
N-acetylglucosaminyltransferase V attenuates myocardial infarction by mediating the insulin-like growth factor 1 receptor signaling pathway
-
Tianqi Chang

 and
Ming Xu
and
Ming Xu

Abstract
Background and Objectives
N-glycosylation, a crucial post-translational modification, is well-recognized for its pivotal role in cardiovascular functions. N-acetylglucosaminyltransferase V (GnT-V) is one of the major glycosyltransferases that determine the complexity of N-glycans in N-glycosylation modification. This study aimed to explore the role of GnT-V in myocardial infarction (MI).
Methods
Proteomics and N-glycoproteomic analysis were performed on myocardial tissues for the N-glycosylation profile after MI. Adeno-associated virus (AAV) with a mouse cTnT promoter was utilized to induce overexpression of GnT-V in the heart for the role of GnT-V in MI. Echocardiography and histological analysis were used to evaluate the effect of GnT-V on MI. For the potential mechanisms of GnT-V, proteomic analysis was performed on cardiomyocytes that were subjected to GnT-V overexpression and hypoxic stress. The results were validated by western blot, lectin blot and immunoprecipitation assays, and confirmed with PNGase F and tunicamycin treatment.
Results
N-glycosylation of protein was significantly reduced after MI, which could be related to a decrease in the expression levels of GnT-V and its target glycans. Targeted GnT-V overexpression in the heart by using AAV improved cardiac function and reduced the infarct size after MI. Further, proteomics analysis of cardiomyocytes revealed that insulin-like growth factor-binding protein 3 (IGFBP3) was targeted by GnT-V and induced degradation through the lysosome pathway. Consequently, the insulin-like growth factor 1 receptor (IGF1R) signaling pathway was activated through overexpression of GnT-V.
Conclusion
Our findings suggest that promoting the IGF1R signaling cascades by regulating the N-glycosylation of certain proteins in the signaling pathway, especially through GnT-V, may act as a promising strategy for treating MI.
Introduction
Myocardial infarction (MI), the leading cause of worldwide mortality,[1] involves inflammation, apoptosis, sur vival, angiogenesis, and fibrosis, among which post-translational modifications (PTMs) play a pivotal role.[2,3] PTMs modulate protein activity, stability, interactions, and intracellular localization by altering targeted protein regions, thus facilitating metabolic pathways in response to acute ischemic and hypoxic conditions.[4,5] In myocardial remodeling after MI, PTMs regulate the modification of extracellular matrix (ECM) proteins, which modulate their secretion and degradation and sustain the integrity of heart structure and function.[6,7] The dynamic and flexible nature of PTMs makes them a critical mediator of ischemic heart and a promising target in the therapeutic strategies for MI.
N-glycosylation, one of the most abundant PTMs, involves the covalent attachment of glycans to the asparagine (Asn) residue within an Asn-X-Ser/Thr amino acid sequence (X is not proline).[8] N-glycosylation is vital for regulating the biological integrity of multiple proteins that maintain the proper cardiomyocyte structure and functions, and the N-glycosylation profile of cardiomyocytes also alters as development and aging happen.[9] In diabetic cardiomyopathy, alterations in N-glycosylation are strongly associated with pathological processes, such as chronic inflammation and cardiac fibrosis, which influence cardiac function. The N-glycosylation level of Calsequestrin 2 in the cardiomyocyte, which is one of the major calcium regulating proteins, varies as heart failure post various models progress.[10,11] Furthermore, N-glycosylation increases the complexity and diversity of glycan structures and regulates both the cell-cell interactions and cell-ECM interactions, which are essential for vascular inflammation and cardiac remodeling.[12] N-glycosylation is crucial in the development and progression of cardiovascular diseases. However, the role of N-glycosylation in MI remains largely unexplored and requires further investigation.
N-glycan biosynthesis is primarily regulated by a series of glycosyltransferases in the Golgi apparatus. Among these, N-acetylglucosaminyltransferase V (GnT-V, encoded by Mgat5) is one of the major glycosyltransferases that determine the complexity of N-glycan structures. GnT-V transfers the N-acetylglucosamine (GlcNAc) residue to the core α1, 6-mannose branch of the N-glycan via β1, 6-linkage, thereby generating tri- or tetra-antennary N-glycans containing β1, 6-GlcNAc.[13] By modifying the branching patterns of N-glycans, GnT-V increases the structural diversity of N-glycans and provides extended functional properties, which include regulating signaling pathways, cell adhesion, and cell migration. GnT-V has been reported to modify the N-glycans on cell surface receptors and influences receptor-ligand interactions. For example, GnT-V facilitates the formation of N-glycans on the epidermal growth factor receptor (EGFR) and enhances the affinity of ligands for galectins, promoting lattice formation which suppresses EGFR endocytosis and prolongs EGFR signaling pathways.[14] Whether GnT-V is involved in the pathology of MI and the underlying mechanism, however, remain unclear.
In this study, we explored the biological function of GnT-V in MI. The expression and activities of GnT-V were inhibited after MI in mice, and myocardium-specific GnT-V overexpression alleviated MI injuries. Additionally, we explored the mechanism of GnT-V in cardiomyocytes and its effect on insulin-like growth factor-binding protein 3 (IGFBP3). Our findings provide novel insights into the therapeutic potential of GnT-V in MI.
Materials and methods
Animal procedures
C57BL/6 mice (10–12 weeks old, 20–25 g) were used in this study. All procedures involving animals were carried out in compliance with the Guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of Peking University Health Science Center (BCJH0213). The mice were housed in standard facilities with controlled conditions of a temperature of 20–26 ℃ and a humidity of 50%–60% under a 12/12 h light-dark cycle.
To overexpression GnT-V in the heart, cTnT promoter was used in Adeno-associated virus serotype 9 (AAV9) delivery system for the myocardium-specific expression. AAV9 encoding mouse Mgat5 with a Flag tag provided by Hanheng Biotechnology (Shanghai, China) was employed. Each mouse was administered with 1.5×1011vector genomes (vg) of either AAV-Mgat5 or AAV-null, injected intravenously into the tail vein. Subsequently, 4 weeks after the injection, the mice were anesthetized with 2% isoflurane and then the following procedures were performed.
The left anterior descending (LAD) branch of the coronary artery was permanently ligated about 2 mm away from its origin to induce MI as previously described.[15] The mice were euthanized 7 days after MI surgery, and hearts were harvested after PBS perfusion for further analysis.
Echocardiographic analysis
Transthoracic echocardiography was performed with a 30 MHz transducer to measure mouse systolic function. Left ventricular ejection fraction (LVEF) was calculated based on B-mode from the parasternal long axis. Left ventricular fractional shortening (LVFS) was determined from M-mode images of the short axis of the heart at mid-papillary muscle level. Vevo Lab v3.2.0 software was used for echocardiography analysis.
Histological analysis
The hearts were harvested 7 days after MI. For Triphenyl tetrazolium chloride (TTC) staining, the hearts were cut into 1-mm-thick sections and incubated with 2% TTC reagent (Solarbio, G3005) for 30 min at room temperature to visualize the infarct size. For Masson trichrome staining, the middle segments of heart were embedded in paraffin and cut into 5-μm-thick sections and stained with Masson trichrome assay kit (Solarbio, G1340), according to the standard protocol. The percentage of infarction size was evaluated with Image-Pro Plus software (version 6.0, Media Cybernetics, Inc.).
Immunofluorescence staining
Anti-GnT-V (Immunoway, YN2492), anti-cTnT (Thermo Fisher Scientific, MA5-12960), and anti-Vimentin (Abcam, ab24525) antibodies were used to explore the expression of GnT-V in cardiac fibroblasts and cardiomyocytes. Anti-GST (Abcepta, AP90035) and anti-LAMP2 (Abcam, ab25631) antibodies were used to determine the localization of IGFBP3 in lysosomes. Cells were fixed with paraformaldehyde and incubated with primary antibodies overnight at 4 ℃, and then incubated with secondary antibodies for 1 h at room temperature. TUNEL Apoptosis Assay Kit (Beyotime, C1089) was employed as the manufacturer’s instruction to measure apoptotic cells in the sections of MI hearts.
Isolation and treatment of primary cardiomyocytes
Cardiomyocytes and cardiac fibroblasts were isolated from 1-day-old neonatal wild-type C57BL/6 mice with the Neonatal Heart Dissociation Kit (Miltenyi Biotec, 130098-373) as previously described.[16,17] Cardiomyocytes were cultured in DMEM medium containing 10% fetal bovine serum (FBS) with 5% CO2. Adenoviruses expressing mouse Mgat5 cDNA (AD-Mgat5) or mouse Igfbp3 cDNA (AD-Igfbp3) were constructed by Hanheng Biotechnology (Shanghai, China) and employed on cardiomyocytes 24 h after plating. For hypoxic conditions, cardiomyocytes were then cultured in 5% CO2 (O2 concentration was 1%) with serum deprivation.
