1097-2765(00)80146-x.Suche in Google Scholar PubMed
21. Colleaux, L, D’Auriol, L, Galibert, F, Dujon, B. Recognition and cleavage site of the intron-encoded omega transposase. Proc Natl Acad Sci U S A 1988;85:6022–6. https://doi.org/10.1073/pnas.85.16.6022.Suche in Google Scholar PubMed PubMed Central
22. Strathern, JN, Klar, AJ, Hicks, JB, Abraham, JA, Ivy, JM, Nasmyth, KA, et al.. Homothallic switching of yeast mating type cassettes is initiated by a double-stranded cut in the MAT locus. Cell 1982;31:183–92. https://doi.org/10.1016/0092-8674(82)90418-4.Suche in Google Scholar PubMed
23. Epinat, JC, Arnould, S, Chames, P, Rochaix, P, Desfontaines, D, Puzin, C, et al.. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res 2003;31:2952–62. https://doi.org/10.1093/nar/gkg375.Suche in Google Scholar PubMed PubMed Central
24. Rudin, N, Haber, JE. Efficient repair of HO-induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol Cell Biol 1988;8:3918–28. https://doi.org/10.1128/mcb.8.9.3918-3928.1988.Suche in Google Scholar PubMed PubMed Central
25. Rouet, P, Smih, F, Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A 1994;91:6064–8. https://doi.org/10.1073/pnas.91.13.6064.Suche in Google Scholar PubMed PubMed Central
26. Rouet, P, Smih, F, Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol 1994;14:8096–106. https://doi.org/10.1128/mcb.14.12.8096.Suche in Google Scholar
27. Urnov, FD, Rebar, EJ, Holmes, MC, Zhang, HS, Gregory, PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 2010;11:636–46. https://doi.org/10.1038/nrg2842.Suche in Google Scholar PubMed
28. Miller, J, McLachlan, AD, Klug, A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J 1985;4:1609–14. https://doi.org/10.1002/j.1460-2075.1985.tb03825.x.Suche in Google Scholar PubMed PubMed Central
29. Smith, J, Bibikova, M, Whitby, FG, Reddy, AR, Chandrasegaran, S, Carroll, D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res 2000;28:3361–9. https://doi.org/10.1093/nar/28.17.3361.Suche in Google Scholar PubMed PubMed Central
30. Greisman, HA, Pabo, CO. A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science 1997;275:657–61. https://doi.org/10.1126/science.275.5300.657.Suche in Google Scholar PubMed
31. Kim, YG, Cha, J, Chandrasegaran, S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 1996;93:1156–60. https://doi.org/10.1073/pnas.93.3.1156.Suche in Google Scholar PubMed PubMed Central
32. Bibikova, M, Golic, M, Golic, KG, Carroll, D. Targeted chromosomal cleavage and mutagenesis in drosophila using zinc-finger nucleases. Genetics 2002;161:1169–75. https://doi.org/10.1093/genetics/161.3.1169.Suche in Google Scholar PubMed PubMed Central
33. Morton, J, Davis, MW, Jorgensen, EM, Carroll, D. Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc Natl Acad Sci U S A 2006;103:16370–5. https://doi.org/10.1073/pnas.0605633103.Suche in Google Scholar PubMed PubMed Central
34. Doyon, Y, McCammon, JM, Miller, JC, Faraji, F, Ngo, C, Katibah, GE, et al.. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 2008;26:702–8. https://doi.org/10.1038/nbt1409.Suche in Google Scholar PubMed PubMed Central
35. Meng, X, Noyes, MB, Zhu, LJ, Lawson, ND, Wolfe, SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol 2008;26:695–701. https://doi.org/10.1038/nbt1398.Suche in Google Scholar PubMed PubMed Central
36. Urnov, FD, Miller, JC, Lee, YL, Beausejour, CM, Rock, JM, Augustus, S, et al.. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005;435:646–51. https://doi.org/10.1038/nature03556.Suche in Google Scholar PubMed
37. Ramirez, CL, Foley, JE, Wright, DA, Müller-Lerch, F, Rahman, SH, Cornu, TI, et al.. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 2008;5:374–5. https://doi.org/10.1038/nmeth0508-374.Suche in Google Scholar PubMed PubMed Central
38. Porteus, MH, Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003;300:763. https://doi.org/10.1126/science.1078395.Suche in Google Scholar PubMed
39. Kim, HJ, Lee, HJ, Kim, H, Cho, SW, Kim, JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 2009;19:1279–88. https://doi.org/10.1101/gr.089417.108.Suche in Google Scholar PubMed PubMed Central
40. Pattanayak, V, Ramirez, CL, Joung, JK, Liu, DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods 2011;8:765–70. https://doi.org/10.1038/nmeth.1670.Suche in Google Scholar PubMed PubMed Central
41. Gabriel, R, Lombardo, A, Arens, A, Miller, JC, Genovese, P, Kaeppel, C, et al.. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 2011;29:816–23. https://doi.org/10.1038/nbt.1948.Suche in Google Scholar PubMed
42. Moscou, MJ, Bogdanove, AJ. A simple cipher governs DNA recognition by TAL effectors. Science 2009;326:1501. https://doi.org/10.1126/science.1178817.Suche in Google Scholar PubMed
43. Boch, J, Scholze, H, Schornack, S, Landgraf, A, Hahn, S, Kay, S, et al.. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009;326:1509–12. https://doi.org/10.1126/science.1178811.Suche in Google Scholar PubMed
44. Cong, L, Zhou, R, Kuo, YC, Cunniff, M, Zhang, F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun 2012;3:968. https://doi.org/10.1038/ncomms1962.Suche in Google Scholar PubMed PubMed Central
45. Miller, JC, Tan, S, Qiao, G, Barlow, KA, Wang, J, Xia, DF, et al.. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 2011;29:143–8. https://doi.org/10.1038/nbt.1755.Suche in Google Scholar PubMed
46. Cermak, T, Doyle, EL, Christian, M, Wang, L, Zhang, Y, Schmidt, C, et al.. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 2011;39:e82. https://doi.org/10.1093/nar/gkr218.Suche in Google Scholar PubMed PubMed Central
47. Mussolino, C, Alzubi, J, Fine, EJ, Morbitzer, R, Cradick, TJ, Lahaye, T, et al.. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res 2014;42:6762–73. https://doi.org/10.1093/nar/gku305.Suche in Google Scholar PubMed PubMed Central
48. Cade, L, Reyon, D, Hwang, WY, Tsai, SQ, Patel, S, Khayter, C, et al.. Highly efficient generation of heritable zebrafish gene mutations using homo- and heterodimeric TALENs. Nucleic Acids Res 2012;40:8001–10. https://doi.org/10.1093/nar/gks518.Suche in Google Scholar PubMed PubMed Central
49. Gaj, T, Gersbach, CA, Barbas, CF3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013;31:397–405. https://doi.org/10.1016/j.tibtech.2013.04.004.Suche in Google Scholar PubMed PubMed Central
50. Mussolino, C, Morbitzer, R, Lütge, F, Dannemann, N, Lahaye, T, Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res 2011;39:9283–93. https://doi.org/10.1093/nar/gkr597.Suche in Google Scholar PubMed PubMed Central
51. Becker, S, Boch, J. TALE and TALEN genome editing technologies. Gene Genome Ed 2021;2:100007. https://doi.org/10.1016/j.ggedit.2021.100007.Suche in Google Scholar
52. Kim, Y, Kweon, J, Kim, A, Chon, JK, Yoo, JY, Kim, HJ, et al.. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 2013;31:251–8. https://doi.org/10.1038/nbt.2517.Suche in Google Scholar PubMed
53. Hubbard, BP, Badran, AH, Zuris, JA, Guilinger, JP, Davis, KM, Chen, L, et al.. Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat Methods 2015;12:939–42. https://doi.org/10.1038/nmeth.3515.Suche in Google Scholar PubMed PubMed Central
54. Gaj, T, Sirk, SJ, Shui, SL, Liu, J. Genome-editing technologies: principles and applications. Cold Spring Harbor Perspect Biol 2016;8:a023754. https://doi.org/10.1101/cshperspect.a023754.Suche in Google Scholar PubMed PubMed Central
55. Ishino, Y, Shinagawa, H, Makino, K, Amemura, M, Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in escherichia coli, and identification of the gene product. J Bacteriol 1987;169:5429–33. https://doi.org/10.1128/jb.169.12.5429-5433.1987.Suche in Google Scholar PubMed PubMed Central
