Lithium fluoroarylsilylamides and their structural features
-
Sakshi Mohan


Abstract
In order to probe the role of Li⋯F interactions toward the stabilisation of low-coordinate lithium complexes, the four fluoroarylsilylamides [LiN(SiMe3)(2-C6H4F)] (1-Li), [LiN(SiMe3)(2,6-C6H3F2)] (2-Li), [LiN(SiMe3)(C6F5)] (3-Li), and [LiN(SiMe2H)(2-C6H4F)] (4-Li) have been synthesised in high yields by deprotonation of the parent amines with nBuLi. They have been comprehensively characterised by multinuclear NMR spectroscopy, and complete assignments were achieved with the help of 2D NMR data. The molecular solid-state structures of [(2-Li)2]∞, [3-Li·Et 2 O]2, and [4-Li]8 were determined by single-crystal X-ray diffraction. They feature unusual coordination patterns, notably for the formation of the polymeric [(2-Li)2]∞ and a unique octagonal, crown-like [4-Li]8. In both structures, the role of Li⋯F non-covalent interactions was the key toward the building of the final architecture. It is shown that Li–F and C–F interatomic distances, along with |1 J C,F| coupling constants, can be used as qualitative tools for the evaluation of the presence and relative strength of Li⋯F contacts.
1 Introduction
Fluorine-containing substituents and molecules are attracting much interest, in particular on account of the remarkable chemical properties of C–F bonds. They have become a common motif in agrochemical, pharmaceutical, and bioactive compounds. The incorporation of fluorine allows for the modulation of ligand binding affinities by establishing electrostatic or hydrogen-bonding effects that are not found in the nonfluorinated congeners (Fujiwara and O’Hagan, 2014; Meanwell, 2018; Purser et al., 2008).
The organometallic chemistry of early main group alkali and large alkaline-earth metals is gaining momentum, in particular because they represent affordable, widespread, and non-toxic alternatives to transition metals in homogeneous catalysis. These elements have been used to great effect for the production of molecular catalysts that display high reactivity and selectivity in a broadening range of metal-catalysed organic transformations, e.g. for hydrofunctionalisation, dehydrocoupling, and polymerisation reactions (Harder, 2020). Low-coordinate, highly electrophilic complexes of the large alkaline earths (Ca, Sr, Ba) have been shown to be extremely versatile and effective catalysts. Among the most prominent features, the need for a coordinatively unsaturated metal centre, that is, with a low coordination number, appears to be essential to achieve unique reactivity (Rösch et al., 2021; Rösch and Harder, 2021; Wilson et al., 2017). As established earlier for rare-earth compounds, the syntheses of stable alkaline-earth complexes with coordination numbers as low as two or three have been made possible through the implementation of reliable synthetic strategies, relying either on the kinetic stabilisation imparted by bulky and specifically designed ancillary ligands, or via the deliberate inclusion of secondary interactions that bring additional thermodynamic stability (Le Coz et al., 2018; Chapple and Sarazin, 2020). Among these non-covalent interactions, agostic (Brookhart et al., 2007), π–π (Salonen et al., 2011), cation–π (Dougherty, 2013), and dispersion force interactions (Liptrot and Power, 2017) are the most salient and well-known ones. A review article in 2010 highlighted the importance of weak interactions in alkaline-earth chemistry (Buchanan et al., 2010), and they have certainly been essential to many of the breakthroughs that the field has witnessed since. Interactions between a metal and fluorine atoms, for instance in fluoroalkoxides, are also very effective at stabilising electron-deficient complexes (Roşca et al., 2014; Sarazin et al., 2011). Our group and that of Harder have recently demonstrated how low coordinate, stable, and yet highly reactive alkaline-earth amides [(ligand)MN(C6F5)2] could be generated by exploitation of M⋯F interactions for M = Ca, Sr, Ba (Fischer et al., 2019; Roueindeji et al., 2019).
Organolithium compounds are remarkably useful and versatile reagents with synthetic applications in organic and organometallic chemistry that populate every synthetic chemistry laboratory (Hevia et al., 2022; Luisi and Capriati, 2014; Luisi et al., 2022; Wakefield, 1988). Despite being known for a century, these seemingly simple species are still attracting sustained attention, notably because the fascinating and often unpredictable structure–reactivity relationships they display are far from being fully comprehended (Barozzino-Consiglio et al., 2017; Hédouin et al., 2023). A recent article from the Arnold group elegantly described how NMR data, and most prominently 1 J C,F couplings, could be used to gauge the strength of Li⋯F interactions in a lithiated m-terphenyl ligand bearing fluorine atoms at the ortho positions (Skeel et al., 2020). As part of our interest in low-coordinate s-block metals stabilised by secondary interactions, we report here on the utilisation of fluoroarylsilylamines to prepare non-solvated lithiated fluoroarylsilylamides. The solid-state structures of several of these complexes, which display varying patterns of intramolecular and intermolecular Li⋯F interactions, as well as their NMR data, are discussed.
2 Results and discussion
N-(2-Fluorophenyl)-trimethylsilylamine (1-H), N-(2,6-difluorophenyl)-trimethylsilylamine (2-H), N-(pentafluorophenyl)-trimethylsilylamine (3-H), and N-(2-Fluorophenyl)-1,1-dimethylsilylamine (4-H) were isolated in 79–93% yields as colourless liquids upon deprotonation of the appropriate fluoroaniline with nBuLi, followed by quenching with Me3SiCl or, for 4-H, with Me2SiHCl (Scheme 1). After initial work-up, the compounds were obtained as analytically pure samples following trap-to-trap transfer. Note that 1-H (Yin et al., 2016) and 3-H (Oliver and Graham, 1969) have already been described; we propose here adapted procedures and full characterisations.
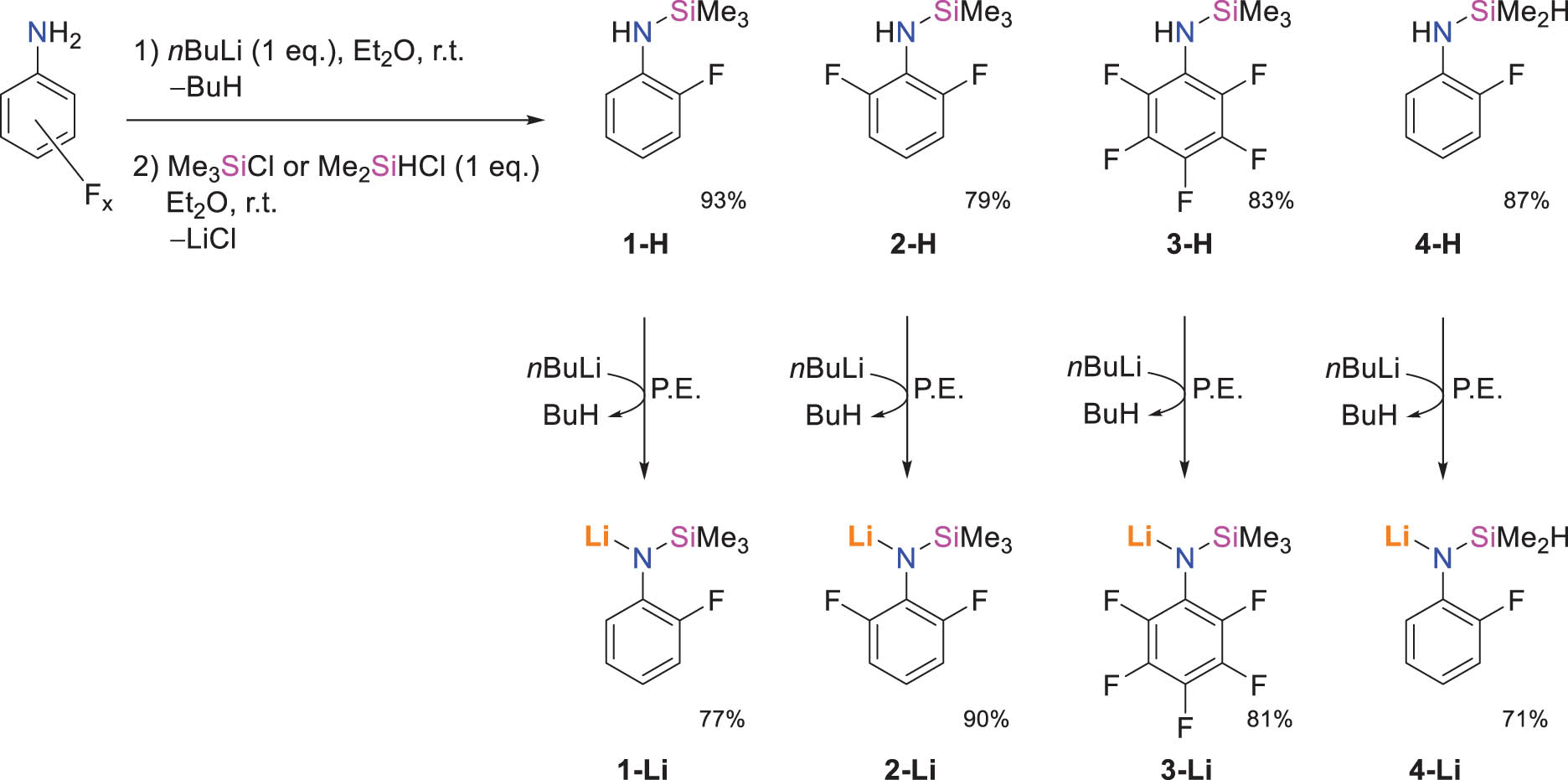
Synthesis of the fluoroarylamines 1-H – 4-H and their lithiated derivatives 1-Li – 4-Li (isolated yields).