Cell line and culture
HEK293A cells were used in this study and were cultured in DMEM medium containing 10% FBS. The expression vector pcDNA3.1 with Human Mgat5, Igfbp3 and Igf1r cloned, were designed and supplied by YouBio Biotechnology (Hunan, China). HEK293A cells were transfected with plasmids with the utilization of Lipofectamine 3000 transfection reagent (Invitrogen, L3000015). 5 μM cycloheximide (MedChemExpresss, HY-12320) was employed on HEK293A cells for 1 h, 2 h, 3 h to evaluate the IGFBP3 stability. To explore the degradation pathway of IGBP3, HEK293A cells were treated with 10 μM chloroquine (CQ) (Selleck, S6999), 10 μM MG132 (MedChemExpress, HY-13259) or 200 nM Bafilomycin A1 (Selleck, S1413) for 6 h.
Proteomic analysis
Protein samples were extracted from cardiac tissue or cardiomyocytes through trypsin digestion. Subsequently, the peptides were separated with EASY-nLC 1200 UPLC system and analyzed in Orbitrap Exploris 480. Proteome Discoverer search engine (v. 2.4) was employed for data processing. With a P value below 0.05, a differential expression alteration greater than 1.5-fold was regarded as the threshold for significant up-regulation proteins, while a change lower than 1/1.5-fold was considered as the threshold for significant down-regulation proteins.
Western blot
The proteins were extracted from cardiac tissue or cells with RIPA buffer. Protein samples were separated via SDS-PAGE (Biotides, WB1102) and then transferred to Biotrace nitrocellulose membranes (Cytiva, 10600001). The membranes were blocked for 1 h and incubated with primary antibodies overnight at 4 ℃. Then the membranes were incubated with HRP-conjugated anti-rabbit or HRP-conjugated anti-mouse IgG secondary antibodies for 1 h at room temperature. The membranes were visualized by enhanced chemiluminescence (ECL) reagents. Anti-GnT-V (Abcam, ab87977), anti-IGFBP3 (Abcam, ab220429), anti-IGF1R (Abcam, ab182408), anti-phosphorylated-IGF1R (CST, #3024), anti-Akt (CST, #4685), anti-phosphorylated-Akt (CST, #4060), anti-Flag (CST, #14793), anti-GST (CST, #2625), anti-HA (CST, #2367), anti-GAPDH (CST, #5174), and anti-β-actin (Proteintech, 66009–1) antibodies were used in this study.
A volume of 1 μL PNGase F (New England Biolabs, P0704s) was added into 20 μg proteins to cleave N-glycans at Asn residues. Deglycosylated proteins were then denatured with SDS loading buffer and then determined with SDS-PAGE.
For immunoprecipitation, cell extracts were obtained with NP-40 lysis buffer. 10 μg antibody was added into 1 mg protein samples overnight at 4 ℃. The homogenates were then immunoprecipitated with protein A/ G magnetic beads (Thermo Fisher Scientific, 88802). Eluted proteins were released with loading buffer and detected with SDS-PAGE.
Lectin blot
The glycoprotein samples were prepared with RIPA buffer and loading buffer, and then separated with SDS-PAGE. After the transfer of glycoprotein samples from the gel to membranes, the membranes were blocked, and incubated in biotinylated Phaseolus vulgaris Leucoagglutinin (PHA-L, Vector laboratories, B-1115-2) or biotinylated Concanavalin A (ConA, Vector laboratories, B-1005–5) overnight at 4 ℃. The membranes were incubated for 1 h with HRP-conjugated streptavidin, and visualized using a chemiluminescence imaging system.
Statistical analysis
GraphPad Prism 9 (version 9.4.1, GraphPad Software Inc.) was used to analyze the experimental data, which was then presented as mean ± standard error of mean (SEM). Shapiro-Wilk test was used to evaluate the normal distribution of the data. Two-tailed student’s t-test and Mann-Whitney test were employed to analyze the differences in normally distributed data and non-normally distributed data between two groups, respectively. One-way ANOVA or two-way ANOVA was used for comparisons involving multiple groups with normally distributed data, followed by Tukey’s multiple comparison post hoc test. Kruskal-Wallis test was performed for multiple groups with non-normally distributed data, followed by Dunn’s post hoc test. P < 0.05 was considered statistically significant.
Results
N-glycosylation was reduced in the heart and GnT-V activity was suppressed in MI
We first performed proteomic and N-glycoproteomic analysis on myocardial tissue from mice to interpret the N-glycosylation profile after MI. Results suggest that the level of N-glycosylation was significantly reduced in MI after normalized to total protein levels (Figure 1A). As key subsequential glycosyltransferases in the biosynthesis and branching of N-glycans, the expression levels of GnT-I, GnT-II, GnT-III, GnT-IV and GnT-V were assessed in various organ tissues (Supplementary Figure S1). Among these, GnT-V was predominantly and specifically expressed in heart tissue, exhibiting significantly higher levels compared to other tissues (Supplementary Figure S1E). Next, we evaluated the expression level and enzymatic activity of GnT-V in mice after MI. As shown in Figure 1B and C, GnT-V expression was markedly reduced after MI. PHA-L, specifically identifying β1, 6-GlcNAc branched structure induced by GnT-V, was reduced after MI, indicating the decrease of GnT-V’s products (Figure 1B and D). ConA, characterizing the substrate of GnT-V, was found to be accumulated and suggested that MI led to a significant reduction in the enzymatic activity of GnT-V (Figure 1B and E). The alteration in the expression level and enzymatic activity of GnT-V in MI indicated that N-glycosylation played important roles and GnT-V may have an underexplored function in MI.
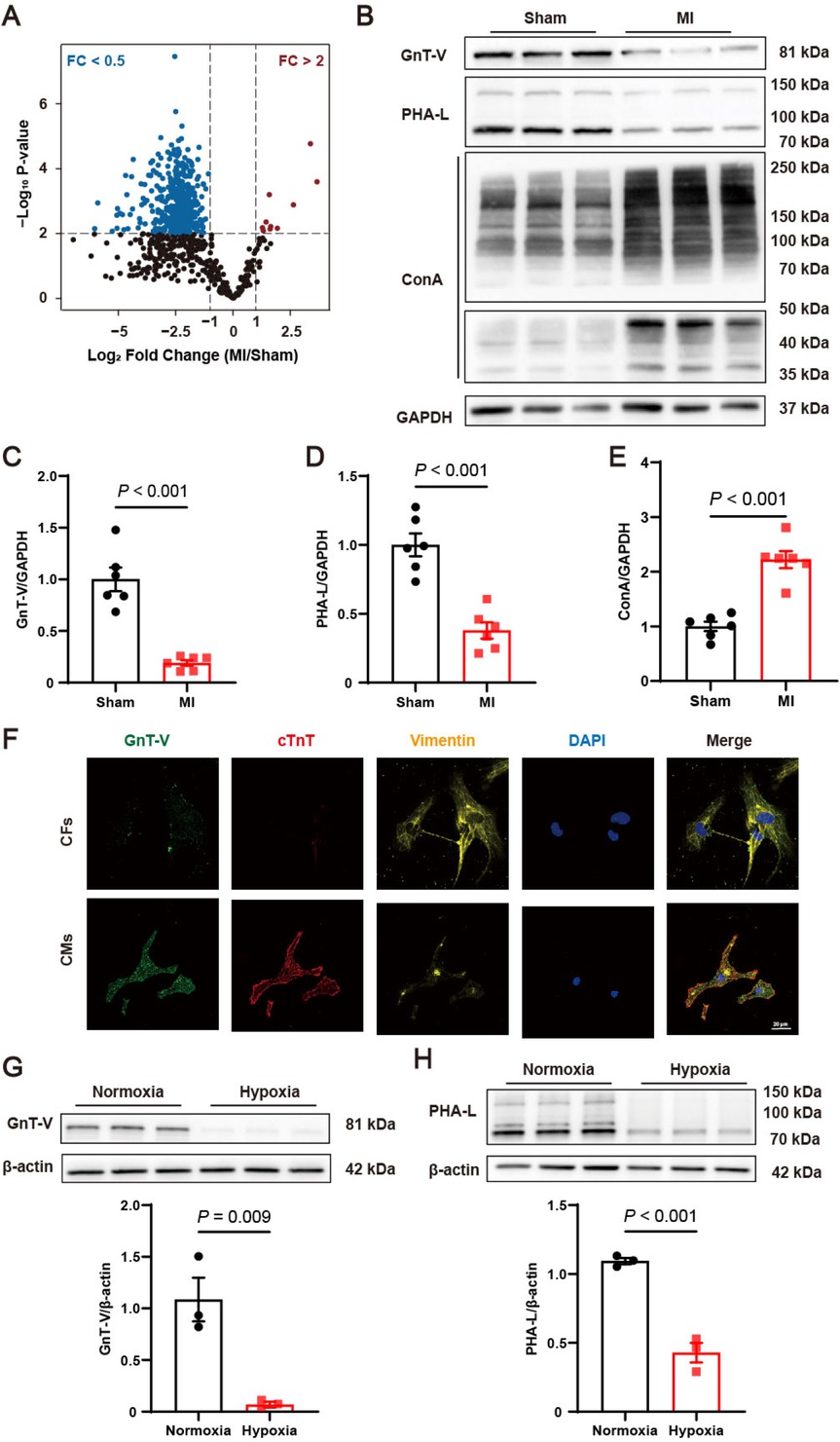
Protein N-glycosylations were reduced and N-acetylglucosaminyltransferase V (GnT-V) expression was suppressed in myocardial infarction (MI). (A) Alterations in the N-glycosylation profile following MI (n = 6 in Sham group, n = 5 in MI group). (B) Representative images and (C–E) quantitative results of the protein expression, products (PHA-L) and substrates (ConA) of GnT-V in MI mice (n = 6). (F) Representative immunofluorescence images of GnT-V (green), cardiac Troponin T (cTnT)(red), and vimentin (yellow) in primary cardiac fibroblasts (CFs) and cardiomyocytes (CMs). The nucleus was stained with DAPI (blue, 1: 5000). Scale bar = 20 μm. (G) Representative western blot images and quantitative results of GnT-V expression in cardiomyocytes exposed to hypoxia (n = 3). (H) Representative lectin blot images and quantitative results of β1, 6-GlcNAc branched structure in cardiomyocytes exposed to hypoxia (n = 3). The results are presented as mean ± SEM. Two-tailed student’s t-test.