56. Jansen, R, JDAv, E, Gaastra, W, Schouls, LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 2002;43:1565–75. https://doi.org/10.1046/j.1365-2958.2002.02839.x.Suche in Google Scholar PubMed
57. Bolotin, A, Quinquis, B, Sorokin, A, Ehrlich, SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005;151:2551–61. https://doi.org/10.1099/mic.0.28048-0.Suche in Google Scholar PubMed
58. Pourcel, C, Salvignol, G, Vergnaud, G. CRISPR elements in yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005;151:653–63. https://doi.org/10.1099/mic.0.27437-0.Suche in Google Scholar PubMed
59. Haft, DH, Selengut, J, Mongodin, EF, Nelson, KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol 2005;1:e60. https://doi.org/10.1371/journal.pcbi.0010060.Suche in Google Scholar PubMed PubMed Central
60. Barrangou, R, Fremaux, C, Deveau, H, Richards, M, Boyaval, P, Moineau, S, et al.. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007;315:1709–12. https://doi.org/10.1126/science.1138140.Suche in Google Scholar PubMed
61. Makarova, KS, Wolf, YI, Iranzo, J, Shmakov, SA, Alkhnbashi, OS, Brouns, SJJ, et al.. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 2020;18:67–83. https://doi.org/10.1038/s41579-019-0299-x.Suche in Google Scholar PubMed PubMed Central
62. Shah, SA, Erdmann, S, Mojica, FJ, Garrett, RA. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol 2013;10:891–9. https://doi.org/10.4161/rna.23764.Suche in Google Scholar PubMed PubMed Central
63. Li, G, Li, X, Zhuang, S, Wang, L, Zhu, Y, Chen, Y, et al.. Gene editing and its applications in biomedicine. Sci China Life Sci 2022;65:660–700. https://doi.org/10.1007/s11427-021-2057-0.Suche in Google Scholar PubMed PubMed Central
64. Jinek, M, Chylinski, K, Fonfara, I, Hauer, M, Doudna, JA, Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–21. https://doi.org/10.1126/science.1225829.Suche in Google Scholar PubMed PubMed Central
65. Gasiunas, G, Barrangou, R, Horvath, P, Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 2012;109:E2579–86. https://doi.org/10.1073/pnas.1208507109.Suche in Google Scholar PubMed PubMed Central
66. Doudna, JA, Charpentier, E. The new Frontier of genome engineering with CRISPR-Cas9. Science 2014;346:1–9. https://doi.org/10.1126/science.1258096.Suche in Google Scholar PubMed
67. Qi, LS, Larson, MH, Gilbert, LA, Doudna, JA, Weissman, JS, Arkin, AP, et al.. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013;152:1173–83. https://doi.org/10.1016/j.cell.2013.02.022.Suche in Google Scholar PubMed PubMed Central
68. Mali, P, Yang, L, Esvelt, KM, Aach, J, Guell, M, DiCarlo, JE, et al.. RNA-guided human genome engineering via Cas9. Science 2013;339:823–6. https://doi.org/10.1126/science.1232033.Suche in Google Scholar PubMed PubMed Central
69. Cong, L, Ran, FA, Cox, D, Lin, S, Barretto, R, Habib, N, et al.. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–23. https://doi.org/10.1126/science.1231143.Suche in Google Scholar PubMed PubMed Central
70. Jinek, M, East, A, Cheng, A, Lin, S, Ma, E, Doudna, J. RNA-programmed genome editing in human cells. Elife 2013;2:e00471. https://doi.org/10.7554/elife.00471.Suche in Google Scholar PubMed PubMed Central
71. Liu, G, Lin, Q, Jin, S, Gao, C. The CRISPR-Cas toolbox and gene editing technologies. Mol Cell 2022;82:333–47. https://doi.org/10.1016/j.molcel.2021.12.002.Suche in Google Scholar PubMed
72. Kleinstiver, BP, Prew, MS, Tsai, SQ, Topkar, VV, Nguyen, NT, Zheng, Z, et al.. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015;523:481–98. https://doi.org/10.1038/nature14592.Suche in Google Scholar PubMed PubMed Central
73. Hu, JH, Miller, SM, Geurts, MH, Tang, W, Chen, L, Sun, N, et al.. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018;556:57–63. https://doi.org/10.1038/nature26155.Suche in Google Scholar PubMed PubMed Central
74. Nishimasu, H, Shi, X, Ishiguro, S, Gao, L, Hirano, S, Okazaki, S, et al.. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 2018;361:1259–62. https://doi.org/10.1126/science.aas9129.Suche in Google Scholar PubMed PubMed Central
75. Miller, SM, Wang, T, Randolph, PB, Arbab, M, Shen, MW, Huang, TP, et al.. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol 2020;38:471–81. https://doi.org/10.1038/s41587-020-0412-8.Suche in Google Scholar PubMed PubMed Central
76. Walton, RT, Christie, KA, Whittaker, MN, Kleinstiver, BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020;368:290–6. https://doi.org/10.1126/science.aba8853.Suche in Google Scholar PubMed PubMed Central
77. Zetsche, B, Gootenberg, JS, Abudayyeh, OO, Slaymake, IM, Makarova, KS, Essletzbichler, P, et al.. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell 2015;163:1–13. https://doi.org/10.1016/j.cell.2015.09.038.Suche in Google Scholar PubMed PubMed Central
78. Pausch, P, Al-Shayeb, B, Bisom-Rapp, E, Tsuchida, CA, Li, Z, Cress, BF, et al.. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 2020;369:333–7. https://doi.org/10.1126/science.abb1400.Suche in Google Scholar PubMed PubMed Central
79. Karvelis, T, Bigelyte, G, Young, JK, Hou, Z, Zedaveinyte, R, Budre, K, et al.. PAM recognition by miniature CRISPR-Cas12f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Res 2020;48:5016–23. https://doi.org/10.1093/nar/gkaa208.Suche in Google Scholar PubMed PubMed Central
80. Tsai, SQ, Zheng, Z, Nguyen, NT, Liebers, M, Topkar, VV, Thapar, V, et al.. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 2015;33:187–97. https://doi.org/10.1038/nbt.3117.Suche in Google Scholar PubMed PubMed Central
81. Kim, D, Bae, S, Park, J, Kim, E, Kim, S, Yu, HR, et al.. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 2015;12:237–43. 1 p following 43. https://doi.org/10.1038/nmeth.3284.Suche in Google Scholar PubMed
82. Ran, FA, Hsu, PD, Lin, CY, Gootenberg, JS, Konermann, S, Trevino, AE, et al.. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013;154:1380–9. https://doi.org/10.1016/j.cell.2013.09.040.Suche in Google Scholar
83. Mali, P, Aach, J, Stranges, PB, Esvelt, KM, Moosburner, M, Kosuri, S, et al.. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 2013;31:833–8. https://doi.org/10.1038/nbt.2675.Suche in Google Scholar PubMed PubMed Central
84. Bolukbasi, MF, Gupta, A, Oikemus, S, Derr, AG, Garber, M, Brodsky, MH, et al.. DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat Methods 2015;12:1150–6. https://doi.org/10.1038/nmeth.3624.Suche in Google Scholar PubMed PubMed Central
85. Tsai, SQ, Wyvekens, N, Khayter, C, Foden, JA, Thapar, V, Reyon, D, et al.. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 2014;32:569–76. https://doi.org/10.1038/nbt.2908.Suche in Google Scholar PubMed PubMed Central
86. Kleinstiver, BP, Pattanayak, V, Prew, MS, Tsai, SQ, Nguyen, NT, Zheng, Z, et al.. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016;529:490–5. https://doi.org/10.1038/nature16526.Suche in Google Scholar PubMed PubMed Central
87. Slaymaker, IM, Gao, L, Zetsche, B, Scott, DA, Yan, WX, Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016;351:84–8. https://doi.org/10.1126/science.aad5227.Suche in Google Scholar PubMed PubMed Central
88. Chen, JS, Dagdas, YS, Kleinstiver, BP, Welch, MM, Sousa, AA, Harrington, LB, et al.. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017;550:407–10. https://doi.org/10.1038/nature24268.Suche in Google Scholar PubMed PubMed Central
89. Kulcsár, PI, Tálas, A, Huszár, K, Ligeti, Z, Tóth, E, Weinhardt, N, et al.. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol 2017;18:190. https://doi.org/10.1186/s13059-017-1318-8.Suche in Google Scholar PubMed PubMed Central