The identity of the fluoroarylsilylamines was established by NMR spectroscopy in benzene-d 6 (1H, 13C, 19F, and 2D spectra) and mass spectrometry. In the 19F NMR spectra, the amines were characterised by single, well-defined resonances at δ F –135.2 ppm for 1-H, –135.0 ppm for 2-H, and −135.4 ppm for 4-H, with corresponding integrations of 1F, 2F, and 1F. The pertaining |1 J C,F| coupling constants, determined from the 13C{1H} NMR spectra, are 236.5, 238.5, and 241.5 Hz, respectively. These latter values are typically in the range found for other Carene–F bonds (Skeel et al., 2020). The 19F NMR spectrum for 3-H exhibits the expected resonances at –159.0 (o-F), –165.4 (m-F), and –173.7 (p-F) ppm. The NMR data for 2-H were also recorded in thf-d 8; they confirmed that the coupling constants, most notably the J C,F ones, are, within the limits of the detection method, not affected by the nature of the solvent, and that the 19F NMR spectrum is identical to that in benzene-d 6.
The equimolar reaction between the fluoroarylamines and nBuLi in petroleum ether at room temperature afforded the corresponding solvent-free lithiated N-(2-fluorophenyl)-trimethylsilylamide [LiN(SiMe3)(2-C6H4F)] (1-Li), N-(2,6-difluorophenyl)-trimethylsilylamide [LiN(SiMe3)(2,6-C6H3F2)] (2-Li), N-(pentafluorophenyl)-trimethylsilylamide [LiN(SiMe3)(C6F5)] (3-Li), and N-(2-fluorophenyl)-1,1-dimethylsilylamide [LiN(SiMe2H)(2-C6H4F)] (4-Li) in good to high isolated yields (71–90%). The four lithiated amides were obtained as colourless powders. They are sparingly soluble in hydrocarbons, but they dissolve readily in Et2O and in thf at room temperature; they decompose rapidly in chlorinated solvents. Due to the limited solubility of all four complexes in aromatic hydrocarbons, their NMR data were recorded in thf-d 8. Full assignment of all resonances in the 1H and 13C{1H} spectra was achieved using standard combinations of 1H, 13C{1H}, and 2D (COSY 1H–1H, HSQC, and HMBC 1H–13C) data. Note that 4-Li is not stable in solution. For instance, an initially colourless solution of the complex in thf-d8 will gradually turn red over the course of 12 h, with the release of a non-identified compound; in its 19F NMR spectrum, this is concomitant with the appearance of a (yet unidentified) resonance of growing intensity at δ F –140.8 ppm, whereas the resonance for the complex itself is found at –145.2 ppm. The 7Li NMR spectra for 1-Li, 2-Li, and 4-Li were characterised by a singlet resonance in the range δ Li 1.18–1.27 ppm; by comparison, the resonance for 3-Li at 0.90 ppm was slightly more shielded. The use of thf-d 8 as the NMR solvent instead of non-coordinating alternatives precluded useful DOSY NMR analysis regarding the actual aggregation of these species.
The representative 13C{1H} NMR spectrum of compound 2-Li recorded in thf-d 8 is displayed in Figure 1. It shows in particular a doublet of doublets at δ C 159.9–158.0 ppm (|1 J C,F| = 226.4 Hz and |3 J C,F| = 13.8 Hz) diagnostic of the two ortho-CF carbon atoms (aka C 2), along with two 1:2:1 triplets centred on δ C 139.1 and 104.2 ppm for the ipso (aka C 1; |2 J C,F| = 17.6 Hz) and para (aka C 4; |3 J C,F| = 10.7 Hz) C-atoms, respectively. In addition, the spectrum also features a multiplet centred on δ C 110.3 ppm for carbon atoms in meta position of the aromatic ring (aka C 3), and a 1:2:1 triplet at 3.3 ppm with a scalar coupling constant |5 J C,F| = 3.8 Hz for the CH3 atoms coupled to the fluorine atoms in ortho positions. Similar scalar couplings with ortho fluorines, with comparable intensities, were observed in several cases, both for the fluoroarylamines and for the corresponding lithiated complexes. In these instances, the 5 J C,F scalar nature of the coupling, instead of a through-space dipolar one, was unambiguously demonstrated by a 19F–13C HMBC experiment (Figure S17 in the Supplementary material).
![Figure 1
13C{1H} NMR spectrum (thf-d
8, 125.77 MHz, 27°C) of [LiN(SiMe3)(2,6-C6H3F2)] (2-Li). * = residual solvent (petroleum ether, Et2O, and benzene).](/document/doi/10.1515/mgmc-2023-0012/asset/graphic/j_mgmc-2023-0012_fig_001.jpg)
13C{1H} NMR spectrum (thf-d 8, 125.77 MHz, 27°C) of [LiN(SiMe3)(2,6-C6H3F2)] (2-Li). * = residual solvent (petroleum ether, Et2O, and benzene).
The unsolvated 2-Li is crystallised from a concentrated solution in petroleum ether. Due to the presence of multiple Li⋯F interactions, it forms the solid-state infinite zigzag monodimensional chains of coordination polymer. The repetitive motif is a centro-symmetric dimer formed by two molecules of 2-Li with bridging N atoms forming a planar Li2N2 core, and the crystalline solid-state structure of the compound is hence best depicted as [(2-Li)2]∞ (Figures 2 and 3). With two Li⋯F interactions per metal, the geometry about each lithium atom forms a distorted tetrahedron with τ 4 = 0.75 (Yang et al., 2007). The Li1–N1 and Li1–N1#2 interatomic distances of 2.068(9) and 2.047(8) Å are unexceptional. They compare well with those found in similar lithium amides, for instance in the trimeric, solvent-free [LiN(SiMe3)2]3 (2.00(2) Å) or in the Et2O-solvated dimer [LiN(SiMe3)2·Et2O]2 (2.06(1) Å) (Lappert et al., 1983). The intramolecular Li1–F2 interatomic distance of 1.985(8) Å is at the low end of values reported for similar interactions and is substantially shorter than the intermolecular one (Li#1–F1 = 2.107(9) Å). For comparison, Li–F distances were in the range 1.972(8)–2.000(9) Å in the heterobimetallic complex C{SiMe2N(2-C6H4F)}3Sn(Li)] (Lutz et al., 2003). The Li–F distances in the known tmeda adduct [2-Li.tmeda] (2.082(5) Å) is also longer (Aharonovich et al., 2009) than the Li1–F2 one, but both distances in [(2-Li)2]∞ are in the range of those measured in the dimeric [{LiN(C6F5)2}2·(o-C6H3F2)] solvated by a single molecule of difluorobenzene (1.986(3)–2.139(3) Å; Schorpp et al., 2020). By contrast, Li–F interatomic distances were shorter (1.925(4) 1.945(5) Å) in [TerphF-Li]2, where TerphF is 2,6-bis(2,6-difluoro-4-trimethylsilylphenyl)-4-tert-butylphenyl (Skeel et al., 2020). The C5–F1 and C9–F2 bond lengths of 1.389(5) and 1.384(5) Å in [(2-Li)2]∞ are conventional and match those in the three aforementioned reported lithium complexes.
![Figure 2
Representation of the molecular solid-state structure of the dimeric [LiN(SiMe3)(2,6-C6H3F2)]2([2-Li]2). H atoms are omitted for clarity. Li⋯F intermolecular interactions within the 1D-coordination network shown in dotted lines. Selected bond lengths (Å) and angles (°): Li1–N1 = 2.068(9), Li1–N1#2 = 2.047(8), Li1–F2 = 1.985(8), L1#1–F1 = 2.107(9), C5–F1 = 1.389(5), C9–F2 = 1.384(5); N1–Li1–N1#2 = 104.2(4), Li1–N1–Li1#2 = 75.8(4). Symmetry transformations: #1: x, y, z, T = [0, −1, 0]; #2: −x, −y, −z, T = [0, 2, 0]. Colour code: C, black; F, green; Li, orange; N, blue; Si, beige.](/document/doi/10.1515/mgmc-2023-0012/asset/graphic/j_mgmc-2023-0012_fig_002.jpg)
Representation of the molecular solid-state structure of the dimeric [LiN(SiMe3)(2,6-C6H3F2)]2([2-Li]2). H atoms are omitted for clarity. Li⋯F intermolecular interactions within the 1D-coordination network shown in dotted lines. Selected bond lengths (Å) and angles (°): Li1–N1 = 2.068(9), Li1–N1#2 = 2.047(8), Li1–F2 = 1.985(8), L1#1–F1 = 2.107(9), C5–F1 = 1.389(5), C9–F2 = 1.384(5); N1–Li1–N1#2 = 104.2(4), Li1–N1–Li1#2 = 75.8(4). Symmetry transformations: #1: x, y, z, T = [0, −1, 0]; #2: −x, −y, −z, T = [0, 2, 0]. Colour code: C, black; F, green; Li, orange; N, blue; Si, beige.