To better understand the function of GnT-V in the heart, we next determined the expression of GnT-V in cardiac fibroblasts and cardiomyocytes. Immunofluorescence showed that GnT-V was mainly co-localized with cardiac Troponin T (cTnT), a biomarker of cardiomyocytes (Figure 1F). Consistent with immunofluorescence, the mRNA and protein levels demonstrated that GnT-V was primarily expressed in cardiomyocytes (Supplementary Figure S2A and B). Cardiomyocytes were stimulated by hypoxia stress for 24 h, commonly used to simulate injury after MI. It showed that both the mRNA and protein expression of GnT-V were restrained in response to hypoxia (Supplementary Figure S2C and Figure 1G). Products of GnT-V represented by PHA-L were correspondingly reduced in hypoxia cardiomyocytes (Figure 1H). Collectively, these data suggested that GnT-V was significantly downregulated in MI, which was primarily reflected in cardiomyocytes.
GnT-V overexpression alleviated cardiac injury after MI
To investigate the role of GnT-V in MI, AAV with a mouse cTnT promoter was utilized to induce overexpression of GnT-V in the heart. 4 weeks after the injection of AAV-Mgat5 (the gene encoded GnT-V), we confirmed the overexpressed protein levels of GnT-V in the heart (Supplementary Figure S2D), and performed LAD ligation surgery on the mice for the MI model thereafter. At 7 days after MI, echocardiography was carried out to evaluate the severity of MI injury, and it showed that overexpression of GnT-V improved LVEF and LVFS (Figure 2A), indicating a marked enhancement in cardiac function. TTC staining revealed a significant reduction in the infarct area after MI with GnT-V overexpression (Figure 2B). Masson trichrome staining indicated a decrease in cardiac fibrosis after overexpression of GnT-V (Figure 2C). Additionally, cell apoptosis in infarct border zone was notably reduced after GnT-V overexpression by TUNEL staining (Figure 2D). These findings suggested that GnT-V exerted a protective effect in the progression of MI.
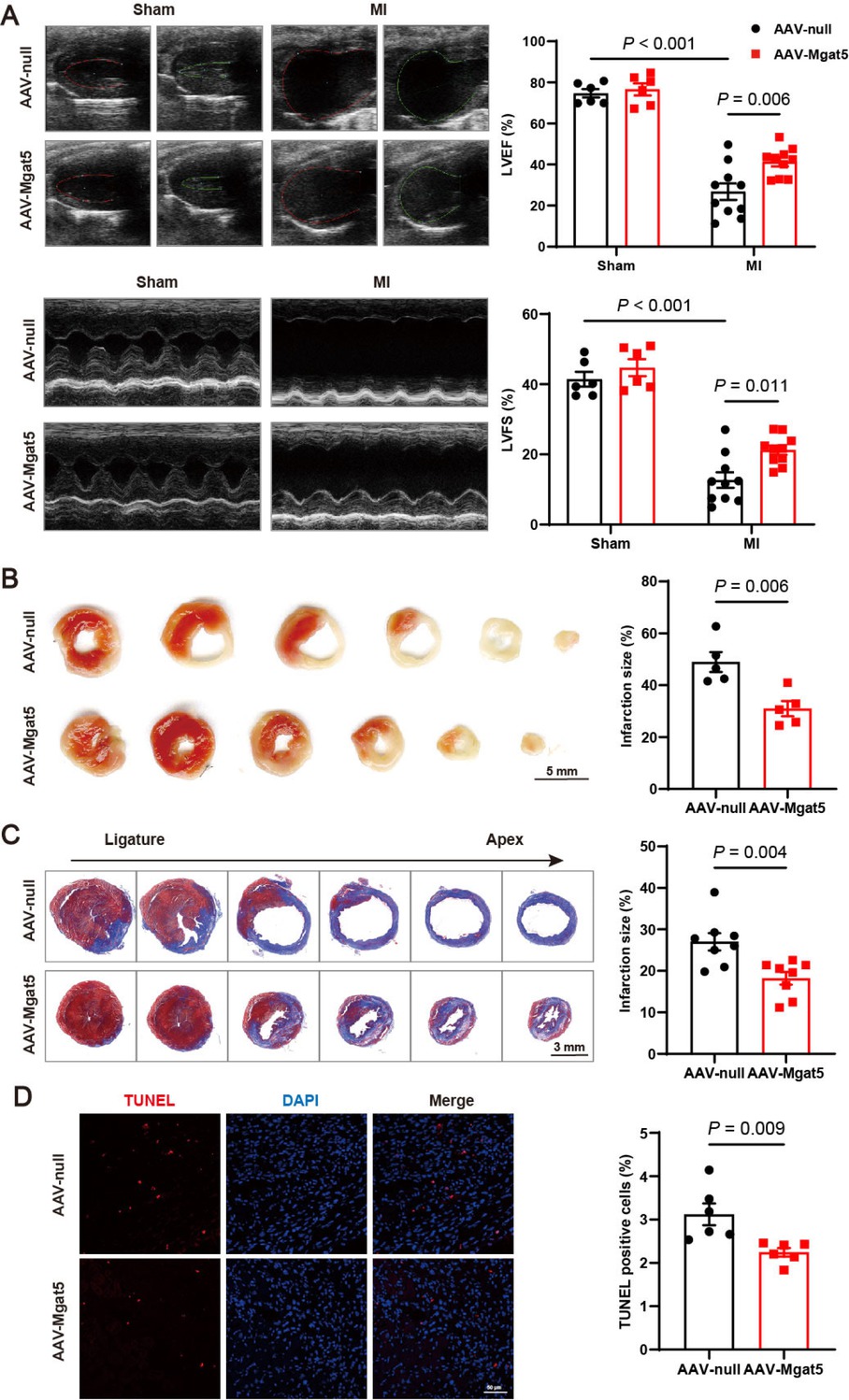
GnT-V overexpression mitigated cardiac injury post-MI. (A) Representative echocardiography images of mice in various groups 7 days after surgery, and quantitative results of left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) (n = 6 in Sham group, n = 10 in MI group). (B) Representative images and quantitative results of TTC staining (n = 5). Scale bar = 5 mm. (C) Representative images and quantitative results of Masson trichrome staining 7 days after MI (n = 8). Scale bar = 3 mm. (D) Representative images and quantitative results of TUNEL staining in infarct area after MI (n = 6). Scale bar = 50 μm. The results are presented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparison test (A), two-tailed student’s t-test (B–D).
GnT-V targeted IGFBP3 and promoted the insulin-like growth factor 1 receptor (IGF1R) signaling pathway
To explore the underlying mechanism of GnT-V’s effects in MI, we performed proteomic analysis on cardiomyocytes treated with hypoxia and overexpressed GnT-V to identify potential downstream targets of GnT-V. We first overexpressed GnT-V in cardiomyocytes with adenoviral delivery (Supplementary Figure S2E). Considering the protective effects of GnT-V in MI, we focused on proteins whose expression could be modulated by GnT-V to counteract hypoxia stress. Among these, IGFBP3, a protein that binds to insulin growth factor 1 (IGF1) and modulates IGF1R signaling pathway, exhibited the strongest correlation (Figure 3A). Western blot analysis confirmed that IGFBP3 was elevated after hypoxia and markedly reduced following GnT-V overexpression (Figure 3B and C). The downstream IGF1R and Akt signaling pathways were accordingly activated upon GnT-V overexpression (Figure 3B, D and E). Notably, even under normoxia conditions, GnT-V presented the effects on the phosphorylation of IGF1R, a phenomenon that was further demonstrated in myocardial tissues.