90. Casini, A, Olivieri, M, Petris, G, Montagna, C, Reginato, G, Maule, G, et al.. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol 2018;36:265–71. https://doi.org/10.1038/nbt.4066.Suche in Google Scholar PubMed PubMed Central
91. Vakulskas, CA, Dever, DP, Rettig, GR, Turk, R, Jacobi, AM, Collingwood, MA, et al.. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med 2018;24:1216–24. https://doi.org/10.1038/s41591-018-0137-0.Suche in Google Scholar PubMed PubMed Central
92. Lee, JK, Jeong, E, Lee, J, Jung, M, Shin, E, Kim, YH, et al.. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun 2018;9:3048. https://doi.org/10.1038/s41467-018-05477-x.Suche in Google Scholar PubMed PubMed Central
93. Kocak, DD, Josephs, EA, Bhandarkar, V, Adkar, SS, Kwon, JB, Gersbach, CA. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat Biotechnol 2019;37:657–66. https://doi.org/10.1038/s41587-019-0095-1.Suche in Google Scholar PubMed PubMed Central
94. Perez, AR, Pritykin, Y, Vidigal, JA, Chhangawala, S, Zamparo, L, Leslie, CS, et al.. GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol 2017;35:347–9. https://doi.org/10.1038/nbt.3804.Suche in Google Scholar PubMed PubMed Central
95. Hiranniramol, K, Chen, Y, Liu, W, Wang, X. Generalizable sgRNA design for improved CRISPR/Cas9 editing efficiency. Bioinformatics 2020;36:2684–9. https://doi.org/10.1093/bioinformatics/btaa041.Suche in Google Scholar PubMed PubMed Central
96. Mendell, JR, Al-Zaidy, SA, Rodino-Klapac, LR, Goodspeed, K, Gray, SJ, Kay, CN, et al.. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther 2021;29:464–88. https://doi.org/10.1016/j.ymthe.2020.12.007.Suche in Google Scholar PubMed PubMed Central
97. Wu, Z, Yang, H, Colosi, P. Effect of genome size on AAV vector packaging. Mol Ther 2010;18:80–6. https://doi.org/10.1038/mt.2009.255.Suche in Google Scholar PubMed PubMed Central
98. Ran, FA, Cong, L, Yan, WX, Scott, DA, Gootenberg, JS, Kriz, AJ, et al.. In vivo genome editing using staphylococcus aureus Cas9. Nature 2015;520:186–91. https://doi.org/10.1038/nature14299.Suche in Google Scholar PubMed PubMed Central
99. Zetsche, B, Volz, SE, Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol 2015;33:139–42. https://doi.org/10.1038/nbt.3149.Suche in Google Scholar PubMed PubMed Central
100. Wang, D, Li, J, Song, CQ, Tran, K, Mou, H, Wu, PH, et al.. Cas9-mediated allelic exchange repairs compound heterozygous recessive mutations in mice. Nat Biotechnol 2018;36:839–42. https://doi.org/10.1038/nbt.4219.Suche in Google Scholar PubMed PubMed Central
101. Kim, E, Koo, T, Park, SW, Kim, D, Kim, K, Cho, HY, et al.. In vivo genome editing with a small Cas9 orthologue derived from campylobacter jejuni. Nat Commun 2017;8:14500. https://doi.org/10.1038/ncomms14500.Suche in Google Scholar PubMed PubMed Central
102. Xu, X, Chemparathy, A, Zeng, L, Kempton, HR, Shang, S, Nakamura, M, et al.. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol Cell 2021;81:4333–45.e4. https://doi.org/10.1016/j.molcel.2021.08.008.Suche in Google Scholar PubMed
103. Höijer, I, Emmanouilidou, A, Östlund, R, van Schendel, R, Bozorgpana, S, Tijsterman, M, et al.. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat Commun 2022;13:627. https://doi.org/10.1038/s41467-022-28244-5.Suche in Google Scholar PubMed PubMed Central
104. Papathanasiou, S, Markoulaki, S, Blaine, LJ, Leibowitz, ML, Zhang, CZ, Jaenisch, R, et al.. Whole chromosome loss and genomic instability in mouse embryos after CRISPR-Cas9 genome editing. Nat Commun 2021;12:5855. https://doi.org/10.1038/s41467-021-26097-y.Suche in Google Scholar PubMed PubMed Central
105. Zuccaro, MV, Xu, J, Mitchell, C, Marin, D, Zimmerman, R, Rana, B, et al.. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell 2020;183:1650–64.e15. https://doi.org/10.1016/j.cell.2020.10.025.Suche in Google Scholar PubMed
106. Kosicki, M, Tomberg, K, Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 2018;36:765–71. https://doi.org/10.1038/nbt.4192.Suche in Google Scholar PubMed PubMed Central
107. Ihry, RJ, Worringer, KA, Salick, MR, Frias, E, Ho, D, Theriault, K, et al.. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 2018;24:939–46. https://doi.org/10.1038/s41591-018-0050-6.Suche in Google Scholar PubMed
108. Haapaniemi, E, Botla, S, Persson, J, Schmierer, B, Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 2018;24:927–30. https://doi.org/10.1038/s41591-018-0049-z.Suche in Google Scholar PubMed
109. Rees, HA, Liu, DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 2018;19:770–88. https://doi.org/10.1038/s41576-018-0059-1.Suche in Google Scholar PubMed PubMed Central
110. Komor, AC, Kim, YB, Packer, MS, Zuris, JA, Liu, DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016;533:420–4. https://doi.org/10.1038/nature17946.Suche in Google Scholar PubMed PubMed Central
111. Gaudelli, NM, Komor, AC, Rees, HA, Packer, MS, Badran, AH, Bryson, DI, et al.. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017;551:464–71. https://doi.org/10.1038/nature24644.Suche in Google Scholar PubMed PubMed Central
112. Kurt, IC, Zhou, R, Iyer, S, Garcia, SP, Miller, BR, Langner, LM, et al.. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat Biotechnol 2021;39:41–6. https://doi.org/10.1038/s41587-020-0609-x.Suche in Google Scholar PubMed PubMed Central
113. Zhao, D, Li, J, Li, S, Xin, X, Hu, M, Price, MA, et al.. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat Biotechnol 2021;39:35–40. https://doi.org/10.1038/s41587-020-0592-2.Suche in Google Scholar PubMed
114. Grünewald, J, Zhou, R, Lareau, CA, Garcia, SP, Iyer, S, Miller, BR, et al.. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat Biotechnol 2020;38:861–4. https://doi.org/10.1038/s41587-020-0535-y.Suche in Google Scholar PubMed PubMed Central
115. Zhang, X, Zhu, B, Chen, L, Xie, L, Yu, W, Wang, Y, et al.. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat Biotechnol 2020;38:856–60. https://doi.org/10.1038/s41587-020-0527-y.Suche in Google Scholar PubMed
116. Sakata, RC, Ishiguro, S, Mori, H, Tanaka, M, Tatsuno, K, Ueda, H, et al.. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat Biotechnol 2020;38:865–9. https://doi.org/10.1038/s41587-020-0509-0.Suche in Google Scholar PubMed
117. Porto, EM, Komor, AC, Slaymaker, IM, Yeo, GW. Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov 2020;19:839–59. https://doi.org/10.1038/s41573-020-0084-6.Suche in Google Scholar PubMed PubMed Central
118. Li, Q, Li, Y, Yang, S, Huang, S, Yan, M, Ding, Y, et al.. CRISPR–Cas9-mediated base-editing screening in mice identifies DND1 amino acids that are critical for primordial germ cell development. Nat Cell Biol 2018;20:1315–25. https://doi.org/10.1038/s41556-018-0202-4.Suche in Google Scholar PubMed
119. Zhao, T, Li, Q, Zhou, C, Lv, X, Liu, H, Tu, T, et al.. Small-molecule compounds boost genome-editing efficiency of cytosine base editor. Nucleic Acids Res 2021;49:8974–86. https://doi.org/10.1093/nar/gkab645.Suche in Google Scholar PubMed PubMed Central
120. Hess, GT, Tycko, J, Yao, D, Bassik, MC. Methods and applications of CRISPR-mediated base editing in eukaryotic genomes. Mol Cell 2017;68:26–43. https://doi.org/10.1016/j.molcel.2017.09.029.Suche in Google Scholar PubMed PubMed Central
121. Nishida, K, Arazoe, T, Yachie, N, Banno, S, Kakimoto, M, Tabata, M, et al.. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016;353:1248–55. https://doi.org/10.1126/science.aaf8729.Suche in Google Scholar PubMed