![Figure 3
Representation of the molecular solid-state structure of [{LiN(SiMe3)(2,6-C6H3F2)}2]∞([(2-Li)2]∞), showing the one-dimensional zigzag arrangement through the presence of intermolecular Li⋯F interactions. H atoms are omitted for clarity. Li⋯F interactions within the coordination network are shown in dotted lines. Colour code: C, black; F, green; Li, orange; N, blue; Si, beige.](/document/doi/10.1515/mgmc-2023-0012/asset/graphic/j_mgmc-2023-0012_fig_003.jpg)
Representation of the molecular solid-state structure of [{LiN(SiMe3)(2,6-C6H3F2)}2]∞([(2-Li)2]∞), showing the one-dimensional zigzag arrangement through the presence of intermolecular Li⋯F interactions. H atoms are omitted for clarity. Li⋯F interactions within the coordination network are shown in dotted lines. Colour code: C, black; F, green; Li, orange; N, blue; Si, beige.
The creation of coordination polymers through the formation of intermolecular Li⋯F interactions, as encountered in [(2-Li)2]∞, is already documented. A number of patterns in organometallic and inorganic examples have been described, e.g. as in [Li(diglyme)][BF4] (Andreev et al., 2005) and in [Li(Et2O)][B(C6F5)4] (Kuprat et al., 2010) or, more relevantly to our work, in the monodimensional [LiN(SiMe3)2·p-C6H4F2]∞ which presents short Li–F interatomic distances (1.999(8) Å; Williard and Liu, 1994). Non-covalent interactions, such as Li⋯F ones in our case, are now an established synthetic tool frequently used to synthesise highly electrophilic complexes with low coordination numbers and devoid of coordinated Lewis base. With s-block oxophilic metals, the M⋯F interaction has been shown to be essentially ionic, although it also possesses a non-negligible covalent character (Roueindeji et al., 2019). It can be safely assumed that it is because of the absence of additional donor onto the lithium centres that [(2-Li)2]∞ forms a polymer in the solid state. Coordination of, for instance, Et2O molecules, would be expected to disrupt many if not all of the Li⋯F contacts.
Single crystals of 1-Li and 3-Li suitable for X-ray diffraction could not be obtained despite repeated attempts. However, X-ray quality crystals of [3-Li·Et 2 O] were grown from a solution of 3-Li in Et2O. The solvated complex exists as a centro-symmetric dimer with four-coordinated metal centres as a result of coordination by two N-atoms and one O-atom along with a single Li⋯F interaction (Figure 4). The bridging positions are occupied by the nitrogen atoms, and as a result the dimer exhibits a planar Li2N2 core. The geometry about the metal atoms forms a distorted tetrahedra (τ 4 = 0.31). The Li–N interatomic distances, in the range 2.061(3)–2.075(3) Å, are unremarkable; they match those found in [(2-Li)2]∞. Despite the presence of a coordinated Et2O molecule in [3-Li·Et 2 O], the complex still shows one Li⋯F interaction per metal, albeit the pertaining Li1–F1 = 2.106(3) Å distance suggests its intensity is lesser than those in [(2-Li)2]∞. There is no significant elongation of the C–F bond length as a result of the (weak) interaction of the fluorine (aka F1) with the metal (aka Li1), as indicated by the very similar C2–F1 and C6–F5 interatomic distances of 1.3643(18) and 1.3541(19) Å, respectively.
![Figure 4
Representation of the molecular solid-state structure of the dimer [LiN(SiMe3)(C6F5)·Et2O]2([3-Li·Et2O]2). H atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Li1–F1 = 2.106(3), Li1–N1 = 2.061(3), Li1–N1#1 = 2.075(3), Li1–O1 = 1.910(3), C2–F1 = 1.3643(18), C6–F5 = 1.3541(19); N1–Li1–F1 = 81.81(11), N1#1–Li1–F1 = 108.69(12), N1–Li1–N1#1 = 103.33(12), N1–Li1–O1 = 128.96(14), N1#1–Li1–O1 = 125.20(14), O1–Li1–F1 = 95.29(12), Li1–N1–Li1#1 = 76.67(12). Symmetry operations: −x, −y, −z, T = [1, 1, 1]. Colour code: C, black; F, green; Li, orange; N, blue; O, red; Si, beige.](/document/doi/10.1515/mgmc-2023-0012/asset/graphic/j_mgmc-2023-0012_fig_004.jpg)
Representation of the molecular solid-state structure of the dimer [LiN(SiMe3)(C6F5)·Et2O]2([3-Li·Et2O]2). H atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Li1–F1 = 2.106(3), Li1–N1 = 2.061(3), Li1–N1#1 = 2.075(3), Li1–O1 = 1.910(3), C2–F1 = 1.3643(18), C6–F5 = 1.3541(19); N1–Li1–F1 = 81.81(11), N1#1–Li1–F1 = 108.69(12), N1–Li1–N1#1 = 103.33(12), N1–Li1–O1 = 128.96(14), N1#1–Li1–O1 = 125.20(14), O1–Li1–F1 = 95.29(12), Li1–N1–Li1#1 = 76.67(12). Symmetry operations: −x, −y, −z, T = [1, 1, 1]. Colour code: C, black; F, green; Li, orange; N, blue; O, red; Si, beige.
The molecular structure of the octamer [4-Li]8 is depicted in Figure 5. The crystals were obtained by recrystallisation from a concentrated toluene solution, in the hope that both C–F and Si–H motifs found in 4-Li would induce an unusual structural arrangement in the molecular solid state through the presence of Li⋯F or Li⋯H–Si non-covalent interactions (Roşca et al., 2016). The unit cell contains two identical molecules of [4-Li]8, one of which only is shown in Figure 5, and two non-interacting toluene molecules. The octamer adopts a unique crown-like arrangement. It features four inequivalent pairs of lithium ions and a crystallographic C 4-symmetry axis orthogonal through its centre to the ring delineated by the eight metal centres (yet, the structure was solved in the monoclinic P2/n space group of lower symmetry due to the presence of lattice toluene molecules). Each lithium is in a highly distorted tetrahedral environment, with two Li–N bonds and two very different Li⋯F interactions; there is no anagostic contact between the metal ions and the Si–H moieties. In fact, the SiMe2H groups are pushed away from the Li8 ring, with Li⋯H–Si distances well over 3 Å.
![Figure 5
(top) Representation of the molecular solid-state structure of [LiN(SiMe2H)(2-C6H4F)]8 ([4-Li]8). Non-interacting toluene molecules and H atoms except those of Si atoms omitted for clarity. Only one of the two independent and identical molecules in the unit cell is depicted. Selected bond lengths (Å): Li1–F50#1 = 2.044(6), Li1–F20 = 2.301(6), Li1–N13 = 1.981(6), Li1–N23 = 2.011(6), Li2–F20 = 1.975(5), Li2–F30 = 2.259(5), Li2–N23 = 1.984(6), Li2–N33 = 2.009(6), Li3–F30 = 2.022(5), Li3–F40 = 2.240(5), Li3–N33 = 1.965(5), Li3–N43 = 1.977(5), Li4–F40 = 2.044(5), Li4–F50 = 2.227(5), Li4–N13#1 = 2.021(6), Li4–N43 = 1.993(6), C19–F20 = 1.400(3), C29–F30 = 1.403(3), C39–F40 = 1.410(3), C49–F50 = 1.407(3). Symmetry operations: −x + 1/2, −y, −z + 1/2, T = [1, 0, 0]. (bottom) Top view of the crown-like molecular solid-state structure of [LiN(SiMe2H)(2-C6H4F)]8([4-Li]8). Non-interacting toluene molecules, aromatic N-bound rings, and H atoms except those of Si atoms omitted for clarity. Colour code: C, black; F, green; H, white; Li, orange; N, blue; Si, beige.](/document/doi/10.1515/mgmc-2023-0012/asset/graphic/j_mgmc-2023-0012_fig_005.jpg)
(top) Representation of the molecular solid-state structure of [LiN(SiMe2H)(2-C6H4F)]8 ([4-Li]8). Non-interacting toluene molecules and H atoms except those of Si atoms omitted for clarity. Only one of the two independent and identical molecules in the unit cell is depicted. Selected bond lengths (Å): Li1–F50#1 = 2.044(6), Li1–F20 = 2.301(6), Li1–N13 = 1.981(6), Li1–N23 = 2.011(6), Li2–F20 = 1.975(5), Li2–F30 = 2.259(5), Li2–N23 = 1.984(6), Li2–N33 = 2.009(6), Li3–F30 = 2.022(5), Li3–F40 = 2.240(5), Li3–N33 = 1.965(5), Li3–N43 = 1.977(5), Li4–F40 = 2.044(5), Li4–F50 = 2.227(5), Li4–N13#1 = 2.021(6), Li4–N43 = 1.993(6), C19–F20 = 1.400(3), C29–F30 = 1.403(3), C39–F40 = 1.410(3), C49–F50 = 1.407(3). Symmetry operations: −x + 1/2, −y, −z + 1/2, T = [1, 0, 0]. (bottom) Top view of the crown-like molecular solid-state structure of [LiN(SiMe2H)(2-C6H4F)]8([4-Li]8). Non-interacting toluene molecules, aromatic N-bound rings, and H atoms except those of Si atoms omitted for clarity. Colour code: C, black; F, green; H, white; Li, orange; N, blue; Si, beige.