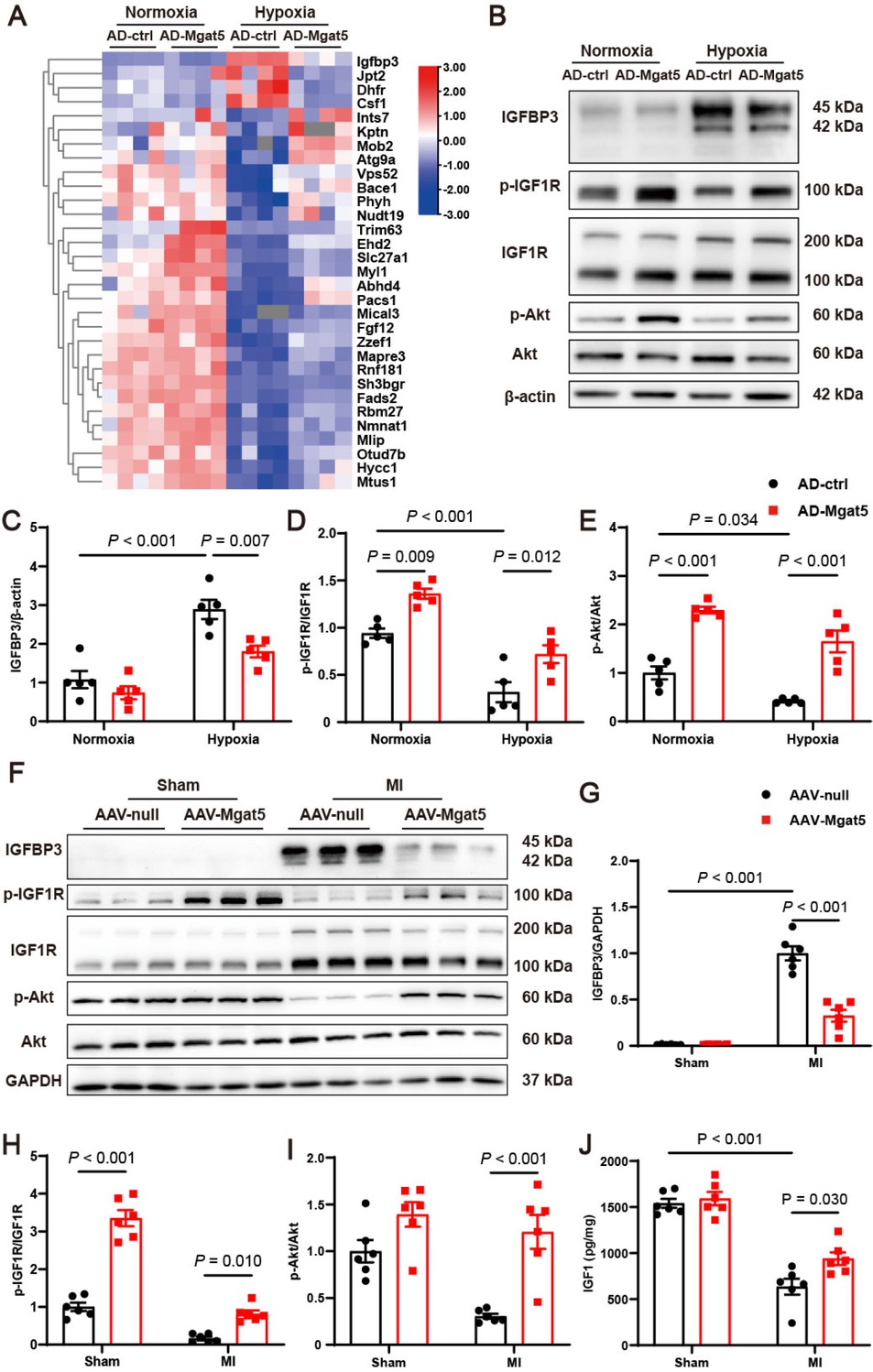
GnT-V overexpression induced downregulation of insulin-like growth factor-binding protein 3 (IGFBP3) and activation of the insulin-like growth factor 1 receptor (IGF1R) signaling pathway. (A) Heatmap of the proteomics showed differentially expressed proteins in cardiomyocytes with the stimulation of hypoxia and overexpression of GnT-V (n = 4). (B) Representative western blot images and (C–E) quantitative results of IGFBP3 and the IGF1R signaling pathway in cardiomyocytes after hypoxia and GnT-V overexpression (n = 5). (F) Representative western blot images and (G–I) quantitative results of IGFBP3 and the IGF1R signaling pathway in the myocardium after MI with GnT-V overexpression (n = 6). (J) Quantification of IGF1 levels in cardiac tissues after MI with GnT-V overexpression (n = 6). The results are presented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparison test.
Alterations in the IGFBP3/IGF1R/Akt signaling pathway were further validated in myocardial tissues after MI. IGFBP3 was significantly increased after MI, and GnT-V overexpression induced a marked reduction in IGFBP3 (Figure 3F and G). The decreased phosphorylation of IGF1R and Akt was also rescued in MI tissues following GnT-V overexpression (Figure 3F, H and I). IGF1, the primary modulator of IGFBP3 and the essential ligand for IGF1R was then assessed in myocardial tissues. The results showed that IGF1 levels were significantly reduced in infarcted cardiac tissues but increased after GnT-V overexpression (Figure 3J). Taken together, these results indicated that IGFBP3 and the IGF1R signaling pathway functioned as the critical downstream effectors of GnT-V in MI.
N-glycosylation was curcial for the effect of GnT-V on the IGF1R signaling pathway
The interactions between GnT-V and IGFBP3, GnT-V and IGF1R were investigated through co-immunoprecipitation (Co-IP) and pull-down assays. The interaction between GnT-V and IGFBP3 in cardiomyocytes was confirmed by the co-precipitation of GnT-V using an IGFBP3 antibody (Figure 4A). Pull-down assays with GST-tagged IGFBP3 revealed that GnT-V was successfully captured from HEK293A cell lysates, indicating an interaction between IGFBP3 and GnT-V (Figure 4B). The direct interaction of GnT-V with IGFBP3 was further confirmed using GnT-V with Flag as the bait protein (Figure 4C). Co-precipitation of GnT-V with the IGF1R antibody in cardiomyocytes provides evidence for the interaction between GnT-V and IGF1R (Figure 4D). IGF1R was tagged with HA and used to pull down GnT-V. The direct binding interaction between GnT-V and IGF1R was demonstrated and further confirmed using GnT-V as the bait (Figure 4E and F). Of note, GnT-V specifically interacted with pro-IGF1R.
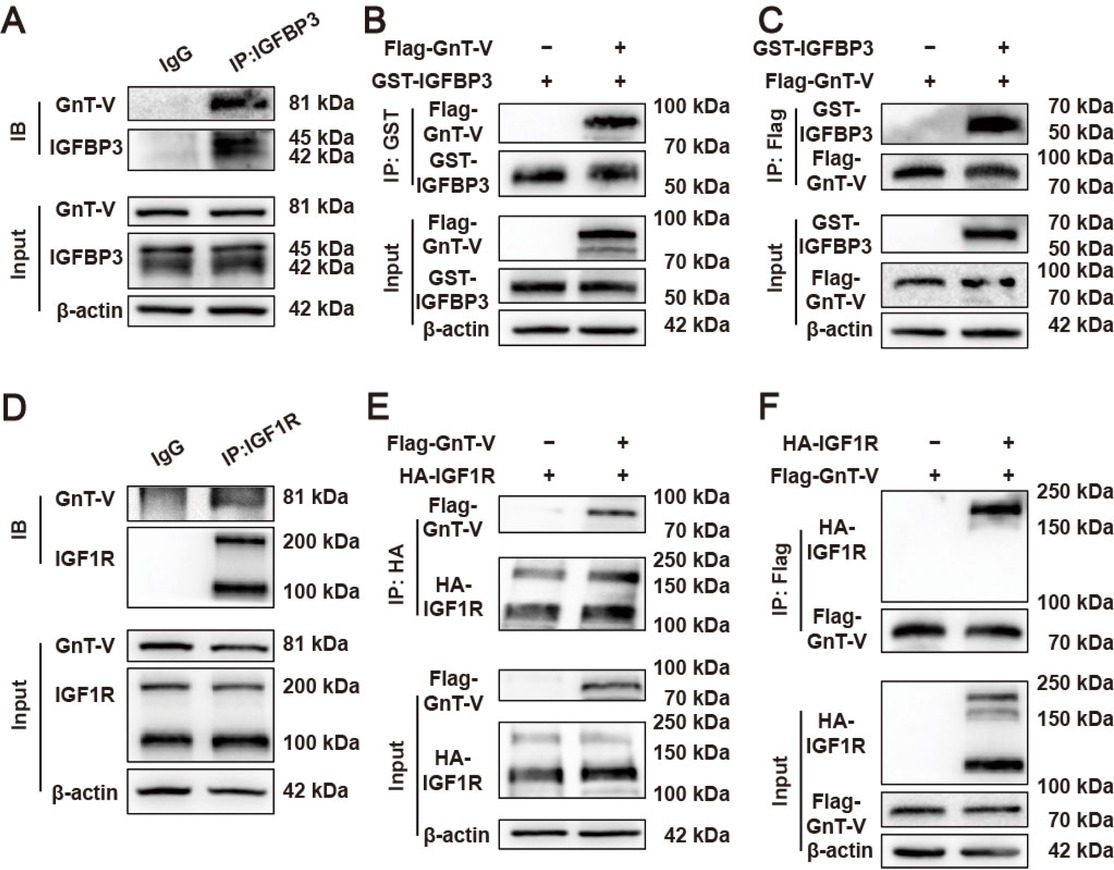
Interactions between GnT-V and IGFBP3, GnT-V and IGF1R. (A) Representative images of the interaction between GnT-V and IGFBP3 in cardiomyocytes with co-immunoprecipitation (Co-IP) assay. (B and C) Representative images of the interaction between GnT-V and IGFBP3 in HEK293A cells with pull-down assays. (D) Representative images of the interaction between GnT-V and IGF1R in cardiomyocytes with Co-IP assay. (E and F) Representative images of the interaction between GnT-V and IGF1R in HEK293A cells with pull-down assays.