122. Ma, Y, Zhang, J, Yin, W, Zhang, Z, Song, Y, Chang, X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat Methods 2016;13:1029–35. https://doi.org/10.1038/nmeth.4027.Suche in Google Scholar PubMed
123. Gehrke, JM, Cervantes, O, Clement, MK, Wu, Y, Zeng, J, Bauer, DE, et al.. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat Biotechnol 2018;36:977–82. https://doi.org/10.1038/nbt.4199.Suche in Google Scholar PubMed PubMed Central
124. Wang, X, Li, J, Wang, Y, Yang, B, Wei, J, Wu, J, et al.. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat Biotechnol 2018;36:946–9. https://doi.org/10.1038/nbt.4198.Suche in Google Scholar PubMed
125. Komor, AC, Zhao, KT, Packer, MS, Gaudelli, NM, Waterbury, AL, Koblan, LW, et al.. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci Adv 2017;3:eaao4774. https://doi.org/10.1126/sciadv.aao4774.Suche in Google Scholar PubMed PubMed Central
126. Lee, S, Ding, N, Sun, Y, Yuan, T, Li, J, Yuan, Q, et al.. Single C-to-T substitution using engineered APOBEC3G-nCas9 base editors with minimum genome- and transcriptome-wide off-target effects. Sci Adv 2020;6:eaba1773. https://doi.org/10.1126/sciadv.aba1773.Suche in Google Scholar PubMed PubMed Central
127. Kim, YB, Komor, AC, Levy, JM, Packer, MS, Zhao, KT, Liu, DR. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 2017;35:371–6. https://doi.org/10.1038/nbt.3803.Suche in Google Scholar PubMed PubMed Central
128. Zuo, E, Sun, Y, Wei, W, Yuan, T, Ying, W, Sun, H, et al.. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 2019;364:289. https://doi.org/10.1126/science.aav9973.Suche in Google Scholar PubMed PubMed Central
129. Jin, S, Zong, Y, Gao, Q, Zhu, Z, Wang, Y, Qin, P, et al.. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 2019;364:292–5. https://doi.org/10.1126/science.aaw7166.Suche in Google Scholar PubMed
130. Grünewald, J, Zhou, R, Garcia, SP, Iyer, S, Lareau, CA, Aryee, MJ, et al.. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019;569:433–7. https://doi.org/10.1038/s41586-019-1161-z.Suche in Google Scholar PubMed PubMed Central
131. Zhou, C, Sun, Y, Yan, R, Liu, Y, Zuo, E, Gu, C, et al.. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 2019;571:275–8. https://doi.org/10.1038/s41586-019-1314-0.Suche in Google Scholar PubMed
132. Villiger, L, Grisch-Chan, HM, Lindsay, H, Ringnalda, F, Pogliano, CB, Allegri, G, et al.. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat Med 2018;24:1519–25. https://doi.org/10.1038/s41591-018-0209-1.Suche in Google Scholar PubMed
133. Jeong, YK, Song, B, Bae, S. Current status and challenges of DNA base editing tools. Mol Ther 2020;28:1938–52. https://doi.org/10.1016/j.ymthe.2020.07.021.Suche in Google Scholar PubMed PubMed Central
134. Zafra, MP, Schatoff, EM, Katti, A, Foronda, M, Breinig, M, Schweitzer, AY, et al.. Optimized base editors enable efficient editing in cells, organoids and mice. Nat Biotechnol 2018;36:888–93. https://doi.org/10.1038/nbt.4194.Suche in Google Scholar PubMed PubMed Central
135. Wang, L, Li, X, Xue, W, Wei, J, Yan, L, Chen, M, et al.. Enhanced base editing by co-expression of free uracil DNA glycosylase inhibitor. Cell Res 2017;27:1289–92. https://doi.org/10.1038/cr.2017.111.Suche in Google Scholar PubMed PubMed Central
136. Li, X, Wang, Y, Liu, Y, Yang, B, Wang, X, Wei, J, et al.. Base editing with a Cpf1-cytidine deaminase fusion. Nat Biotechnol 2018;36:324–7. https://doi.org/10.1038/nbt.4102.Suche in Google Scholar PubMed
137. Thuronyi, BW, Koblan, LW, Levy, JM, Yeh, WH, Zheng, C, Newby, GA, et al.. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat Biotechnol 2019;37:1070–9. https://doi.org/10.1038/s41587-019-0193-0.Suche in Google Scholar PubMed PubMed Central
138. Doman, JL, Raguram, A, Newby, GA, Liu, DR. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat Biotechnol 2020;38:620–8. https://doi.org/10.1038/s41587-020-0414-6.Suche in Google Scholar PubMed PubMed Central
139. Zuo, E, Sun, Y, Yuan, T, He, B, Zhou, C, Ying, W, et al.. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat Methods 2020;17:600–4. https://doi.org/10.1038/s41592-020-0832-x.Suche in Google Scholar PubMed
140. Yu, Y, Leete, TC, Born, DA, Young, L, Barrera, LA, Lee, SJ, et al.. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat Commun 2020;11:2052. https://doi.org/10.1038/s41467-020-15887-5.Suche in Google Scholar PubMed PubMed Central
141. Li, A, Mitsunobu, H, Yoshioka, S, Suzuki, T, Kondo, A, Nishida, K. Cytosine base editing systems with minimized off-target effect and molecular size. Nat Commun 2022;13:4531. https://doi.org/10.1038/s41467-022-32157-8.Suche in Google Scholar PubMed PubMed Central
142. Koblan, LW, Doman, JL, Wilson, C, Levy, JM, Tay, T, Newby, GA, et al.. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol 2018;36:843–6. https://doi.org/10.1038/nbt.4172.Suche in Google Scholar PubMed PubMed Central
143. Huang, TP, Zhao, KT, Miller, SM, Gaudelli, NM, Oakes, BL, Fellmann, C, et al.. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat Biotechnol 2019;37:626–31. https://doi.org/10.1038/s41587-019-0134-y.Suche in Google Scholar PubMed PubMed Central
144. Fu, J, Li, Q, Liu, X, Tu, T, Lv, X, Yin, X, et al.. Human cells based directed evolution of adenine base editors with improved efficiency. Nat Commun 2021;12:5897. https://doi.org/10.1038/s41467-021-26211-0.Suche in Google Scholar PubMed PubMed Central
145. Richter, MF, Zhao, KT, Eton, E, Lapinaite, A, Newby, GA, Thuronyi, BW, et al.. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol 2020;38:883–91. https://doi.org/10.1038/s41587-020-0453-z.Suche in Google Scholar PubMed PubMed Central
146. Gaudelli, NM, Lam, DK, Rees, HA, Solá-Esteves, NM, Barrera, LA, Born, DA, et al.. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol 2020;38:892–900. https://doi.org/10.1038/s41587-020-0491-6.Suche in Google Scholar PubMed
147. Grünewald, J, Zhou, R, Iyer, S, Lareau, CA, Garcia, SP, Aryee, MJ, et al.. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat Biotechnol 2019;37:1041–8. https://doi.org/10.1038/s41587-019-0236-6.Suche in Google Scholar PubMed PubMed Central
148. Rees, HA, Wilson, C, Doman, JL, Liu, DR. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci Adv 2019;5:eaax5717. https://doi.org/10.1126/sciadv.aax5717.Suche in Google Scholar PubMed PubMed Central
149. Kim, HS, Jeong, YK, Hur, JK, Kim, JS, Bae, S. Adenine base editors catalyze cytosine conversions in human cells. Nat Biotechnol 2019;37:1145–8. https://doi.org/10.1038/s41587-019-0254-4.Suche in Google Scholar PubMed
150. Jeong, YK, Lee, S, Hwang, GH, Hong, SA, Park, SE, Kim, JS, et al.. Adenine base editor engineering reduces editing of bystander cytosines. Nat Biotechnol 2021;39:1426–33. https://doi.org/10.1038/s41587-021-00943-2.Suche in Google Scholar PubMed
151. Chen, S, Liu, Z, Xie, W, Yu, H, Lai, L, Li, Z. Compact Cje3Cas9 for efficient in vivo genome editing and adenine base editing. CRISPR J 2022;5:472–86. https://doi.org/10.1089/crispr.2021.0143.Suche in Google Scholar PubMed
152. Davis, JR, Wang, X, Witte, IP, Huang, TP, Levy, JM, Raguram, A, et al.. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base. Nat Biomed Eng 2022. https://doi.org/10.1038/s41551-022-00911-4. In press.Suche in Google Scholar PubMed PubMed Central
153. Anzalone, AV, Randolph, PB, Davis, JR, Sousa, AA, Koblan, LW, Levy, JM, et al.. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019;576:149–57. https://doi.org/10.1038/s41586-019-1711-4.Suche in Google Scholar PubMed PubMed Central
154. Choi, J, Chen, W, Suiter, CC, Lee, C, Chardon, FM, Yang, W, et al.. Precise genomic deletions using paired prime editing. Nat Biotechnol 2022;40:218–26. https://doi.org/10.1038/s41587-021-01025-z.Suche in Google Scholar PubMed PubMed Central