The Li8 octagon is held together by bridging N- and F-atoms. The Li–N interatomic distances to the bridging N-atoms, in the range 1.965(5)–2.021(6), are all within the same narrow range. They are noticeably shorter than those measured in the polymeric [(2-Li)2]∞ (2.047(8)–2.068(9) Å) and in the dimer [3-Li·Et 2 O]2 (2.061(3)–2.075(3) Å). For each Li centre, one Li⋯F interaction is very much stronger than the other, as indicated by the largely different Li–F interatomic distances: Li1–F50#1 = 2.044(6) vs Li1–F20 = 2.301(6) Å, Li2–F20 = 1.975(5) vs Li2–F30 = 2.259(5) Å, Li3–F30 = 2.022(5) vs Li3–F40 = 2.240(5) Å, and Li4–F40 = 2.044(5) vs Li4–F50 = 2.227(5) Å. In fact, one may question whether the set of weak Li⋯F contacts are truly significant, even if they are much shorter than the sum of van der Waals radii for Li and F (1.82 and 1.47 Å, respectively); note however that, perhaps more relevantly, the tabulated ionic radius of the tetra-coordinated Li+ ion is 0.59 Å (Shannon, 1976). The shorter set of Li–F distances, in the range 1.975(5)–2.044(5) Å, compare well with those in the already discussed complexes [(2-Li)2]∞ (1.985(8)–2.107(9) Å) and C{SiMe2N(2-C6H4F)}3Sn(Li)] (1.972(8)–2.000(9) Å; Lutz et al., 2003). We note that the C–F bonds in [4-Li]8, with interatomic distances in the range 1.400(3)–1.410(3) Å, are slightly stretched compared to those in [(2-Li)2]∞ (1.389(5)–1.384(5) Å) and [3-Li·Et 2 O]2 (1.3541(19)–1.3643(18) Å). By comparison, the C–F bond lengths are also shorter (1.371(5) Å) in the titanium(III) triamidotriamine complex bearing the 1,4,7-{N(2-C6H4F)SiMe2}3-1,4,7-triazacyclononane ligand and where no Ti⋯F interaction was detected (Martins et al., 2005). Overall, we interpret the increase of the C–F bond lengths in crystalline [(2-Li)2]∞ as a consequence (and an evidence) of the Li⋯F interactions and resulting slight weakening of the pertaining C–F bonds. This hypothesis also agrees with the NMR data for 4-Li, which witnesses a very tangible reduction of the |1 J C,F| coupling constant on moving from 4-H (241.5 Hz) to the lithiated complex (217.3 Hz). Similar decreases were also observed for 2-Li (|1 J C,F| = 226.4 vs 238.5 Hz in 2-H) and 3-Li (|1 J C,F| = 221.3 vs 243.5 Hz in 3-H). Although there is no direct correlation between the C–F interatomic distances and corresponding values of the |1 J C,F| couplings, it seems legitimate to see both types of physical data only as valuable qualitative tools to evaluate the presence of Li⋯F interactions in the lithium fluoroarylsilylamides. On the other hand, variations of NMR chemical shifts from amine to amide are of little relevance, as these will mostly be affected by the developing negative charge on the nitrogen atom as a result of the deprotometallation of the amine. A more thorough interpretation of these data and structural analysis for the complexes would require in-depth analysis of NMR data in a non-coordinating solvent (including DOSY NMR analysis to probe whether the crystalline structures are retained in solution and 6/7Li NMR), but such studies were unfortunately prohibited by the poor solubility in the compounds in aromatic hydrocarbons and their instability in chlorinated solvents. A summary of the most salient properties for complexes 2-Li, 3-Li, and 4-Li is collated in Table 1.
Representative NMR and single-crystal XRD data for complexes 1-Li–4-Li
| 1-Li | 2-Li | 3-Li | 4-Li | ||
|---|---|---|---|---|---|
| [LiN(SiMe3)(2-F-C6H4)] | [LiN(SiMe3)(2,6-F2-C6H3)] | [LiN(SiMe3)(C6F5)] | [LiN(SiMe2H)(2-F-C6H4)] | ||
| NMR | |1 J C,F| (Hz) | ||||
| Parent amine | 236.5 | 238.5 | 243.5 | 241.5 | |
| Complex | 216.3 | 226.4 | 221.3 | 217.3 | |
| Δ(|1 J C,F|) (Hz) | 20.2 | 12.1 | 22.2 | 24.2 | |
| δ 7Li (ppm) | 1.27 | 1.20 | 0.90 | 1.18 | |
| XRD | Structure | n/a | [(2-Li)2]∞ | [3-Li·Et 2 O]2 | [4-Li]8 |
| d(Li-F) (Å) | n/a | 1.985(8)–2.107(9) | 2.106(3) | 1.975(5)–2.044(5) | |
| 2.227(5)–2.301(6) | |||||
| d(C-F) (Å) | n/a | 1.389(5)–1.384(5) | 1.354(2)–1.364(2) | 1.400(3)–1.410(3) |
3 Conclusion
A set of lithium fluoroarylsilylamides have been prepared in high yields upon deprotometallation of the parent amines with nBuLi in petroleum ether. The complexes have been comprehensively characterised by all available analytical tools, and detailed NMR data are provided. Three of the complexes, namely [LiN(SiMe3)(2,6-C6H3F2)] (2-Li), [LiN(SiMe3)(C6F5)] (3-Li), and [LiN(SiMe2H)(2-C6H4F)] (4-Li) were crystallised under different experimental conditions, and the molecular solid-state structures of the corresponding [(2-Li)2]∞, [3-Li·Et 2 O]2, and [4-Li]8 were solved. In all cases, medium to very strong Li⋯F interactions were detected. These secondary interactions are the key toward the construction of unusual arrangements, such as the coordination polymer [(2-Li)2]∞ or the octagonal [4-Li]8. The presence of Li⋯F interactions could be related to the elongation of the pertaining C–F bond along with a decrease of the corresponding |1 J C,F| coupling constants in the 13C{1H} NMR spectra of the complexes, although no direct relationship between d(Li–F), d(C–F), and Δ(|1 J C,F|) could be drawn. The results presented herein constitute another evidence that M⋯F interactions can be used to obtain unorthodox molecular structure for low-coordinate oxophilic elements, and we will endeavour to further exploit this feature in our ongoing research program.
Experimental section
General procedures
All manipulations were performed under an inert atmosphere by using standard Schlenk techniques or in a dry, solvent‐free glovebox (Jacomex; O2 <1 ppm, H2O <3 ppm). Petroleum ether, toluene, and diethyl ether were collected from MBraun SPS‐800 purification alumina columns and thoroughly degassed with argon before being stored on 4 Å molecular sieves; thf was distilled under argon from Na/benzophenone prior to use. Deuterated solvents (Eurisotop, Saclay, France) were stored in sealed ampoules over activated 4 Å molecular sieves or a potassium mirror and degassed by a minimum of three freeze–thaw cycles. NMR spectra were recorded with Bruker AM-300, AM‐400, or AM‐500 spectrometers. All chemical shifts (δ) [ppm] were determined relative to the residual signal of the deuterated solvent in benzene-d 6 or thf-d 8 or to FCCl3 for 19F NMR. Assignments of signals were carried out using 1D and 2D NMR experiments. Reliable and reproducible elemental analyses for the compounds could not be obtained, most probably on account of their high hydrolytic sensitivity.
Synthesis of N-(2-fluorophenyl)-trimethylsilylamine (1-H)
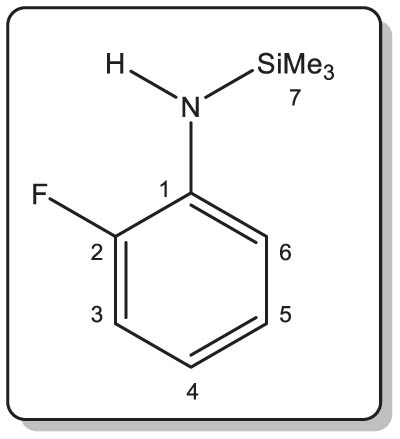
The synthesis of 1 was adapted from a known protocol (Yin et al., 2016). In a round-bottom flask, nBuLi (18.6 mL of a 1.57 M solution in hexanes, 29.2 mmol) was added dropwise (ca. 30 min) at –78°C to a solution of o-F-C6H4-NH2 (2.8 mL, 3.2 g, 28.8 mmol) in diethyl ether (50 mL). The clear and colourless solution was then allowed to warm to room temperature. A solution of Me3SiCl (3.8 mL, 3.2 g, 29.9 mmol) in diethyl ether (50 mL) was added in two portions. The resulting slurry was stirred overnight at room temperature. The precipitate was then eliminated by filtration. The volatiles were pumped off under dynamic vacuum to give the title compound as an analytically pure colourless liquid. Isolated yield: 4.9 g (93%). MS ASAP for [M + H]+, calcd m/z 184.09523. Found 184.0950 (1 ppm).
1H NMR (400.16 MHz, 27°C, benzene-d 6): δ 6.93–6.88 (ddd, |1 J H,H| = 12.0 Hz, |2 J H,H| = 8.0 Hz, |3 J H,H| = 1.6, 1H, C3 H), 6.83–6.79 (m, 1H, C5 H), 6.76–6.72 (m, 1H, C6 H), 6.53–6.47 (m, 1H, C4 H), 3.59 (br s, 1H, NH), 0.07 (s, 9H, CH 3) ppm.