Since GnT-V acts as a crucial enzyme in N-glycosylation, we next investigated whether N-glycosylation contributes to the downstream effects of GnT-V. The molecular weights of IGFBP3 and IGF1R were significantly altered following the removal of N-glycans by PNGase F, indicating that both IGFBP3 and IGF1R could be modified by N-glycosylation (Figure 5A and B). The specific N-glycosylation of IGFBP3 was significantly increased upon GnT-V overexpression (Figure 5C and D), while the substrate glycans of GnT-V on IGFBP3 were notably reduced (Figure 5C and E). To further investigate the role of N-glycosylation in the IGF1R signaling pathway, tunicamycin, an inhibitor of N-glycosylation, was administered to cardiomyocytes with hypoxia. Upon tunicamycin treatment, IGFBP3 in cardiomyocytes exhibited a significant deglycosylation with alterations in molecular weight, and a notable increase in protein levels (Figure 5F and G). The reduction in IGFBP3 levels induced by GnT-V overexpression was abolished by tunicamycin treatment. Additionally, tunicamycin treatment resulted in the deglycosylation of pro-IGF1R and the elimination of GnT-V’s effects on IGF1R and Akt phosphorylation (Figure 5F, H and I). These results suggested that GnT-V facilitated the reduction of IGFBP3 and the activation of the IGF1R signaling pathway in an N-glycosylation-dependent manner.
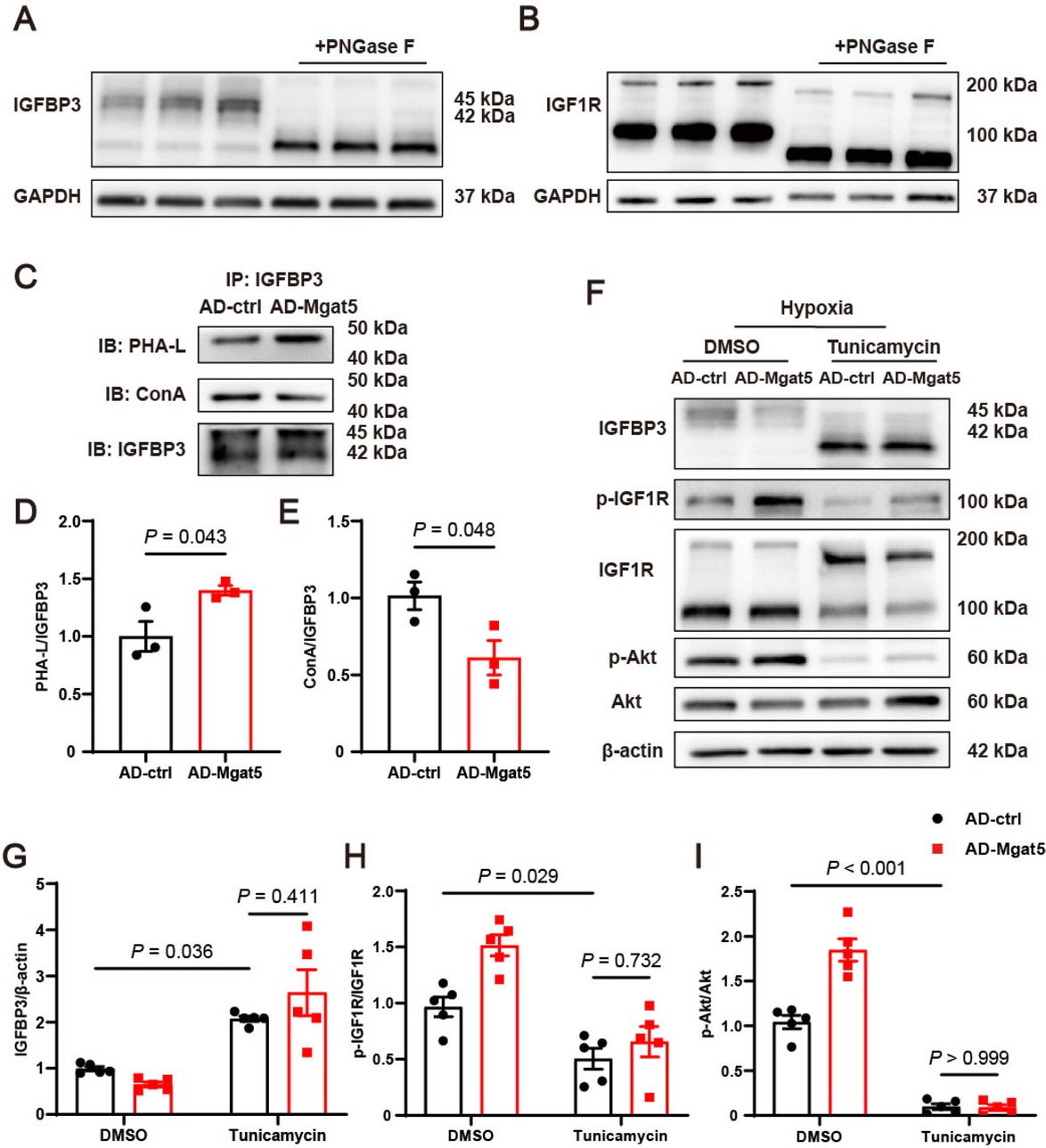
GnT-V activated the IGF1R signaling pathway in an N-glycosylation-dependent manner. Representative western blot images of (A) IGFBP3 and (B) IGF1R in cardiomyocytes lysates with and without PNGase F (n = 3). (C) Representative immunoprecipitation images and (D and E) quantitative results of the N-glycosylation modification of GnT-V on IGFBP3 in cardiomyocytes. (F) Representative western blot images and (G–I) quantitative results of IGFBP3 and the IGF1R signaling pathway in cardiomyocytes treated with tunicamycin for 24 h (n = 5). The results are presented as mean ± SEM. Two-tailed student’s t-test (D, E), two-way ANOVA followed by Tukey’s multiple comparison test (G–I).
GnT-V mediated the lysosome-dependent degradation of IGFBP3
Considering GnT-V overexpression greatly decreased IGFBP3 protein expression without influencing IGFBP3 mRNA expression (Supplementary Figure S2F), we inferred that GnT-V inhibited IGFBP3 protein expression by promoting its degradation. To investigate the effect of GnT-V on IGFBP3 protein stability, the cycloheximide (CHX) chase assay was performed and revealed that GnT-V overexpression notably accelerated IGFBP3 degradation (Figure 6A and B). We then explored the pathway of GnT-V mediated-degradation of IGBP3 using the proteasome inhibitor MG132, the lysosomal inhibitor CQ, and the autophagy inhibitor Bafilomycin A1 (Baf A1). CQ treatment effectively abolished the effects of GnT-V on IGFBP3, leading to a significant increase in IGFBP3 protein expression (Figure 6C). Immunofluorescence analysis demonstrated that GnT-V overexpression enhanced the localization of IGFBP3 to lysosomes (Figure 6D). These results indicated that GnT-V promoted the lysosome-dependent degradation of IGFBP3. When IGFBP3 was overexpressed in cardiomyocytes, the activation of IGF1R and Akt induced by GnT-V overexpression was significantly inhibited (Figure 6E–G). Thus, our findings suggested that GnT-V modulated the IGF1R signaling pathway by facilitating the lysosomal degradation of IGFBP3.