155. Lin, Q, Zong, Y, Xue, C, Wang, S, Jin, S, Zhu, Z, et al.. Prime genome editing in rice and wheat. Nat Biotechnol 2020;38:582–5. https://doi.org/10.1038/s41587-020-0455-x.Suche in Google Scholar PubMed
156. Liu, Y, Li, X, He, S, Huang, S, Li, C, Chen, Y, et al.. Efficient generation of mouse models with the prime editing system. Cell Discov 2020;6:27. https://doi.org/10.1038/s41421-020-0165-z.Suche in Google Scholar PubMed PubMed Central
157. Petri, K, Zhang, W, Ma, J, Schmidts, A, Lee, H, Horng, JE, et al.. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat Biotechnol 2022;40:189–93. https://doi.org/10.1038/s41587-021-00901-y.Suche in Google Scholar PubMed PubMed Central
158. Kim, HK, Yu, G, Park, J, Min, S, Lee, S, Yoon, S, et al.. Predicting the efficiency of prime editing guide RNAs in human cells. Nat Biotechnol 2021;39:198–206. https://doi.org/10.1038/s41587-020-0677-y.Suche in Google Scholar PubMed
159. Chow, RD, Chen, JS, Shen, J, Chen, S. A web tool for the design of prime-editing guide RNAs. Nat Biomed Eng 2021;5:190–4. https://doi.org/10.1038/s41551-020-00622-8.Suche in Google Scholar PubMed PubMed Central
160. Nelson, JW, Randolph, PB, Shen, SP, Everette, KA, Chen, PJ, Anzalone, AV, et al.. Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol 2022;40:402–10. https://doi.org/10.1038/s41587-021-01039-7.Suche in Google Scholar PubMed PubMed Central
161. Li, X, Zhou, L, Gao, BQ, Li, G, Wang, X, Wang, Y, et al.. Highly efficient prime editing by introducing same-sense mutations in pegRNA or stabilizing its structure. Nat Commun 2022;13:1669. https://doi.org/10.1038/s41467-022-29339-9.Suche in Google Scholar PubMed PubMed Central
162. Chen, PJ, Hussmann, JA, Yan, J, Knipping, F, Ravisankar, P, Chen, PF, et al.. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 2021;184:5635–52.e29. https://doi.org/10.1016/j.cell.2021.09.018.Suche in Google Scholar PubMed PubMed Central
163. Doman, JL, Sousa, AA, Randolph, PB, Chen, PJ, Liu, DR. Designing and executing prime editing experiments in mammalian cells. Nat Protoc 2022. https://doi.org/10.1038/s41596-022-00724-4. In press.Suche in Google Scholar PubMed PubMed Central
164. Strecker, J, Ladha, A, Gardner, Z, Schmid-Burgk, JL, Makarova, KS, Koonin, EV, et al.. RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019;365:48–53. https://doi.org/10.1126/science.aax9181.Suche in Google Scholar PubMed PubMed Central
165. Klompe, SE, Vo, PLH, Halpin-Healy, TS, Sternberg, SH. Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 2019;571:219–25. https://doi.org/10.1038/s41586-019-1323-z.Suche in Google Scholar PubMed
166. Rybarski, JR, Hu, K, Hill, AM, Wilke, CO, Finkelstein, IJ. Metagenomic discovery of CRISPR-associated transposons. Proc Natl Acad Sci U S A 2021;118:e2112279118. https://doi.org/10.1073/pnas.2112279118.Suche in Google Scholar PubMed PubMed Central
167. Yang, S, Zhang, Y, Xu, J, Zhang, J, Zhang, J, Yang, J, et al.. Orthogonal CRISPR-associated transposases for parallel and multiplexed chromosomal integration. Nucleic Acids Res 2021;49:10192–202. https://doi.org/10.1093/nar/gkab752.Suche in Google Scholar PubMed PubMed Central
168. Halpin-Healy, TS, Klompe, SE, Sternberg, SH, Fernández, IS. Structural basis of DNA targeting by a transposon-encoded CRISPR-Cas system. Nature 2020;577:271–4. https://doi.org/10.1038/s41586-019-1849-0.Suche in Google Scholar PubMed
169. Saito, M, Ladha, A, Strecker, J, Faure, G, Neumann, E, Altae-Tran, H, et al.. Dual modes of CRISPR-associated transposon homing. Cell 2021;184:2441–53.e18. https://doi.org/10.1016/j.cell.2021.03.006.Suche in Google Scholar PubMed PubMed Central
170. Ma, W, Xu, YS, Sun, XM, Huang, H. Transposon-associated CRISPR-cas system: a powerful DNA insertion tool. Trends Microbiol 2021;29:565–8. https://doi.org/10.1016/j.tim.2021.01.017.Suche in Google Scholar PubMed
171. Yang, J, Yang, J, Zhang, Y, Yang, S, Zhang, J, Jiang, Y, et al.. CRISPR-associated transposase system can insert multiple copies of donor DNA into the same target locus. CRISPR J 2021;4:789–98.10.1089/crispr.2021.0019Suche in Google Scholar PubMed
172. Kato, GJ, Piel, FB, Reid, CD, Gaston, MH, Ohene-Frempong, K, Krishnamurti, L, et al.. Sickle cell disease. Nat Rev Dis Prim 2018;4:18010. https://doi.org/10.1038/nrdp.2018.10.Suche in Google Scholar PubMed
173. Shah, FT, Sayani, F, Trompeter, S, Drasar, E, Piga, A. Challenges of blood transfusions in beta-thalassemia. Blood Rev 2019;37:100588. https://doi.org/10.1016/j.blre.2019.100588.Suche in Google Scholar PubMed
174. Eapen, M, Brazauskas, R, Walters, MC, Bernaudin, F, Bo-Subait, K, Fitzhugh, CD, et al.. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol 2019;6:e585–96. https://doi.org/10.1016/s2352-3026(19)30154-1.Suche in Google Scholar PubMed PubMed Central
175. Gluckman, E, Cappelli, B, Bernaudin, F, Labopin, M, Volt, F, Carreras, J, et al.. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017;129:1548–56. https://doi.org/10.1182/blood-2016-10-745711.Suche in Google Scholar PubMed PubMed Central
176. Baronciani, D, Angelucci, E, Potschger, U, Gaziev, J, Yesilipek, A, Zecca, M, et al.. Hemopoietic stem cell transplantation in thalassemia: a report from the European society for blood and bone marrow transplantation hemoglobinopathy registry, 2000-2010. Bone Marrow Transplant 2016;51:536–41. https://doi.org/10.1038/bmt.2015.293.Suche in Google Scholar PubMed
177. Xu, J, Peng, C, Sankaran, VG, Shao, Z, Esrick, EB, Chong, BG, et al.. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 2011;334:993–6. https://doi.org/10.1126/science.1211053.Suche in Google Scholar PubMed PubMed Central
178. Lettre, G, Bauer, DE. Fetal haemoglobin in sickle-cell disease: from genetic epidemiology to new therapeutic strategies. Lancet 2016;387:2554–64. https://doi.org/10.1016/s0140-6736(15)01341-0.Suche in Google Scholar
179. Germino-Watnick, P, Hinds, M, Le, A, Chu, R, Liu, X, Uchida, N. Hematopoietic stem cell gene-addition/editing therapy in sickle cell disease. Cells 2022;11:1843. https://doi.org/10.3390/cells11111843.Suche in Google Scholar PubMed PubMed Central
180. Paz-Priel, I. A phase I/II study of GPH101 in autologous CD34+ hematopoietic stem cells to convert HbS to HbA for treating severe sickle cell disease: ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04819841?term=gene&cond=Sickle+cell+disease&draw=3&rank=11 [Accessed 29 Jun 2022].Suche in Google Scholar
181. Walters, M. Transplantation of CRISPRCas9 corrected hematopoietic stem cells (CRISPR_SCD001) in patients with severe sickle cell disease: ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04774536?term=CRISPR_SCD001&cond=Sickle+cell+disease&draw=2&rank=1 [Accessed 27 Apr 2022].Suche in Google Scholar
182. Eisenstein, M. Graphite bio: gene editing blood stem cells for sickle cell disease. Nat Biotechnol 2021. https://doi.org/10.1038/d41587-021-00010-w.Suche in Google Scholar PubMed
183. Lattanzi, A, Camarena, J, Lahiri, P, Segal, H, Srifa, W, Vakulskas, CA, et al.. Development of beta-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci Transl Med 2021;13:eabf2444. https://doi.org/10.1126/scitranslmed.abf2444.Suche in Google Scholar PubMed PubMed Central
184. Zhang, X. A safety and efficacy study of β-globin restored autologous hematopoietic stem cells for β-thalassemia major patients with CVS-654 mutation: ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04205435?term=NCT04205435&draw=2&rank=1 [Accessed 11 Apr 2022].Suche in Google Scholar