13C{1H} NMR (100.63 MHz, 27°C, benzene-d 6): δ 154.4–152.1 (d, |1 J C,F| = 236.5 Hz, C 2), 136.2–136.1 (d, |2 J C,F| = 12.1 Hz, C 1), 124.7 (d, |4 J C,F| = 3.0 Hz, C 5), 117.8 (d, |3 J C,F| = 7.0 Hz, C 4), 116.9 (d, |3 J C,F| = 3.0 Hz, C 6), 115.5–115.3 (d, |2 J C,F| = 19.1 Hz, C 3), –0.2 (t, |1 J Si,C| = 57.3 Hz, CH3) ppm.
19F NMR (376.47 MHz, 25°C, benzene-d 6): δ –135.1 (s, 1F) ppm.
1H NMR (400.16 MHz, 25°C, thf-d 8): δ 6.93–6.80 (m, 3H, arom-CH), 6.59–6.54 (m, 1H, arom-CH), 6.53–6.48 (m, 1H, arom-CH), 4.21 (br s, 1H, NH), 0.26 (s, 9H, CH 3) ppm.
19F NMR (376.47 MHz, 25°C, thf-d 8); δ –135.2 (s, 1F) ppm.
Synthesis of N-(2,6-difluorophenyl)-trimethylsilylamine (2-H)
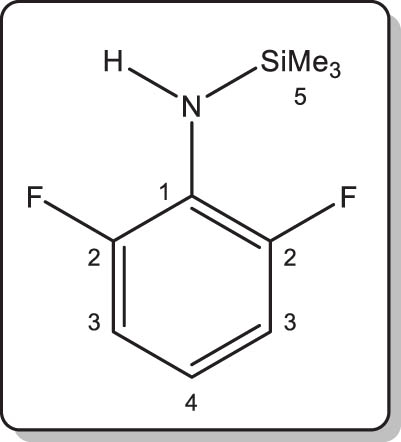
In a protocol identical to that described for 1, the title compound was obtained by reaction of 2,6-difluoroaniline (2.2 mL, 2.6 g, 20 mmol), nBuLi (12.6 mL of a 1.57 M solution in hexanes, 19.8 mmol), and Me3SiCl (2.6 mL, 2.2 g, 20.0 mmol). Compound 2 was isolated as a colourless liquid. Isolated yield: 3.2 g (79%). MS ASAP for [M + H]+, calcd m/z 202.08581. Found 202.0856 (1 ppm).
1H NMR (400.13 MHz, 24°C, benzene-d 6): δ 6.62–6.58 (m, 2H, C3 H), 6.23–6.18 (m, 1H, C4 H), 3.44 (br s, 1H, NH), 0.15 (t, |5 J H,F| = 1.4 Hz, 9H, CH 3) ppm.
13C{1H} NMR (100.62 MHz, 26°C, benzene-d 6): δ 154.8–152.4 (dd, |1 J C,F| = 238.5 Hz, |3 J C,F| = 8.0 Hz, C 2), 116.4 (t, |3 J C,F| = 9.0 Hz, C 4), 111.6–111.2 (dd, |2 J C,F| = 27.2 Hz, |4 J C,F| = 4.0 Hz, C 3), 111.5 (m, C 1), 0.07 (t, |1 J Si,C| = 58.4 Hz, |5 J C,F| = 3.0 Hz, CH 3) ppm.
19F NMR (282.36 MHz, 25°C, benzene-d 6): δ –128.5 (s, 2F) ppm.
1H NMR (400.16 MHz, 27°C, thf-d 8): δ 6.84–6.77 (m, 2H, C3 H), 6.60–6.53 (m, 1H, C4 H), 4.01 (br s, 1H, NH), 0.24 (s, 9H, CH 3) ppm.
13C{1H} NMR (100.63 MHz, 27°C, thf-d 8): δ 155.3–152.9 (dd, |1 J C,F| = 238.5 Hz, |3 J C,F| = 8.2 Hz, C 2), 125.9 (t, |2 J C,F| = 16.3 Hz, C 1), 116.7 (t, |3 J C,F| = 9.4 Hz, C 4), 111.8–111.4 (m, C 3), 0.70 (t, |1 J Si,C| = 58.3 Hz, |5 J C,F| = 3.4 Hz, CH 3) ppm.
19F NMR (376.47 MHz, 27°C, thf-d 8): δ –128.8 (s, 2F) ppm.
Synthesis of N-(pentafluorophenyl)-trimethylsilylamine (3-H)
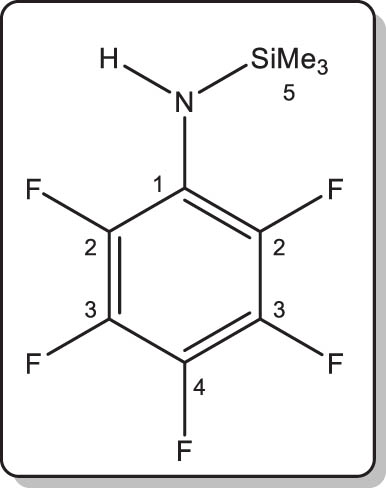
In a protocol identical to that described for 1, the title compound was obtained by reaction of pentafluoroaniline (3.0 g, 16.5 mmol), nBuLi (10.5 mL of a 1.57 M solution in hexanes, 16.5 mmol), and Me3SiCl (2.1 mL, 1.8 g, 16.5 mmol). Compound 3 was isolated as a colourless liquid. Isolated yield: 3.5 g (83%). Anal. found: C, 42.4; H, 4.1; N, 5.8. Calcd for C9H10F5NSi (255.26 g·mol–1): C, 42.35; H, 3.95; N, 5.49. MS ASAP for [M + H]+, calcd m/z 256.05754. Found 256.0573 (1 ppm).
1H NMR (400.16 MHz, 27°C, benzene-d 6): δ 3.04 (br s, 1H, NH), 0.05 (t, |5 J H,F| = 1.4 Hz, 9H, CH 3) ppm.
13C{1H} NMR (100.62 MHz, 26°C, benzene-d 6): δ 139.9–139.4 and 137.5–137.2 (overlapping dm, C 2, and C 3) 134.8–132.0 (dtt, |1 J C,F| = 243.5 Hz, |2 J C,F| = 14.1 Hz, |3 J C,F| = 5.0 Hz, C 4), 123.5–123.2 (t, |2 J C,F| = 13.1 Hz, C 1), 0.30 (t, |1 J Si,C| = 58.4 Hz, |5 J C,F| = 3.1 Hz, CH3) ppm.
19F NMR (376.47 MHz, 25°C, benzene-d 6): δ –159.0 (d, |3 J F,F| = 18.8 Hz, 2F, o-F), –165.4 (m, |3 J F,F| = 18.8 Hz, 2F, m-F), –173.7 (m, |3 J F,F| = 18.8 Hz, 1F, p-F) ppm.
N-(2-fluorophenyl)-1,1-dimethylsilylamine (4-H)
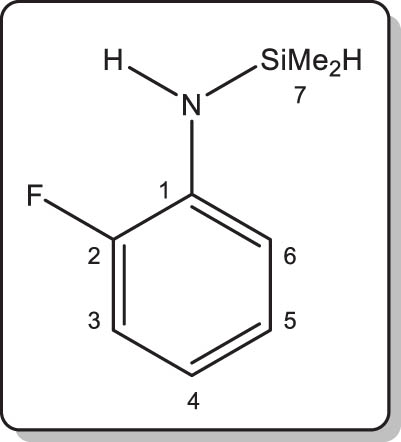
A solution of 2-fluoroaniline (4.4 mL, 5.0 g, 45.4 mmol) in diethyl ether (100 mL) was cooled down to 0°C, and nBuLi (29.3 mL of a 1.55 M solution in hexanes, 45.4 mmol) was then added dropwise over 30 min. The reaction mixture was then left to gently warm up to room temperature for 1 h. A solution of chlorodimethylsilane (5.1 mL, 4.3 g, 45.8 mmol) in Et2O (70 mL) was then added dropwise, and the milky suspension was stirred at room temperature for 16 h. The solution was then filtered to eliminate the precipitate, and the volatiles were removed under vacuum. The amine was isolated as analytically pure colourless oil by trap-to-trap transfer. Isolated yield: 6.7 g (87%). MS ASAP for [M + H]+, calcd m/z 170.07958. Found 170.0795 (0 ppm).
1H NMR (400.16 MHz, 27°C, benzene-d 6): δ 6.93–6.83 (m, 1H, C3 H), 6.81–6.78 (m, 2H, C5 H, and C6 H), 6.53–6.47 (m, 1H, C4 H), 4.77 (m, with |1 J Si,H| = 204.0 Hz and |3 J H,H| = 1.6 Hz, 1H, SiH), 3.51 (1H, NH), 0.02 (d, |3 J H,H| = 3.2 Hz, 6H, CH 3) ppm.
13C{1H} NMR (100.62 MHz, 26°C, benzene-d 6): δ 154.4–152.0 (d, |1 J C,F| = 241.5 Hz, C 2), 135.9 (d, |2 J C,F| = 13.1 Hz, C 1), 124.9 (d, |4 J C,F| = 4.0 Hz, C 5), 118.3 (d, |3 J C,F| = 7.0 Hz, C 4), 116.7 (d, |3 J C,F| = 4.0 Hz, C 6), 115.4–115.2 (d, |2 J C,F| = 20.1 Hz, C 3), –2.4 (s, |1 J Si,C| = 57.3 Hz, CH3) ppm.