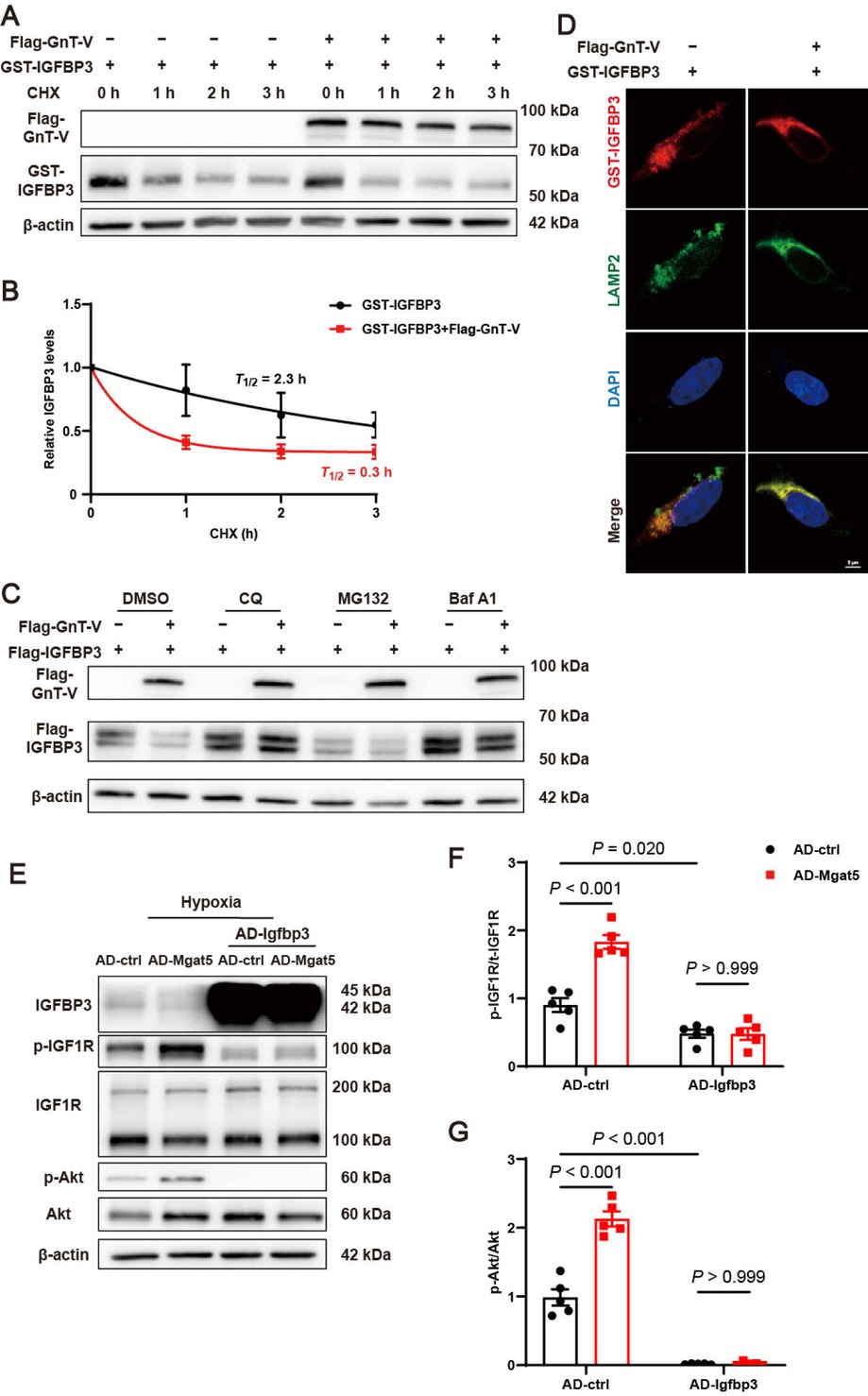
GnT-V facilitated the activation of the IGF1R signaling pathway via the lysosome-dependent degradation of IGFBP3. (A) Representative western blot images and (B) quantitative results of IGFBP3 protein stability in HEK293A cells with or without the effect of GnT-V (n = 4). (C) Representative western blot images of HEK293A cells, which were transfected with Flag-GnT-V and Flag-IGFBP3 expression plasmid, and then treated with CQ (10 μmol/L), MG132 (10 μmol/L) or Bafilomycin A1 (Baf A1, 200 nM) for 6 h. (D) Representative immunofluorescence images of GST (red), LAMP2 (green) and DAPI (blue) in HEK293A cells transfected with or without Flag-GnT-V. Scale bar = 5 μm. (E) Representative western blot images and (F and G) quantitative results of IGFBP3 and the IGF1R signaling pathway in cardiomyocytes with GnT-V or IGFBP3 overexpression (n = 5). The results are presented as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparison test (F, G).
Discussion
Protein glycosylation is a ubiquitous protein modification that regulates diverse homeostatic functions. Approximately 20% of patients with congenital disorders of glycosylation (CDGs) have comorbidities with major cardiac pathologies.[18] N-glycosylation, a highly conserved and pivotal modification, determines the structural diversity of N-glycans and has garnered growing recognition for its involvement in maintaining multiple cellular functions in the cardiovascular system, which includes regulating ion channel activity, modulating vascular inflammation and mediating cardiac remodeling.[19] However, the roles and mechanisms of glycosyltransferases in the pathophysiological processes of cardiovascular diseases remain poorly elucidated, especially in MI. In this study, we first interpreted that protein N-glycosylations were significantly reduced after MI, and identified the crucial role of GnT-V, a key glycosyltransferase responsible for complex N-glycans, in MI. It was already known that GnT-I, GnT-II, GnT-III, GnT-IV and GnT-V collectively determine the core N-glycans structure of proteins in the Golgi apparatus. We determined that tissue-specific expression of only GnT-V in the heart, and further proved it predominantly expressed in cardiomyocytes, strongly co-localizing with cTnT. GnT-V displayed a significant reduction in its protein expression level and enzymatic activity following MI. The branched glycans, induced by GnT-V, could specifically bind to galectin, and thereby modulate receptor dimerization and signaling, which was mainly involved in studies for tumor metabolism and liver fibrosis.[20,21] We demonstrated that the maintenance of N-glycosylation, especially the function of GnT-V in the heart, improved cardiac function, reduced apoptosis, and minimized the infarcted area after MI, highlighting its protective role in the development of MI.
Further investigation by proteomics revealed that the expression of multiple proteins was altered under the condition of hypoxia and was reversed by GnT-V overexpression, among which IGFBP3 was one of the most significant. Insulin-like Growth Factor Binding Protein (IGFBP) family, the carriers for IGFs, facilitates the transport and prolongs the half-life of IGFs, playing a crucial role in regulating IGF actions.[22] IGF1, a critical factor in cardiovascular biology, regulates several processes in cardiovascular diseases, including metabolism, contractility, autophagy, apoptosis and hypertrophy.[23] IGF1 binds to and activates IGF1R, thereby initiating the activation of canonical pathways, including the phosphatidylinositol-3 kinase (PI3K)/Akt pathway and the extracellular signal-regulated kinase (ERK) pathway. With a high binding affinity for IGF1, IGFBP3 can prevent the binding of IGF1 to IGF1R, thus inhibiting the IGF1R signaling pathway.[24] Conversely, IGFBP3 can also serve as an IGF1 reservoir, gradually releasing IGF1 and thereby sustaining IGF1R signaling over time.[25] Thus, IGFBP3 exhibits a dual effect on IGF1 action. Low IGFBP3 expression augments the activation of IGF1/IGF1R signaling, whereas high IGFBP3 expression decreases free IGF1 levels, dampening IGF1 action. IGFBP3 protein expression has been reported a marked elevation in response to hypoxia, oxidative stress and doxorubicin treatment.[26,27] Our present work also confirmed the upregulation of IGFBP3 and the inhibition of IGF1R signaling cascades during MI.
The IGFBP family comprises six primary members, with IGFBP3 and IGFBP4 having well-characterized N-glycosylation sites.[28] Three N-glycosylation sites have been reported in human IGFBP3 protein, Asn89, Asn109, and Asn172.[29] Native IGFBP3 displays two distinct bands of approximately 40–45 kDa, which arise from differences in N-glycan chains at these sites.[30] In our study, we observed a significant reduction in the molecular weight of IGFBP3 following either direct deglycosylation with PNGase F or inhibition of N-glycans biosynthesis with tunicamycin. GnT-V increased N-glycosylation of IGFBP3 and facilitated its lysosomal degradation, therefore promoting the IGF1R signaling pathway.
It is worth noting that a non-glycosylated form of IGFBP3 is more rapidly degraded by proteases in vivo, leaving the glycosylated form,[31] underscoring the importance of N-glycosylation for IGFBP3 stability. These findings align with the observed molecular weight of IGFBP3 in our work. Ubiquitin-proteasome pathway, endosome-lysosome pathway and autophagy-lysosome pathway predominantly engage in protein degradation within mammalian cells, collectively maintaining protein stability and cellular homeostasis.[32] Our research revealed that the degradation of glycosylated IGFBP3 was independent of the ubiquitin-proteasome pathway. Moreover, the degradation of IGFBP3 induced by GnT-V after their direct binding was mediated by lysosomes, which was further verified by the observation that GnT-V facilitated the localization of IGFBP3 onto lysosomes. However, direct evidence confirming the functional role of N-glycosylation in IGFBP3 remains elusive. Our study demonstrated an interaction between GnT-V and IGFBP3, and revealed that modification by GnT-V may have a biological impact on IGFBP3.