185. Sankaran, VG, Menne, TF, Xu, J, Akie, TE, Lettre, G, Van Handel, B, et al.. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008;322:1839–42. https://doi.org/10.1126/science.1165409.Suche in Google Scholar PubMed
186. Liu, N, Hargreaves, VV, Zhu, Q, Kurland, JV, Hong, J, Kim, W, et al.. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 2018;173:430–42.e17. https://doi.org/10.1016/j.cell.2018.03.016.Suche in Google Scholar PubMed PubMed Central
187. Canver, MC, Smith, EC, Sher, F, Pinello, L, Sanjana, NE, Shalem, O, et al.. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015;527:192–7. https://doi.org/10.1038/nature15521.Suche in Google Scholar PubMed PubMed Central
188. Bauer, DE, Kamran, SC, Lessard, S, Xu, J, Fujiwara, Y, Lin, C, et al.. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013;342:253–7. https://doi.org/10.1126/science.1242088.Suche in Google Scholar PubMed PubMed Central
189. Psatha, N, Reik, A, Phelps, S, Zhou, Y, Dalas, D, Yannaki, E, et al.. Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with beta-thalassemia major. Mol Ther Methods Clin Dev 2018;10:313–26. https://doi.org/10.1016/j.omtm.2018.08.003.Suche in Google Scholar PubMed PubMed Central
190. Demirci, S, Zeng, J, Wu, Y, Uchida, N, Shen, AH, Pellin, D, et al.. BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J Clin Invest 2020;130:6677–87. https://doi.org/10.1172/jci140189.Suche in Google Scholar
191. Frangoul, H, Altshuler, D, Cappellini, MD, Chen, YS, Domm, J, Eustace, BK, et al.. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N Engl J Med 2021;384:252–60. https://doi.org/10.1056/nejmoa2031054.Suche in Google Scholar
192. Vertex Pharmaceuticals Incorporated CT. A long-term follow-up study of subjects with β-thalassemia or sickle cell disease treated with autologous CRISPR-cas9 modified hematopoietic stem cells (CTX001): ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04208529?term=NCT05329649&cond=Sickle+cell+disease&draw=2&rank=2 [Accessed 5 Jul 2022].Suche in Google Scholar
193. Vertex Pharmaceuticals Incorporated CT. A phase 3 study to evaluate the safety and efficacy of a single dose of CTX001 in pediatric subjects with severe sickle cell disease: ClinicalTrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT05329649?term=NCT05329649&cond=Sickle+cell+disease&draw=2&rank=1 [Accessed 1 Jun 2022].Suche in Google Scholar
194. Pharmaceuticals, N. A first-in-patient phase I/II clinical study to investigate the safety, tolerability and efficacy of genome-edited hematopoietic stem and progenitor cells in subjects with severe complications of sickle cell disease: ClinicalTrials.gov; 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT04443907?term=OTQ923&draw=2&rank=1 [Accessed 21 Jun 2022].Suche in Google Scholar
195. Fu, B, Liao, J, Chen, S, Li, W, Wang, Q, Hu, J, et al.. CRISPR–Cas9-mediated gene editing of the BCL11A enhancer for pediatric β0/β0 transfusion-dependent β-thalassemia. Nat Med 2022;28:1573–80. https://doi.org/10.1038/s41591-022-01906-z.Suche in Google Scholar PubMed
196. Zhang, G. A safety and efficacy study evaluating ET-01 in subjects with transfusion dependent β-thalassaemia (ET-01): ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04925206?term=NCT04925206&draw=2&rank=1 [Accessed 11 May 2022].Suche in Google Scholar
197. Medicine, E. EDIT-301 for autologous HSCT in subjects with severe sickle cell disease: ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/study/NCT04853576 [Accessed 15 Jun 2022].Suche in Google Scholar
198. Medicine, E. Robust pre-clinical results and large-scale manufacturing process for EDIT-301: an autologous cell therapy for the potential treatment of SCD: Editasmedicine.com; 2020. Available from: https://www.editasmedicine.com/wp-content/uploads/2020/12/EDIT-301-ASH-Poster.pdf.Suche in Google Scholar
199. Walters, MC, Smith, AR, Schiller, GJ, Esrick, EB, Williams, DA, Gogoleva, T, et al.. Updated results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent beta thalassemia. Blood 2021;138:3974–4. https://doi.org/10.1182/blood-2021-147907.Suche in Google Scholar
200. Alavi, A, Krishnamurti, L, Abedi, M, Galeon, I, Reiner, D, Smith, SE, et al.. Preliminary safety and efficacy results from precizn-1: an ongoing phase 1/2 study on zinc finger nuclease-modified autologous CD34+ HSPCs for sickle cell disease (SCD). Blood 2021;138:2930–0. https://doi.org/10.1182/blood-2021-151650.Suche in Google Scholar
201. Newby, GA, Yen, JS, Woodard, KJ, Mayuranathan, T, Lazzarotto, CR, Li, Y, et al.. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 2021;595:295–302. https://doi.org/10.1038/s41586-021-03609-w.Suche in Google Scholar PubMed PubMed Central
202. Therapeutics, B. BEACON: a study evaluating the safety and efficacy of BEAM-101 in patients with severe sickle cell disease: ClinicalTrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT05456880?term=BEAM-101&draw=2&rank=1 [Accessed 13 Jul 2022].Suche in Google Scholar
203. Peake, JD, Noguchi, E. Fanconi anemia: current insights regarding epidemiology, cancer, and DNA repair. Hum Genet 2022. https://doi.org/10.1007/s00439-022-02462-9. In press.Suche in Google Scholar PubMed
204. Diez, B, Genovese, P, Roman-Rodriguez, FJ, Alvarez, L, Schiroli, G, Ugalde, L, et al.. Therapeutic gene editing in CD34 (+) hematopoietic progenitors from fanconi anemia patients. EMBO Mol Med 2017;9:1574–88. https://doi.org/10.15252/emmm.201707540.Suche in Google Scholar PubMed PubMed Central
205. Roman-Rodriguez, FJ, Ugalde, L, Alvarez, L, Diez, B, Ramirez, MJ, Risueno, C, et al.. NHEJ-mediated repair of CRISPR-cas9-induced DNA breaks efficiently corrects mutations in HSPCs from patients with fanconi anemia. Cell Stem Cell 2019;25:607–21.e7. https://doi.org/10.1016/j.stem.2019.08.016.Suche in Google Scholar PubMed
206. Zhang, ZY, Thrasher, AJ, Zhang, F. Gene therapy and genome editing for primary immunodeficiency diseases. Genes Dis 2020;7:38–51. https://doi.org/10.1016/j.gendis.2019.07.007.Suche in Google Scholar PubMed PubMed Central
207. Tucci, F, Scaramuzza, S, Aiuti, A, Mortellaro, A. Update on clinical ex vivo hematopoietic stem cell gene therapy for inherited monogenic diseases. Mol Ther 2021;29:489–504. https://doi.org/10.1016/j.ymthe.2020.11.020.Suche in Google Scholar PubMed PubMed Central
208. Genovese, P, Schiroli, G, Escobar, G, Tomaso, TD, Firrito, C, Calabria, A, et al.. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014;510:235–40. https://doi.org/10.1038/nature13420.Suche in Google Scholar PubMed PubMed Central
209. Schiroli, G, Ferrari, S, Conway, A, Jacob, A, Capo, V, Albano, L, et al.. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci Transl Med 2017;9:eaan0820. https://doi.org/10.1126/scitranslmed.aan0820.Suche in Google Scholar PubMed
210. van Haasteren, J, Li, J, Scheideler, OJ, Murthy, N, Schaffer, DV. The delivery challenge: fulfilling the promise of therapeutic genome editing. Nat Biotechnol 2020;38:845–55. https://doi.org/10.1038/s41587-020-0565-5.Suche in Google Scholar PubMed
211. Doudna, JA. The promise and challenge of therapeutic genome editing. Nature 2020;578:229–36. https://doi.org/10.1038/s41586-020-1978-5.Suche in Google Scholar PubMed PubMed Central
212. Jones, RJ, DeBaun, MR. Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both, or neither. Blood 2021;138:942–7. https://doi.org/10.1182/blood.2021011488.Suche in Google Scholar PubMed
213. Yang, Y, Wang, L, Bell, P, McMenamin, D, He, Z, White, J, et al.. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol 2016;34:334–8. https://doi.org/10.1038/nbt.3469.Suche in Google Scholar PubMed PubMed Central