19F NMR (376.44 MHz, 24°C, benzene-d 6): δ −135.4 (s, 1F) ppm. The presence of an impurity (ca. 1 mol%) was detected at –136.0 ppm.
Synthesis of lithium N-(2-fluorophenyl)-trimethylsilylamide (1-Li)
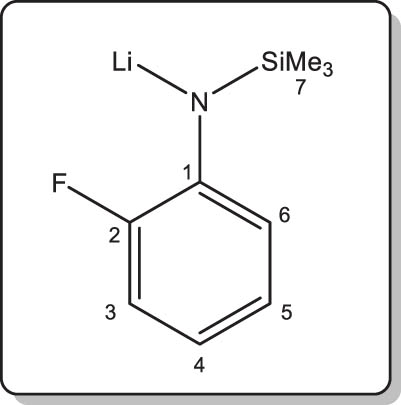
At –78°C, nBuLi (3.5 mL of a 1.57 M solution in hexanes, 5.5 mmol) was added dropwise (ca. 30 min) to a solution of 1-H (1.0 g, 5.5 mmol) in petroleum ether (50 mL). The mixture was allowed to warm to room temperature and was stirred for 4 h, leading to the formation of a white precipitate. The supernatant was removed by filtration, and the title complex was obtained as a white powder after drying the precipitate to constant weight under vacuum. Isolated yield: 0.8 g (77%).
1H NMR (500.13 MHz, 27°C, thf-d 8): δ 6.63–6.50 (overlapping m, 3H, arom-H), 5.86–5.82 (m, 1H, arom-H), 0.04 (s, 9H, CH 3) ppm.
13C{1H} NMR (125.77 MHz, 27°C, thf-d 8): δ 160.6–158.9 (d, |1 J C,F| = 216.3 Hz, C 2), 150.8 (d, |2 J C,F| = 10.1 Hz Hz, C 1), 125.3 (d, |4 J C,F| = 2.5 Hz, C 5), 121.0 ppm (d, |3 J C,F| = 6.3 Hz, C 4), 112.8 (d, |2 J C,F| = 23.4 Hz, C 3), 107.0 (d, |3 J C,F| = 8.8 Hz, C 6), 1.8 (s, |1 J Si,C| = 53.6 Hz, CH3) ppm.
19F NMR (470.52 MHz, 27°C, thf-d 8): δ –145.2 (s, 1F) ppm.
7Li NMR (thf-d 8, 194.37 MHz, 27°C): δ 1.27 (s) ppm.
Synthesis of lithium N-(2,6-difluorophenyl)-trimethylsilylamide (2-Li)
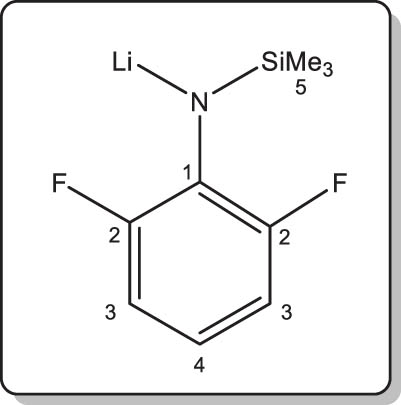
Following the same protocol as that described for 1-Li, amine 2-H (1.0 g, 5.0 mmol) was reacted with nBuLi (3.2 mL of a 1.57 M solution in hexanes, 5.0 mmol) to afford the title compound as a colourless solid. Isolated yield: 0.9 g (90%). X-ray quality crystals of [2-Li]∞ were grown from a concentrated solution in petroleum ether stored at –30°C.
1H NMR (500.13 MHz, 27°C, thf-d 8): δ 6.51–6.47 (overlapping m, 2H, C3 H), 5.79–5.73 (m, 1H, C4 H), 0.02 (s, 9H, CH 3) ppm.
13C{1H} NMR (125.77 MHz, 27°C, thf-d 8): δ 159.9–158.0 (dd, |1 J C,F| = 226.4 Hz, |3 J C,F| = 13.8 Hz, C 2), 139.1 (t, |2 J C,F| = 17.6 Hz, C 1), 110.3 (m, C 3), 104.2 (t, |3 J C,F| = 10.7 Hz, C 4), 3.3 (t, |5 J C,F| = 3.8 Hz, CH3) ppm.
19F NMR (470.52 MHz, 27°C, thf-d 8): δ –135.0 (s, 2F) ppm.
7Li NMR (thf-d 8, 194.37 MHz, 27 °C): δ 1.20 (s) ppm.
Synthesis of lithium N-(pentafluorophenyl)-trimethylsilylamide (3-Li)
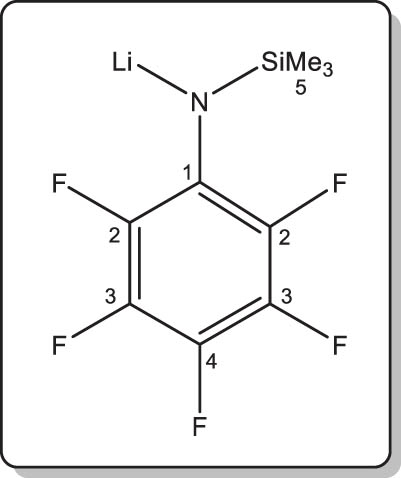
In a similar procedure to that described for 1-Li, the title compound was isolated as a colourless solid by reaction of 3-H (1.0 g, 3.9 mmol) and nBuLi (2.5 mL of a 1.57 M solution in hexanes, 3.9 mmol). Isolated yield: 0.85 g (81%). The NMR data repeatedly showed the presence (ca. 4 mol%) of an unknown unidentified side-product, possibly (C6F5)SiMe3 or (Me3Si)(C6F5)N-N(C6F5)(SiMe3). Single crystals of [3-Li·Et 2 O] suitable for XRD analysis were grown from a solution in Et2O.
1H NMR (500.13 MHz, 27°C, thf-d 8): δ 0.04 (s, 9H, CH 3) ppm. The presence of an unknown product was detected (v. small singlet at 0.06 ppm).
13C{1H} NMR (125.77 MHz, 27°C, thf-d 8): δ 143.7–142.0 (dt, |1 J C,F| = 221.3 Hz, |2 J C,F| = 9.5 Hz, C 4), 140.3–138.4 (dm, |1 J C,F| = 247.7 Hz, C 2 or C 3), 137.3 (t, |2 J C,F| = 14.8 Hz, C 1), 128.1–126.2 (dm, |1 J C,F| = 229.9 Hz, C 2 or C 3), 2.8 (t, |1 J Si,C| = 55.3 Hz, |5 J C,F| = 3.1 Hz, CH3) ppm. The presence of an unknown product was detected by a singlet resonance at δ –0.7 ppm.
19F NMR (470.52 MHz, 27°C, thf-d 8): δ –163.7 (br s, 2F, o-F), –172.0 (t, |3 J F,F| = 21.2 Hz, 2F, m-F), –192.5 (br s, 1F, p-F) ppm. The resonances for an unidentified product were detected at δ –147.9, –163.7, and –166.2 ppm.
7Li NMR (thf-d 8, 155.51 MHz, 25°C): δ 0.90 ppm.
Synthesis of lithium N-(2-fluorophenyl)-1,1-dimethylsilylamide (4-Li)
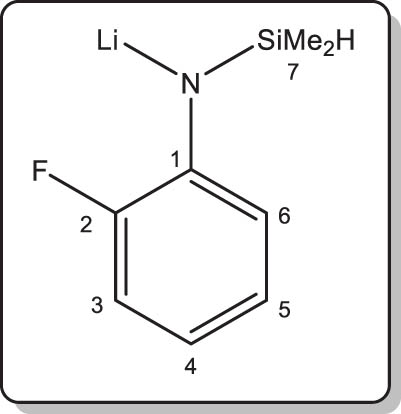
Compound 4-H (0.4 g, 2.4 mmol) was diluted in petroleum ether (15 mL). nBuLi (1.5 mL of a 1.57 M solution in hexanes, 2.4 mmol) was added dropwise at 0°C. The reaction mixture was stirred for 1 h at room temperature; formation of a white precipitate was observed. The solid was isolated by filtration, and dried to constant weight under vacuum. We found that the various samples we prepared always contained residual petroleum ether that could not be easily removed under vacuum. A crop of colourless X-ray quality single crystals of [4-Li]8 was obtained by recrystallisation from a saturated toluene solution. Isolated yield: 0.3 g (71%).
1H NMR (400.13 MHz, 25°C, thf-d 8): δ 6.68–6.56 (m, 3H, C3 H + C4 H + C5 H), 5.92 (m, 1H, C6 H), δ 4.84 (hept, 1H, |3 J H,H| = 2.9 Hz, |1 J Si,H| = 176.0 Hz, SiH), 0.08 (d, |3 J H,H| = 2.9 Hz, 6H, CH 3) ppm.
13C{1H} NMR (100.62 MHz, 26°C, thf-d 8): δ 160.6–158.4 (d, |1 J C,F| = 217.3 Hz, C 2), 150.7 (d, |2 J C,F| = 10.1 Hz, C 1), 125.3 (d, |4 J C,F| = 2.0 Hz, C 5), 120.6 (d, |3 J C,F| = 4.0 Hz, C 4), 112.8 (d, |2 J C,F| = 23.1 Hz, C 3), 108.0 (d, |3 J C,F| = 9.0 Hz, C 6), 0.24 (s, |1 J Si,C| = 54.3 Hz, CH3) ppm.