Notably, overexpression of IGFBP3 abolished the GnT-V-mediated activation of IGF1R signaling in cardiomyocytes. Additionally, our results indicated that GnT-V directly promoted the phosphorylation of IGF1R under physiological conditions, thereby enhancing the activation of the IGF1R signaling pathway. IGF1R possesses 16 potential N-glycosylation sites,[33] and its mature form requires N-glycosylation of pro-IGF1R, which is essential for correct transport to the cell surface.[34] Incorrect glycosylation can lead to the degradation of IGF1R.[34] This is consistent with our observation that tunicamycin resulted in a significant increase in pro-IGF1R expression and a marked reduction in mature IGF1R protein expression. Protein interaction studies further revealed that GnT-V binds to pro-IGF1R, suggesting that GnT-V primarily modulates the N-glycosylation of pro-IGF1R. Additionally, GnT-V promoted the activation of IGF1R in the mice under physiological conditions, both in cardiomyocytes and in the sham group of mice. Importantly, both the mature form and pro-form of IGF1R displayed significant molecular weight shifts upon deglycosylation with PNGase F, indicating that both forms are subject to N-glycosylation. Based on these findings, it was supposed that GnT-V triggered IGF1R phosphorylation and signaling activation through the N-glycosylation on pro-IGF1R.
There are still certain limitations of the study. First of all, the whole study was conducted using the mouse model or cardiomyocytes, with no verification in the human subjects or samples. Since there is quite limited access to myocardial tissues from human subjects after MI, we only did most of our studies with mice, and we planned to make some extensions of the study with human samples in the near future, making the application of regulation of N-glycosylation meaningful in the treatment of MI. Secondly, only one of the glycosyltransferases, GnT-V, was studied in the present study. Since N-glycosylation is a sequential process involving multiple glycosyltransferases, and the functions of other ones, such as GnT-I or GnT-II, were not yet discovered in the MI, and may also remain further investigation. Third, we only focused on the short-term effects of GnT-V overexpression in the heart after MI, for the observation made up to 7 days, and the long-term effects were not evaluated. And we will conduct further studies to investigate the role of GnT-V in cardiac fibrosis, remodeling and chronic heart failure as a long-term evaluation of MI.
By exploring the functions of glycosylation in MI, we first determined the roles and mechanisms of the myocardium-specific glycosyltransferase GnT-V in the heart. With the dual effects of glycosylation on IGFBP3 and IGF1R by GnT-V, it synergistically accelerated the degradation of IGFBP3 through the lysosomal pathway and phosphorylation of IGF1R, thus activating the IGF1R signaling pathway and promoting cardiomyocyte survival. Our findings provide novel insights into the knowledge of N-glycosylation and GnT-V in MI and suppose therapeutic potentials.
Conclusion
This study highlights the pivotal role of N-glycosylation in MI and identifies GnT-V as a positive regulator. We demonstrated that GnT-V enhanced cardiac function and mitigated the detrimental effects in mice after MI. This effect was mediated by the facilitation of lysosomal degradation of IGFBP3, which was elevated in response to MI, thereby promoting the activation of the IGF1R signaling pathway. Furthermore, GnT-V interacted with pro-IGF1R and promoted IGF1R phosphorylation. These findings suggest that targeting GnT-V could serve as a promising therapeutic strategy for MI by restoring proper N-glycosylation.
Supplementary Information
Supplementary materials are only available at the official site of the journal (www.intern-med.com).
Funding statement: This work was supported by the National Natural Science Foundation of China (No. 82270343 to Dr J. Wang), Natural Science Foundation of Beijing (No. L248019 to Dr M. Xu), CAMS Innovation Fund for Medical Sciences (2021-I2M-5-003).
Acknowledgements
The authors also thank Prof. Xing Chen for his guidance in the research and assistance in the experimental techniques.
-
Author Contributions
Tianqi Chang and Jiaxing Wang performed study concept and wrote the paper. Tianqi Chang, and Yangya’nan Jin performed the experiments and analyzed the data. Chenyu Fan, Hu Wang, Ling Jin, Yidan Shi, Houhua Li, Jiaxing Wang and Ming Xu provided acquisition. Jiaxing Wang and Ming Xu supervised the study provided financial support. All authors read and approved the final manuscript.
-
Ethical Approval
All procedures involving animals were carried out in compliance with the Guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of Peking University Health Science Center (BCJH0213).
-
Informed Consent
Not applicable.
-
Conflict of Interest
The authors declare no conflicts of interest.
-
Use of Large Language Models, AI and Machine Learning Tools
None.
-
Data Availability Statement
The data that support the findings of this study are available within the article and its supplementary materials.
References
1 Roth GA, Mensah GA, Fuster V. The Global Burden of Cardiovascular Diseases and Risks: A Compass for Global Action. J Am Coll Cardiol 2020;76:2980–2981.10.1016/j.jacc.2020.11.021Search in Google Scholar PubMed
2 Al-U'datt DGF, Allen BG, Hiram R, Alrabadi N. Current knowledge into the role of the peptidylarginine deiminase (PAD) enzyme family in cardiovascular disease. Eur J Pharmacol 2021;891:173765.10.1016/j.ejphar.2020.173765Search in Google Scholar PubMed
3 Yin Z, Zou Y, Wang D, Huang X, Xiong S, Cao L, et al. Regulation of the Tec family of non-receptor tyrosine kinases in cardiovascular disease. Cell Death Discov 2022;8:119.10.1038/s41420-022-00927-4Search in Google Scholar PubMed PubMed Central
4 Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, et al. Histone Lactylation Boosts Reparative Gene Activation Post-Myocardial Infarction. Circ Res 2022;131:893–908.10.1161/CIRCRESAHA.122.320488Search in Google Scholar PubMed
5 Zhang X, Wang Y, Li H, Wang DW, Chen C. Insights into the post-translational modifications in heart failure. Ageing Res Rev 2024;100:102467.10.1016/j.arr.2024.102467Search in Google Scholar PubMed
6 Tyagi SC, Kumar SG, Haas SJ, Reddy HK, Voelker DJ, Hayden MR, et al. Post-transcriptional regulation of extracellular matrix metalloproteinase in human heart end-stage failure secondary to ischemic cardiomyopathy. J Mol Cell Cardiol 1996;28:1415–1428.10.1006/jmcc.1996.0132Search in Google Scholar PubMed
7 An M, Kwon K, Park J, Ryu DR, Shin JA, Lee Kang J, et al. Extracellular matrix-derived extracellular vesicles promote cardiomyocyte growth and electrical activity in engineered cardiac atria. Biomaterials 2017;146:49–59.10.1016/j.biomaterials.2017.09.001Search in Google Scholar PubMed
8 Lin Y, Lubman DM. The role of N-glycosylation in cancer. Acta Pharm Sin B 2024;14:1098–1110.10.1016/j.apsb.2023.10.014Search in Google Scholar PubMed PubMed Central
9 Ednie AR, Parrish AR, Sonner MJ, Bennett ES. Reduced hybrid/complex N-glycosylation disrupts cardiac electrical signaling and calcium handling in a model of dilated cardiomyopathy. J Mol Cell Cardiol 2019;132:13–23.10.1016/j.yjmcc.2019.05.001Search in Google Scholar PubMed
10 Liu M, Peng T, Hu L, Wang M, Guo D, Qi B, et al. N-glycosylation-mediated CD 147 accumulation induces cardiac fibrosis in the diabetic heart through ALK5 activation. Int J Biol Sci 2023;19:137–155.10.7150/ijbs.77469Search in Google Scholar PubMed PubMed Central
11 Ednie AR, Deng W, Yip KP, Bennett ES. Reduced myocyte complex N-glycosylation causes dilated cardiomyopathy. FASEB J 2019;33:1248–1261.10.1096/fj.201801057RSearch in Google Scholar PubMed PubMed Central
12 Del Monte-Nieto G, Fischer JW, Gorski DJ, Harvey RP, Kovacic JC. Basic Biology of Extracellular Matrix in the Cardiovascular System, Part 1/4: JACC Focus Seminar. J Am Coll Cardiol 2020;75:2169–2188.10.1016/j.jacc.2020.03.024Search in Google Scholar PubMed PubMed Central