214. Charlesworth, CT, Deshpande, PS, Dever, DP, Camarena, J, Lemgart, VT, Cromer, MK, et al.. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med 2019;25:249–54. https://doi.org/10.1038/s41591-018-0326-x.Suche in Google Scholar PubMed PubMed Central
215. Glass, Z, Li, Y, Xu, Q. Nanoparticles for CRISPR-Cas9 delivery. Nat Biomed Eng 2017;1:854–5. https://doi.org/10.1038/s41551-017-0158-x.Suche in Google Scholar PubMed PubMed Central
216. Han, JP, Kim, M, Choi, BS, Lee, JH, Lee, GS, Jeong, M, et al.. In vivo delivery of CRISPR-Cas9 using lipid nanoparticles enables antithrombin gene editing for sustainable hemophilia A and B therapy. Sci Adv 2022;8:eabj6901. https://doi.org/10.1126/sciadv.abj6901.Suche in Google Scholar PubMed PubMed Central
217. Zuris, JA, Thompson, DB, Shu, Y, Guilinger, JP, Bessen, JL, Hu, JH, et al.. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol 2015;33:73–80. https://doi.org/10.1038/nbt.3081.Suche in Google Scholar PubMed PubMed Central
218. Erkut, E, Yokota, T. CRISPR therapeutics for duchenne muscular dystrophy. Int J Mol Sci 2022;23:1832. https://doi.org/10.3390/ijms23031832.Suche in Google Scholar PubMed PubMed Central
219. Gillmore, JD, Gane, E, Taubel, J, Kao, J, Fontana, M, Maitland, ML, et al.. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med 2021;385:493–502. https://doi.org/10.1056/nejmoa2107454.Suche in Google Scholar PubMed
220. Adams, D, Koike, H, Slama, M, Coelho, T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 2019;15:387–404. https://doi.org/10.1038/s41582-019-0210-4.Suche in Google Scholar PubMed
221. Banerji, A, Busse, P, Shennak, M, Lumry, W, Davis-Lorton, M, Wedner, HJ, et al.. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med 2017;376:717–28. https://doi.org/10.1056/nejmoa1605767.Suche in Google Scholar PubMed
222. Kaplan, AP, Joseph, K. The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol 2010;104:193–204. https://doi.org/10.1016/j.anai.2010.01.007.Suche in Google Scholar PubMed
223. Seitzer, J. NTLA-2002: CRISPR/Cas9-Mediated gene knockout of KLKB1 for hereditary angioedema: 2021 AAAAi Annual Meeting; 2021; Virtual meeting.10.1016/j.jaci.2020.12.531Suche in Google Scholar
224. Therapeutics, I. NTLA-2002 in adults with hereditary angioedema (HAE): ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT05120830 [Accessed 20 Jul 2022].Suche in Google Scholar
225. Medicine, E. Single ascending dose study in participants with LCA10: ClinicalTrials.gov; 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT03872479?term=EDIT-101&draw=2&rank=1 [Accessed 15 Jul 2022].Suche in Google Scholar
226. den Hollander, AI, Koenekoop, RK, Yzer, S, Lopez, I, Arends, ML, Voesenek, KE, et al.. Mutations in the CEP290 (NPHP6) gene are a frequent cause of leber congenital amaurosis. Am J Hum Genet 2006;79:556–61. https://doi.org/10.1086/507318.Suche in Google Scholar PubMed PubMed Central
227. Stone, EM, Andorf, JL, Whitmore, SS, DeLuca, AP, Giacalone, JC, Streb, LM, et al.. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 2017;124:1314–31. https://doi.org/10.1016/j.ophtha.2017.04.008.Suche in Google Scholar PubMed PubMed Central
228. Maeder, ML, Stefanidakis, M, Wilson, CJ, Baral, R, Barrera, LA, Bounoutas, GS, et al.. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 2019;25:229–33. https://doi.org/10.1038/s41591-018-0327-9.Suche in Google Scholar PubMed
229. Verve takes base editors into humans. Nat Biotechnol 2022;40:1159.10.1038/s41587-022-01445-5Suche in Google Scholar PubMed
230. Therapeutics, V. A study of VERVE-101 in patients with familial hypercholesterolemia and cardiovascular disease: ClinicalTrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT05398029?term=base+editor&cond=Hereditary+Diseases&draw=2&rank=1 [Accessed 9 Aug 2022].Suche in Google Scholar
231. Brautbar, A, Leary, E, Rasmussen, K, Wilson, DP, Steiner, RD, Virani, S. Genetics of familial hypercholesterolemia. Curr Atherosclerosis Rep 2015;17:491. https://doi.org/10.1007/s11883-015-0491-z.Suche in Google Scholar PubMed
232. Musunuru, K, Chadwick, AC, Mizoguchi, T, Garcia, SP, DeNizio, JE, Reiss, CW, et al.. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021;593:429–34. https://doi.org/10.1038/s41586-021-03534-y.Suche in Google Scholar PubMed
233. Therapeutics, V. Verve therapeutics to present preclinical data from gene editing programs for the treatment of atherosclerotic cardiovascular disease at ASGCT; 2021. Available from: https://www.vervetx.com/press-releases/verve-therapeutics-to-present-preclinical-data-from-gene-editing-programs-for-the-treatment-of-atherosclerotic-cardiovascular-disease-at-asgct/.Suche in Google Scholar
234. Consiglieri, G, Bernardo, ME, Brunetti-Pierri, N, Aiuti, A. Ex vivo and in vivo gene therapy for mucopolysaccharidoses: state of the art. Hematol Oncol Clin N Am 2022;36:865–78. https://doi.org/10.1016/j.hoc.2022.03.012.Suche in Google Scholar PubMed
235. Butterfield, JSS, Hege, KM, Herzog, RW, Kaczmarek, R. A molecular revolution in the treatment of hemophilia. Mol Ther 2020;28:997–1015. https://doi.org/10.1016/j.ymthe.2019.11.006.Suche in Google Scholar PubMed PubMed Central
236. Therapeutics, S. Ascending dose study of genome editing by zinc finger nuclease therapeutic SB-FIX in subjects with severe hemophilia B: ClinicalTrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/study/NCT02695160?term=NCT02695160&draw=2&rank=1 [Accessed 23 Mar 2022].Suche in Google Scholar
237. Therapeutics, S. Ascending dose study of genome editing by the zinc finger nuclease (ZFN) therapeutic SB-913 in subjects with MPS II: ClinicalTrials.gov; 2022. Available from: https://clinicaltrials.gov/ct2/history/NCT03041324?V_23&embedded=true [Accessed 17 Jun 2022].Suche in Google Scholar
238. Therapeutics, S. Ascending dose study of genome editing by the zinc finger nuclease (ZFN) therapeutic SB-318 in subjects with MPS I: ClinicalTrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT02702115?term=NCT02702115&draw=2&rank=1 [Accessed 16 Dec 2021].Suche in Google Scholar
239. Ou, L, Przybilla, MJ, Ahlat, O, Kim, S, Overn, P, Jarnes, J, et al.. A highly efficacious PS gene editing system corrects metabolic and neurological complications of mucopolysaccharidosis type I. Mol Ther 2020;28:1442–54. https://doi.org/10.1016/j.ymthe.2020.03.018.Suche in Google Scholar PubMed PubMed Central
240. YCJ, C, Arudkumar, J, Aartsma-Rus, A, Adikusuma, F, Thomas, PQ. CRISPR applications for Duchenne muscular dystrophy: from animal models to potential therapies. WIREs Mech Dis 2022;e1580. In press.Suche in Google Scholar
241. Aartsma-Rus, A, Fokkema, I, Verschuuren, J, Ginjaar, I, van Deutekom, J, van Ommen, GJ, et al.. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 2009;30:293–9. https://doi.org/10.1002/humu.20918.Suche in Google Scholar PubMed
242. Mbakam, CH, Lamothe, G, Tremblay, G, Tremblay, JP. CRISPR-Cas9 gene therapy for Duchenne muscular dystrophy. Neurotherapeutics 2022;19:931–41. https://doi.org/10.1007/s13311-022-01197-9.Suche in Google Scholar PubMed PubMed Central
243. Baylis, F, Darnovsky, M, Hasson, K, Krahn, TM. Human Germ Line and Heritable Genome Editing: The Global Policy Landscape. CRISPR J 2020;3:365–77. https://doi.org/10.1089/crispr.2020.0082.Suche in Google Scholar PubMed
244. Wolf, DP, Mitalipov, PA, Mitalipov, SM. Principles of and strategies for germline gene therapy. Nat Med 2019;25:890–7. https://doi.org/10.1038/s41591-019-0473-8.Suche in Google Scholar PubMed