19F NMR (376.44 MHz, 24°C, thf-d 8): δ −145.2 (s) ppm. Gradual decomposition in solution was visible by a resonance of growing intensity at –140.8 ppm.
7Li NMR (155.51 MHz, 24°C, thf-d 8): δ 1.18 (s) ppm.
X-ray diffraction crystallography
For [2-Li] ∞ (C9H12F2LiNSi, M = 207.23 g·mol–1), CCDC 2238385. A suitable crystal for X-ray diffraction single crystal experiment (colourless stick, dimensions = 0.300 mm × 0.120 mm × 0.110 mm) was selected and mounted on the goniometer head of a APEXII Kappa-CCD (Bruker-AXS) diffractometer equipped with a CCD plate detector, using Mo-Kα radiation (λ = 0.71073 Å, graphite monochromator) at T = 150(2) K. The crystal structure has been described in monoclinic symmetry and P21/n (I.T.#14) centric space group. Cell parameters have been refined as follows: a = 10.760(2), b = 6.5810(16), c = 14.522(3) Å, β = 91.476(11)°, V = 1,028.0(4) Å3. Number of formula unit Z is equal to 4 and calculated density d and absorption coefficient µ values are 1.339 g·cm–3 and 0.212 mm–1, respectively. The crystal structure was solved by dual-space algorithm using SHELXT (Sheldrick, 2015a), and then refined with full-matrix least-squares methods based on F2 (SHELXL; Sheldrick, 2015b). All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. A final refinement on F 2 with 2,289 unique intensities and 130 parameters converged at ωR F2 = 0.1968 (R F = 0.0834) for 1,344 observed reflections with I > 2σ(I).
For [3-Li·Et 2 O] 2 (C26H38F10Li2N2O2Si2, M = 670.64 g·mol–1), CCDC 2238384. A suitable crystal for X-ray diffraction single crystal experiment (colourless prism, dimensions = 0.600 mm × 0.490 mm × 0.250 mm) was selected and mounted on the goniometer head of a APEXII Kappa-CCD (Bruker-AXS) diffractometer equipped with a CCD plate detector, using Mo-Kα radiation (λ = 0.71073 Å, graphite monochromator) at T = 150(2) K. The crystal structure has been described in triclinic symmetry and P–1 (I.T.#2) centric space group. Cell parameters have been refined as follows: a = 9.0873(7), b = 10.5228(8), c = 10.6719(9) Å, α = 116.451(2), β = 99.726(3), γ = 106.944(2)°, V = 819.10(11) Å3. Number of formula unit Z is equal to 1 and calculated density d and absorption coefficient µ values are 1.360 g·cm–3 and 0.191 mm–1, respectively. The crystal structure was solved by dual-space algorithm using SHELXT (Sheldrick, 2015a), and then refined with full-matrix least-squares methods based on F2 (SHELXL; Sheldrick, 2015b). All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. A final refinement on F2 with 3,713 unique intensities and 204 parameters converged at ωR F2 = 0.1094 (R F = 0.0389) for 3,151 observed reflections with I > 2σ(I).
For [4-Li] 8 (C64H88F8Li8N8Si8·C7H8, M = 1,493.79 g·mol–1), CCDC 2238386. D8 VENTURE Bruker AXS diffractometer equipped with a (CMOS) PHOTON 100 detector, Mo-Kα radiation (λ = 0.71073 Å, multilayer monochromator), T = 150 K; monoclinic P2/n (I.T.#13), a = 16.620(3), b = 12.903(2), c = 19.799(4) Å, β = 89.952(7)°, V = 4,245.6(13) Å3. Z = 2, d = 1.168 g·cm–3, μ = 0.186 mm–1. The structure was solved by dual-space algorithm using the SHELXT program (Sheldrick, 2015a), and then refined with full-matrix least-squares methods based on F2 (SHELXL; Sheldrick, 2015b). All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Except hydrogen atoms linked to silicon atoms that were introduced in the structural model through Fourier difference maps analysis, H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. A final refinement on F2 with 9,756 unique intensities and 479 parameters converged at ωR F2 = 0.1227 (R F = 0.0527) for 8,734 observed reflections with I > 2σ(I).
Acknowledgements
The authors thank the University of Rennes and the CNRS for funding. We thank Dr Elsa Caytan (Institut des Sceinces Chimiques de Rennes, France) for her assistance with 2D 19F–13C HMBC NMR data.
-
Funding information: The research was funded by the University of Rennes and the CNRS.
-
Author contributions: Sakshi Mohan: experimental work; Yahya Al Ayi: experimental work; Savarithai Jenani Louis Anandaraj: experimental work; Marie Cordier: X-ray diffraction analysis; Thierry Roisnel: X-ray diffraction analysis; Jean-François Carpentier: writing completion, fund securing; Yann Sarazin: methodology, writing, and fund securing.
-
Conflict of interest: The corresponding author (Yann Sarazin) is a member of the Editorial Board of Main Group Metal Chemistry.
-
Supplementary material: Supplementary material is available for this publication.
-
Data availability statement: Crystallographic data (CCDC 2238384-2238386) of this article can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Aharonovich S., Botoshanski M., Eisen M.S., Synthesis of lithium N-pentafluorophenyl, N’-trimethylsilyl 2-pyridylamidinate and its cyclization to lithium tetrafluoro-2-(2-pyridyl)benzimidazolate via a Me3SiF elimination. Coordination chemistry, reactivity, and mechanism. Inorg. Chem., 2009, 48, 5269–5278.10.1021/ic9002065Search in Google Scholar PubMed
Andreev Y.G., Seneviratne V., Khan M., Henderson W.A., Frech R.E., Bruce P.G., Crystal structures of poly(ethylene oxide)3:LiBF4 and (diglyme)n:LiBF4 (n = 1, 2). Chem. Mater., 2005, 17, 767–772.10.1021/cm048310uSearch in Google Scholar
Barozzino-Consiglio G., Hamdoun G., Fressigné C., Harrison-Marchand A., Maddaluno J., Oulyadi H., A combined 1H/6Li NMR DOSY strategy finally uncovers the structure of isopropyllithium in THF. Chem. Eur. J., 2017, 23, 12475–12479.10.1002/chem.201702990Search in Google Scholar PubMed
Brookhart M., Green M.L.H., Parkin, G. Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914.10.1073/pnas.0610747104Search in Google Scholar PubMed PubMed Central
Buchanan W.D., Allis D.G., Ruhlandt-Senge K., Synthesis and stabilization-advances in organoalkaline earth metal chemistry. Chem. Commun., 2010, 46, 4449–4465.10.1039/c002600jSearch in Google Scholar PubMed
Chapple P.M., Sarazin Y., Contemporary molecular barium chemistry. Eur. J. Inorg. Chem., 2020, 3321–3346.10.1002/ejic.202000501Search in Google Scholar
Dougherty D.A., The cation–π interaction. Acc. Chem. Res., 2013, 46, 885–893.10.1021/ar300265ySearch in Google Scholar PubMed PubMed Central
Fischer C.A., Rösch A., Elsen H., Ballmann G., Wiesinger M., Langer J., et al. Lewis acidic alkaline earth metal complexes with a perfluorinated diphenylamide ligand. Dalton Trans., 2019, 48, 6757–6766.10.1039/C9DT01272ASearch in Google Scholar PubMed
Fujiwara T., O’Hagan D., Successful fluorine-containing herbicide agrochemicals. J. Fluorine Chem., 2014, 167, 16–29.10.1016/j.jfluchem.2014.06.014Search in Google Scholar
Harder S., Early main group metal catalysis. Concepts and Reactions. Wiley-VCH, Weinheim, 2020.10.1002/9783527818020Search in Google Scholar
Hédouin M., Barthelemy A.L., Vanthuyne N., Besrour H., Maddaluno J., Magnier E., et al., NMR and DFT studies with a doubly labelled 15N/6Li S-trifluoromethyl sulfoximine reveal why a directed ortho-lithiation requires an excess of n-BuLi. Angew. Chem. Int. Ed., 2023, 62, e202214106.10.1002/anie.202214106Search in Google Scholar PubMed PubMed Central
Hevia E., Uzelac M., Borys A.M., Organometallic complexes of the alkali metals. In comprehensive organometallic chemistry IV (Eds-in-Chief: Parkin G., Meyer K., O’Hare D.). Elsevier, Kidlington, 2022, vol. 2, pp. 5–70.10.1016/B978-0-12-820206-7.00160-8Search in Google Scholar
Kuprat M., Lehmann M., Schulz A., Villinger A., Synthesis of pentafluorophenyl silver by means of Lewis acid catalysis: structure of silver solvent complexes. Organometallics, 2010, 29, 1421–1427.10.1021/om901063aSearch in Google Scholar
Lappert M.F., Slade J.S., Singh A., Atwood J.L., Rogers R.D., Shakir R., Structure and reactivity of sterically hindered lithium amides and their diethyl etherates: crystal and molecular structures of [Li|N(SiMe3)2}(OEt2)]2 and [Li(NCMe2CH2CH2CH2CMe2)]4. J. Am. Chem. Soc., 1983, 105, 302–304.10.1021/ja00340a031Search in Google Scholar
Le Coz E., Dorcet V., Roisnel T., Tobisch S., Carpentier J.-F., Sarazin Y., Low-coordinate barium boryloxides: synthesis and dehydrocoupling catalysis for the production of borasiloxanes. Angew. Chem. Int. Ed., 2018, 57, 11747–11751.10.1002/anie.201807297Search in Google Scholar PubMed