13 Nagae M, Kizuka Y, Mihara E, Kitago Y, Hanashima S, Ito Y, et al. Structure and mechanism of cancer-associated N-acetylglucosaminyltransferase-V. Nat Commun 2018;9:3380.10.1038/s41467-018-05931-wSearch in Google Scholar PubMed PubMed Central
14 Cai J, Huang J, Wang W, Zeng J, Wang P. miR-124-3p Regulates FGF2-EGFR Pathway to Overcome Pemetrexed Resistance in Lung Adenocarcinoma Cells by Targeting MGAT5. Cancer Manag Res 2020;12:11597–11609.10.2147/CMAR.S274192Search in Google Scholar PubMed PubMed Central
15 Chang T, Liu C, Yang H, Lu K, Han Y, Zheng Y, et al. Fibrin-based cardiac patch containing neuregulin-1 for heart repair after myocardial infarction. Colloids Surf B Biointerfaces 2022;220:112936.10.1016/j.colsurfb.2022.112936Search in Google Scholar PubMed
16 Wang H, Wang J, Cui H, Fan C, Xue Y, Liu H, et al. Inhibition of fatty acid uptake by TGR5 prevents diabetic cardiomyopathy. Nat Metab 2024;6:1161–1177.10.1038/s42255-024-01036-5Search in Google Scholar PubMed PubMed Central
17 Wang J, Zhang J, Lin X, Wang Y, Wu X, Yang F, et al. DCA-TGR5 signaling activation alleviates inflammatory response and improves cardiac function in myocardial infarction. J Mol Cell Cardiol 2021;151:3–14.10.1016/j.yjmcc.2020.10.014Search in Google Scholar PubMed
18 Chatham JC, Patel RP. Protein glycosylation in cardiovascular health and disease. Nat Rev Cardiol 2024;21:525–544.10.1038/s41569-024-00998-zSearch in Google Scholar PubMed
19 Marques-da-Silva D, Francisco R, Webster D, Dos Reis Ferreira V, Jaeken J, Pulinilkunnil T. Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J Inherit Metab Dis 2017;40:657–672.10.1007/s10545-017-0066-ySearch in Google Scholar PubMed
20 Cong X, Liu X, Dong X, Fang S, Sun Z, Fan J. Silencing GnT-V reduces oxaliplatin chemosensitivity in human colorectal cancer cells through N-glycan alteration of organic cation transporter member 2. Exp Ther Med 2021;21:128.10.3892/etm.2020.9560Search in Google Scholar PubMed PubMed Central
21 Beatrice Greco, Valeria Malacarne, Federica De Girardi, Giulia Maria Scotti, Francesco Manfredi, Elia Angelino, et al. Disrupting N-glycan expression on tumor cells boosts chimeric antigen receptor T cell efficacy against solid malignancies. Sci Transl Med 2022;14:eabg3072.10.1126/scitranslmed.abg3072Search in Google Scholar PubMed
22 Kim H, Fu Y, Hong HJ, Lee SG, Lee DS, Kim HM. Structural basis for assembly and disassembly of the IGF/IGFBP/ALS ternary complex. Nat Commun 2022;13:4434.10.1038/s41467-022-32214-2Search in Google Scholar PubMed PubMed Central
23 Troncoso R, Ibarra C, Vicencio JM, Jaimovich E, Lavandero S. New insights into IGF-1 signaling in the heart. Trends Endocrinol Metab 2014;25:128–137.10.1016/j.tem.2013.12.002Search in Google Scholar PubMed
24 Baxter RC. Nuclear actions of insulin-like growth factor binding protein-3. Gene 2015;569:7–13.10.1016/j.gene.2015.06.028Search in Google Scholar PubMed PubMed Central
25 Lee H, Kim SR, Oh Y, Cho SH, Schleimer RP, Lee YC. Targeting insulinlike growth factor-I and insulin-like growth factor-binding protein-3 signaling pathways. A novel therapeutic approach for asthma. Am J Respir Cell Mol Biol 2014;50:667–677.10.1165/rcmb.2013-0397TRSearch in Google Scholar PubMed PubMed Central
26 Koga T, Endo H, Miyamoto Y, Mukai M, Akira S, Inoue M. IGFBPs contribute to survival of pancreatic cancer cells under severely hypoxic conditions. Cancer Lett 2008;268:82–88.10.1016/j.canlet.2008.03.030Search in Google Scholar PubMed
27 Huang YT, Liu CH, Yang YC, Aneja R, Wen SY, Huang CY, et al. ROS- and HIF1α-dependent IGFBP3 upregulation blocks IGF1 survival signaling and thereby mediates high-glucose-induced cardiomyocyte apoptosis. J Cell Physiol 2019;234:13557–13570.10.1002/jcp.28034Search in Google Scholar PubMed
28 Poreba E, Durzynska J. Nuclear localization and actions of the insulin-like growth factor 1 (IGF-1) system components: Transcriptional regulation and DNA damage response. Mutat Res Rev Mutat Res 2020;784:108307.10.1016/j.mrrev.2020.108307Search in Google Scholar PubMed
29 Paulette M Yamada, Kuk-Wha Lee. Perspectives in mammalian IGFBP-3 biology: local vs. systemic action. Am J Physiol Cell Physiol 2009;296:C954–976.10.1152/ajpcell.00598.2008Search in Google Scholar PubMed
30 Masnikosa R, Baricević I, Lagundzin D, Nedić O. Characterisation of N-glycans bound to IGFBP-3 in sera from healthy adults. Biochimie 2010;92:97–101.10.1016/j.biochi.2009.09.015Search in Google Scholar PubMed
31 Clemmons DR, Sleevi M, Busby WH Jr. Recombinant, nonglycosylated human insulin-like growth factor-binding protein-3 (IGFBP-3) is degraded preferentially after administration to type II diabetics, resulting in increased endogenous glycosylated IGFBP-3. J Clin Endocrinol Metab 2005;90:6561–6568.10.1210/jc.2005-1389Search in Google Scholar PubMed
32 Zhong G, Chang X, Xie W, Zhou X. Targeted protein degradation: advances in drug discovery and clinical practice. Signal Transduct Target Ther 2024;9:308.10.1038/s41392-024-02004-xSearch in Google Scholar PubMed PubMed Central
33 Robajac D, Masnikosa R, Miković Ž, Nedić O. Gestation-associated changes in the glycosylation of placental insulin and insulin-like growth factor receptors. Placenta 2016;39:70–76.10.1016/j.placenta.2016.01.005Search in Google Scholar PubMed
34 Siddals KW, Allen J, Sinha S, Canfield AE, Kalra PA, Gibson JM. Apposite insulin-like growth factor (IGF) receptor glycosylation is critical to the maintenance of vascular smooth muscle phenotype in the presence of factors promoting osteogenic differentiation and mineralization. J Biol Chem 2011;286:16623–16630.10.1074/jbc.M110.202929Search in Google Scholar PubMed PubMed Central
© 2025 Tianqi Chang, Yangya'nan Jin, Chenyu Fan, Hu Wang, Ling Jin, Yidan Shi, Houhua Li, Jiaxing Wang, Ming Xu, published by De Gruyter on behalf of the SMP
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Perspective
- Advances in the diagnosis and treatment of gastrointestinal tumors under the concept of super minimally invasive surgery
- Review Article
- Harnessing the potential of small extracellular vesicle biomarkers for cancer diagnosis and prognosis with advanced analytical technologies
- Current understanding and controversy on brain access of GLP-1 and GLP-1 receptor agonists
- Mitochondrial quality control as a therapeutic target in cardiovascular disease: Mechanistic insights and future directions
- Original Article
- Causal association between gut microbiota composition and the risk of atrial fibrillation
- Artificial intelligence-based predictive model for relapse in acute myeloid leukemia patients following haploidentical hematopoietic cell transplantation
- CircPLK1 upregulates ETS1 to confer anthracycline resistance in triple-negative breast cancer
- N-acetylglucosaminyltransferase V attenuates myocardial infarction by mediating the insulin-like growth factor 1 receptor signaling pathway
- A similar effect of fibrinogen on efficacy and safety of tenecteplase versus alteplase in acute ischemic cerebrovascular events (TRACE II) trial
Articles in the same Issue
- Perspective
- Advances in the diagnosis and treatment of gastrointestinal tumors under the concept of super minimally invasive surgery
- Review Article
- Harnessing the potential of small extracellular vesicle biomarkers for cancer diagnosis and prognosis with advanced analytical technologies
- Current understanding and controversy on brain access of GLP-1 and GLP-1 receptor agonists
- Mitochondrial quality control as a therapeutic target in cardiovascular disease: Mechanistic insights and future directions
- Original Article
- Causal association between gut microbiota composition and the risk of atrial fibrillation
- Artificial intelligence-based predictive model for relapse in acute myeloid leukemia patients following haploidentical hematopoietic cell transplantation
- CircPLK1 upregulates ETS1 to confer anthracycline resistance in triple-negative breast cancer
- N-acetylglucosaminyltransferase V attenuates myocardial infarction by mediating the insulin-like growth factor 1 receptor signaling pathway
- A similar effect of fibrinogen on efficacy and safety of tenecteplase versus alteplase in acute ischemic cerebrovascular events (TRACE II) trial