245. Jin, LF, Li, JS. Generation of genetically modified mice using CRISPR/Cas9 and haploid embryonic stem cell systems. Dongwuxue Yanjiu 2016;37:205–13.Suche in Google Scholar
246. Takehashi, M, Kanatsu-Shinohara, M, Shinohara, T. Generation of genetically modified animals using spermatogonial stem cells. Dev Growth Differ 2010;52:303–10. https://doi.org/10.1111/j.1440-169x.2009.01167.x.Suche in Google Scholar PubMed
247. Wu, Y, Liang, D, Wang, Y, Bai, M, Tang, W, Bao, S, et al.. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013;13:659–62. https://doi.org/10.1016/j.stem.2013.10.016.Suche in Google Scholar PubMed
248. Wu, Y, Zhou, H, Fan, X, Zhang, Y, Zhang, M, Wang, Y, et al.. Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res 2015;25:67–79. https://doi.org/10.1038/cr.2014.160.Suche in Google Scholar PubMed PubMed Central
249. Long, C, McAnally, JR, Shelton, JM, Mireault, AA, Bassel-Duby, R, Olson, EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014;345:1184–8. https://doi.org/10.1126/science.1254445.Suche in Google Scholar PubMed PubMed Central
250. Lu, D, Gong, X, Fang, Y, Guo, X, Chen, Y, Yang, F, et al.. Correction of RNA splicing defect in beta (654)-thalassemia mice using CRISPR/Cas9 gene-editing technology. Haematologica 2022;107:1427–37.10.3324/haematol.2020.278238Suche in Google Scholar PubMed PubMed Central
251. Wang, YH, Yan, M, Zhang, X, Liu, XY, Ding, YF, Lai, CP, et al.. Rescue of male infertility through correcting a genetic mutation causing meiotic arrest in spermatogonial stem cells. Asian J Androl 2021;23:590–9. https://doi.org/10.4103/aja.aja_97_20.Suche in Google Scholar PubMed PubMed Central
252. Liang, P, Xu, Y, Zhang, X, Ding, C, Huang, R, Zhang, Z, et al.. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2015;6:363–72. https://doi.org/10.1007/s13238-015-0153-5.Suche in Google Scholar PubMed PubMed Central
253. Tang, L, Zeng, Y, Du, H, Gong, M, Peng, J, Zhang, B, et al.. CRISPR/Cas9-mediated gene editing in human zygotes using Cas9 protein. Mol Genet Genom 2017;292:525–33. https://doi.org/10.1007/s00438-017-1299-z.Suche in Google Scholar PubMed
254. Ma, H, Marti-Gutierrez, N, Park, SW, Wu, J, Lee, Y, Suzuki, K, et al.. Correction of a pathogenic gene mutation in human embryos. Nature 2017;548:413–9. https://doi.org/10.1038/nature23305.Suche in Google Scholar PubMed
255. Egli, D, Zuccaro, MV, Kosicki, M, Church, GM, Bradley, A, Jasin, M. Inter-homologue repair in fertilized human eggs? Nature 2018;560:E5–7. https://doi.org/10.1038/s41586-018-0379-5.Suche in Google Scholar PubMed
256. Adikusuma, F, Piltz, S, Corbett, MA, Turvey, M, McColl, SR, Helbig, KJ, et al.. Large deletions induced by Cas9 cleavage. Nature 2018;560:E8–9. https://doi.org/10.1038/s41586-018-0380-z.Suche in Google Scholar PubMed
257. Ma, H, Marti-Gutierrez, N, Park, SW, Wu, J, Hayama, T, Darby, H, et al.. Ma et al. reply. Nature 2018;560:E10–23. https://doi.org/10.1038/s41586-018-0381-y.Suche in Google Scholar PubMed
258. Liang, D, Gutierrez, NM, Chen, T, Lee, Y, Park, SW, Ma, H, et al.. Frequent gene conversion in human embryos induced by double strand breaks. bioRxiv 2020;0619:162214.10.1101/2020.06.19.162214Suche in Google Scholar
259. Li, G, Liu, Y, Zeng, Y, Li, J, Wang, L, Yang, G, et al.. Highly efficient and precise base editing in discarded human tripronuclear embryos. Protein Cell 2017;8:776–9. https://doi.org/10.1007/s13238-017-0458-7.Suche in Google Scholar PubMed PubMed Central
260. Zhou, C, Zhang, M, Wei, Y, Sun, Y, Sun, Y, Pan, H, et al.. Highly efficient base editing in human tripronuclear zygotes. Protein Cell 2017;8:772–5. https://doi.org/10.1007/s13238-017-0459-6.Suche in Google Scholar PubMed PubMed Central
261. Liang, P, Ding, C, Sun, H, Xie, X, Xu, Y, Zhang, X, et al.. Correction of beta-thalassemia mutant by base editor in human embryos. Protein Cell 2017;8:811–22. https://doi.org/10.1007/s13238-017-0475-6.Suche in Google Scholar PubMed PubMed Central
262. Zeng, Y, Li, J, Li, G, Huang, S, Yu, W, Zhang, Y, et al.. Correction of the marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol Ther 2018;26:2631–7. https://doi.org/10.1016/j.ymthe.2018.08.007.Suche in Google Scholar PubMed PubMed Central
263. Zhang, M, Zhou, C, Wei, Y, Xu, C, Pan, H, Ying, W, et al.. Human cleaving embryos enable robust homozygotic nucleotide substitutions by base editors. Genome Biol 2019;20:101. https://doi.org/10.1186/s13059-019-1703-6.Suche in Google Scholar PubMed PubMed Central
264. Li, G, Liu, X, Huang, S, Zeng, Y, Yang, G, Lu, Z, et al.. Efficient generation of pathogenic A-to-G mutations in human tripronuclear embryos via ABE-mediated base editing. Mol Ther Nucleic Acids 2019;17:289–96. https://doi.org/10.1016/j.omtn.2019.05.021.Suche in Google Scholar PubMed PubMed Central
265. Lander, ES, Baylis, F, Zhang, F, Charpentier, E, Berg, P, Bourgain, C, et al.. Adopt a moratorium on heritable genome editing. Nature 2019;567:165–8. https://doi.org/10.1038/d41586-019-00726-5.Suche in Google Scholar PubMed
266. Zhang, D, Lie, RK. Ethical issues in human germline gene editing: a perspective from China. Monash Bioeth Rev 2018;36:23–35. https://doi.org/10.1007/s40592-018-0091-0.Suche in Google Scholar PubMed
267. Raguram, A, Banskota, S, Liu, DR. Therapeutic in vivo delivery of gene editing agents. Cell 2022;185:2806–27. https://doi.org/10.1016/j.cell.2022.03.045.Suche in Google Scholar PubMed PubMed Central
268. Lyu, P, Wang, L, Lu, B. Virus-like particle mediated CRISPR/Cas9 delivery for efficient and safe genome editing. Life 2020;10:366. https://doi.org/10.3390/life10120366.Suche in Google Scholar PubMed PubMed Central
269. Kim, D, Lim, K, Kim, ST, Yoon, SH, Kim, K, Ryu, SM, et al.. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol 2017;35:475–80. https://doi.org/10.1038/nbt.3852.Suche in Google Scholar PubMed
270. Kim, D, Kim, DE, Lee, G, Cho, SI, Kim, JS. Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat Biotechnol 2019;37:430–5. https://doi.org/10.1038/s41587-019-0050-1.Suche in Google Scholar PubMed
271. Chapman, JR, Taylor, MR, Boulton, SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012;47:497–510. https://doi.org/10.1016/j.molcel.2012.07.029.Suche in Google Scholar PubMed
272. Yeh, CD, Richardson, CD, Corn, JE. Advances in genome editing through control of DNA repair pathways. Nat Cell Biol 2019;21:1468–78. https://doi.org/10.1038/s41556-019-0425-z.Suche in Google Scholar PubMed
© 2022 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- Editorial
- Exploring the mysteries of reproductive health
- Reviews
- Developmental origins of adult diseases
- Site-specific genome editing in treatment of inherited diseases: possibility, progress, and perspectives
- An update on placental drug transport and its relevance to fetal drug exposure
- Organoid research on human early development and beyond
- Fibroblast growth factor 21 and dietary interventions: what we know and what we need to know next
- Hypothalamic GABAergic neurocircuitry in the regulation of energy homeostasis and sleep/wake control
Artikel in diesem Heft
- Frontmatter
- Editorial
- Exploring the mysteries of reproductive health
- Reviews
- Developmental origins of adult diseases
- Site-specific genome editing in treatment of inherited diseases: possibility, progress, and perspectives
- An update on placental drug transport and its relevance to fetal drug exposure
- Organoid research on human early development and beyond
- Fibroblast growth factor 21 and dietary interventions: what we know and what we need to know next
- Hypothalamic GABAergic neurocircuitry in the regulation of energy homeostasis and sleep/wake control