Liptrot D.J., Power P.P., London dispersion forces in sterically crowded inorganic and organometallic molecules. Nat. Rev. Chem., 2017, 1, 1–12.10.1038/s41570-016-0004Search in Google Scholar
Luisi R., Capriati V., Lithium complexes in organic synthesis: from fundamentals to applications. Wiley‐VCH Verlag GmbH & Co. KGaA, 2014.10.1002/9783527667512Search in Google Scholar
Luisi R., Degennaro L., Colella M., Lithium complexes in organic synthesis. In Comprehensive organometallic chemistry IV. In: Parkin G., Meyer K., O’Hare D. (Eds.). Elsevier, Kidlington, 2022, vol. 11, pp. 2–56.10.1016/B978-0-12-820206-7.00049-4Search in Google Scholar
Lutz M., Haukka M., Pakkanen T.A., McPartlin M., Gade L.H., Lithium cation solvation in the ligand periphery of an ortho-fluorophenyl substituted tripodal triaminostannate. Inorg. Chim. Acta, 2003, 345, 185–189.10.1016/S0020-1693(02)01299-9Search in Google Scholar
Martins A.M., Ascenso J.R., de Azevedo C.G., Dias A.R., Duarte M.T., Ferreira H., et al. Titanium triamidotriamine compounds: syntheses, structures and redox properties. Eur. J. Inorg. Chem., 2005, 1689–1697.10.1002/ejic.200400813Search in Google Scholar
Meanwell N.A., Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem., 2018, 61, 5822–5880.10.1021/acs.jmedchem.7b01788Search in Google Scholar PubMed
Oliver A.J., Graham W.A.G., Preparation and properties of some pentafluorophenyl group IV compounds. J. Organomet. Chem., 1969, 19, 17–27.10.1016/S0022-328X(00)87750-5Search in Google Scholar
Purser S., Moore P.R., Swallow S., Gouverneur V., Fluorine in medicinal chemistry. Chem. Soc. Rev., 2008, 37, 320–330.10.1039/B610213CSearch in Google Scholar PubMed
Rösch B., Gentner T.X., Langer J., Färber C., Eyselein J., Zhao L., et al., Dinitrogen complexation and reduction at low-valent calcium. Science., 2021, 371, 1125–1128.10.1126/science.abf2374Search in Google Scholar PubMed
Rösch B., Harder S., New horizons in low oxidation state group 2 metal chemistry. Chem. Commun., 2021, 57, 9354–9365.10.1039/D1CC04147ASearch in Google Scholar PubMed
Roşca S.-C., Roisnel T., Dorcet V., Carpentier J.-F., Sarazin Y., Potassium and well-defined neutral and cationic calcium fluoroalkoxide complexes: structural features and reactivity. Organometallics, 2014, 33, 5630–5642.10.1021/om500343wSearch in Google Scholar
Roşca S.-C., Dinoi C., Caytan E., Dorcet V., Etienne M., Carpentier J.-F., et al., Alkaline earth-olefin complexes with secondary interactions. Chem. Eur. J., 2016, 22, 6505–6509.10.1002/chem.201601096Search in Google Scholar PubMed
Roueindeji H., Ratsifitahina A., Roisnel T., Dorcet V., Kahlal S., Saillard J.-Y., et al., Metal·F-C bonding in low-coordinate alkaline earth fluoroarylamides. Chem. Eur. J., 2019, 25, 8854–8864.10.1002/chem.201901262Search in Google Scholar PubMed
Salonen L.M., Ellermann M., Diederich F., Aromatic rings in chemical and biological recognition: energetics and structures. Angew. Chem. Int. Ed., 2011, 50, 4808–4842.10.1002/anie.201007560Search in Google Scholar PubMed
Sarazin Y., Liu B., Roisnel T., Maron L., Carpentier J.-F., Discrete, solvent-free alkaline-earth metal cations: metal⋯fluorine interactions and ROP catalytic activity. J. Am. Chem. Soc., 2011, 133, 9069–9087.10.1021/ja2024977Search in Google Scholar PubMed
Schorpp M., Heizmann T., Schmucker M., Rein S., Weber S., Krossing I., Synthesis and application of a perfluorinated ammoniumyl radical cation as a very strong deelectronator. Angew. Chem. Int. Ed., 2020, 59, 9453–9459.10.1002/anie.202002768Search in Google Scholar PubMed PubMed Central
Shannon, R.D., Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst., 1976, A32, 751–767.10.1107/S0567739476001551Search in Google Scholar
Sheldrick, G.M., SHELXT-integrated space-group and crystal-structure determination. Acta Cryst., 2015a, A71, 3–8.10.1107/S2053273314026370Search in Google Scholar PubMed PubMed Central
Sheldrick G.M., Crystal structure refinement with SHELXL. Acta Cryst., 2015b, C71, 3–8.10.1107/S2053229614024218Search in Google Scholar PubMed PubMed Central
Skeel B.A., Boreen M.A., Lohrey T.D., Arnold, J., Perturbation of 1JC,F coupling in carbon-fluorine bonds on coordination to Lewis acids: a structural, spectroscopic, and computational study. Inorg. Chem., 2020, 59, 17259–17267.10.1021/acs.inorgchem.0c02516Search in Google Scholar PubMed
Wakefield, B.H., Organolithium methods. Academic Press Limited, London, 1988.Search in Google Scholar
Williard P.G., Liu Q.-Y., Complexes between fluorobenzenes and lithium hexamethyldisilazide dimer. J. Org. Chem., 1994, 59, 1596–1597.10.1021/jo00086a002Search in Google Scholar
Wilson A.S.S., Hill M.S., Mahon M.F., Dinoi C., Maron L., Organocalcium-mediated nucleophilic alkylation of benzene. Science, 2017, 358, 1168–1171.10.1126/science.aao5923Search in Google Scholar PubMed
Yang L., Powell D.R., Houser, R.P., Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans., 2007, 955–964.10.1039/B617136BSearch in Google Scholar PubMed
Yin H., Carroll P.J., Schelter E.J., Cerium(III) and uranium(IV) complexes of the 2‑fluorophenyl trimethylsilyl amide ligand: C-F → Ln/An interactions that modulate the coordination spheres of f‑block elements. Inorg. Chem., 2016, 55, 5684–5692.10.1021/acs.inorgchem.6b00785Search in Google Scholar PubMed
© 2023 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Research Articles
- Two new zinc(ii) coordination complexes constructed by phenanthroline derivate: Synthesis and structure
- Lithium fluoroarylsilylamides and their structural features
- On computation of neighbourhood degree sum-based topological indices for zinc-based metal–organic frameworks
- Two novel lead(ii) coordination complexes incorporating phenanthroline derivate: Synthesis and characterization
- Thermodynamics of transamination reactions with aminotrimethylsilanes and diaminodimethylsilanes
- From synthesis to biological impact of palladium bis(benzimidazol-2-ylidene) complexes: Preparation, characterization, and antimicrobial and scavenging activity
- Novel ruthenium(ii) N-heterocyclic carbene complexes: Synthesis, characterization, and evaluation of their biological activities
- Entropy measures of the metal–organic network via topological descriptors
- Rapid Communication
- Synthesis and crystal structure of an ionic phenyltin(iv) complex of N-salicylidene-valine
- Special Issue: Theoretical and computational aspects of graph-theoretic methods in modern-day chemistry (Guest Editors: Muhammad Imran and Muhammad Javaid)
- Topological indices for random spider trees
- On distance-based indices of regular dendrimers using automorphism group action
- Retraction
- Retraction to “Aluminium(iii), Fe(ii) Complexes and Dyeing Properties of Apigenin(5,7,4′-trihydroxy flavone)”
Articles in the same Issue
- Research Articles
- Two new zinc(ii) coordination complexes constructed by phenanthroline derivate: Synthesis and structure
- Lithium fluoroarylsilylamides and their structural features
- On computation of neighbourhood degree sum-based topological indices for zinc-based metal–organic frameworks
- Two novel lead(ii) coordination complexes incorporating phenanthroline derivate: Synthesis and characterization
- Thermodynamics of transamination reactions with aminotrimethylsilanes and diaminodimethylsilanes
- From synthesis to biological impact of palladium bis(benzimidazol-2-ylidene) complexes: Preparation, characterization, and antimicrobial and scavenging activity
- Novel ruthenium(ii) N-heterocyclic carbene complexes: Synthesis, characterization, and evaluation of their biological activities
- Entropy measures of the metal–organic network via topological descriptors
- Rapid Communication
- Synthesis and crystal structure of an ionic phenyltin(iv) complex of N-salicylidene-valine
- Special Issue: Theoretical and computational aspects of graph-theoretic methods in modern-day chemistry (Guest Editors: Muhammad Imran and Muhammad Javaid)
- Topological indices for random spider trees
- On distance-based indices of regular dendrimers using automorphism group action
- Retraction
- Retraction to “Aluminium(iii), Fe(ii) Complexes and Dyeing Properties of Apigenin(5,7,4′-trihydroxy flavone)”