NPR2 gene variants in familial short stature: a single-center study
-
Ke Yuan
 ,
Jiao Chen
,
Jiao Chen


Abstract
Objectives
NPR2 variants are associated with various short stature and bone dysplasia, such as acromesomelic dysplasia Maroteaux tyoe, individuals with a phenotype similar to Léri–Weill syndrome (LWD), and idiopathic short stature (ISS). However, few studies have reported on the relationship between familial short stature (FSS) and NPR2 variants. This study aimed to explore the relationship between FSS and NPR2 variants through the detection and identification of NPR2 variants in children with FSS, phenotypic description, clear treatment plan, and follow-up of treatment effect.
Methods
Children who met the FSS diagnostic criteria and had informed consent were included in the study. The trio whole-exome sequencing method (trio-WES) was used to detect and evaluate the NPR2 variants.
Results
A total of 16 children with short stature were included in this study (pretreatment height ≤ −2 standard deviation (SD) in both the patient and the shorter parent, unknown genetic etiology). NPR2 variants were identified in 12.5%(2/16) of the participants. Patient A was a 6-year-old male and 103.7 cm tall (−3.11SD), while Patient B was a 9-year-old female and 123.2 cm tall (−1.88SD). However, their heights increased after recombinant human growth hormone (rhGH) treatment. The height of patient A increased by 0.36SD six months after treatment while that of patient B increased by 1.22SD after one and a half years of treatment.
Conclusions
NPR2 variant causes FSS. The growth rate of children significantly improved after rhGH treatment. However, further follow-up study is needed to determine the final height after long-term treatment.
Introduction
Short stature is described as having less than two standard deviations (SD) of the average height of the same race, age, and gender [1]. Several etiologies of short stature exist, including endocrine diseases, metabolic diseases, chromosomal diseases, intrauterine dysplasia, family factors, institutional factors, nutritional factors, socioeconomic factors, and systemic chronic diseases. While Idiopathic short stature (ISS) accounts for between 60 and 80% of short stature, its specific etiology is unknown [2]. Furthermore, ISS can be divided into familial short stature (FSS) or nonfamilial idiopathic short stature based on the international classification of pediatric endocrine diagnosis [3]. FSS accounts for between 11 and 23% of the total short stature patients [4].
Growth hormone (GH) – insulin-like growth factor (IGF)-I axis is essential for regulating bone formation during growth. GH and growth hormone receptor (GHR) are synthesized and secreted in the pituitary gland. Besides, IGF-1 mediates GH, thus promoting the proliferation and differentiation of cartilage. The abnormal gene at each level of the axis affects the axis, causing short stature [5, 6]. In the past, the GH-IGF-1 axis was considered as the nuclear type axis regulating the growth of children. However, with the rapid development of science and technology, some recent studies have shown that several factors affect growth and development. The genome-wide association studies (GWS) have shown the presence of about 180 genetic loci with several variations associated with human height phenotypes. In recent years, the NPR2 variant has been shown to influence dwarfism, cartilage bone growth, and the classical GH-IGF-1 related gene [7].
Previous studies have shown that several NPR2 variants are associated with acromesomelic dysplasia Maroteaux tyoe, ISS, and individuals with a phenotype similar to Léri–Weill syndrome (LWD), while few are related to FSS. Particularly, a small study has reported the efficacy of recombinant human growth hormone (rhGH) in the treatment of FSS caused by NPR2 variants.
This study aimed to evaluate the frequency, types, and clinical manifestations of NPR2 variants in FSS children after rhGH treatment via the trio whole-exome sequencing (trio-WES). The effectiveness of rhGH therapy was also evaluated.
Materials and methods
Inclusion Criteria were: a) Children with short stature who were diagnosed in the First Affiliated Hospital; b) The diagnostic criteria of familial short stature, such as 1) height less than −2SD or below the third percentile of the mean height of normal children of the same age, sex, and race, 2) both parents or one of them is short (father’s height is less than 160 cm (−2SD) or mother’s height is less than 150 cm (−2SD) and 3) normal birth weight and length during gestational age, and normal body proportion [8]; c) Informed consent from the children and their legal guardians.
Exclusion criteria: a) Short stature with definite etiology (growth hormone deficiency and other genetic diseases); b) short stature caused by other diseases (chronic renal insufficiency and pituitary tumor.); c) other limiting factors (severe organ dysfunction, serious mental disease, combined with other long-term drug control diseases, such as hypertension, precocious puberty, and thyroid dysfunction.
The ethics committee of the First Affiliated Hospital approved this study.
The trio-WES was conducted on all participants via variant screening (high-throughput sequencing technology), gene data analysis (bioinformatics and clinical information analysis technology), and suspected pathogenic variant verification (Sanger sequencing technology). Variant screening: IDT xGen Exome Research Panel v1.0 whole exon capture chip was used for whole-exome sequencing via the Illumina NovaSeq 6000 series sequencer. The target sequencing coverage was not less than 99%. Gene data analysis: The cloud platform system was analyzed and screened for accurate diagnosis of genetic diseases. Molecular biology annotation, biology, genetics, and characteristic clinical analysis, combined with pathogenic variant database, normal human genome database, clinical characteristics database of 4,000 known genetic diseases, and gene data analysis algorithm were used to grade several gene variants and divide them into three groups. The three-factor and American Society for Medical Genetics (ACMG) gene variation grading systems were used. Validation of suspected pathogenic variant: ABI3730 sequencer was used for Sanger sequencing after PCR. The sequence analysis software was then used to validate the results. The genetics of children with NPR2 variants were evaluated.
All children were treated with rhGH after diagnosis with FSS when the NPR2 variants were found, and regular follow-up (every three months) was conducted. The rhGH therapy was adjusted according to the height increase, IGF-1 level, and related examination results.
Results
Analysis was conducted on 16 FSS (10 males, average height of −2.42 ± 0.52SD and six females, the average height of −2.55 ± 0.63SD, the average age was 6.32 ± 2.28 years), and two NPR2 variant cases, accounting for 12.5% of FSS children. The details were shown in Table 1.
Features of patients with NPR2 gene variants.
| Patient A | Patient B | |
|---|---|---|
| Age, years | 6.0 | 9.0 |
| Gender | Male | Female |
| Gene variant | NPR2 | NPR2 |
| Nucleic acid variants | c.2643 G>A | c.2298_2299delGCinsTT |
| Amino acid variants | p.Gln881Gln | p.Glu766_Arg767delinsAspStop |
| Height, SDS | −3.11 | −1.88 |
| Father’s height, SDS | −2.90 | −2.90 |
| Mather’s height, SDS | −1.03 | −0.67 |
| Birth weight, SDS | 0.1 | −0.2 |
| Birth length, SDS | 0.0 | −0.3 |
| Peak GH after stimulating test, ug/L | 7.86 | N/A |
| IGF-1 at baseline, SDS | 1.4 | 0.54 |
| Age at GH therapy onset, years | 7.5 | 9.0 |
| Initial dose of rhGH, mg/kg.day | 0.050 | 0.043 |
| Growth velocity before rhGH treatment, cm/3 months | 1.5 | 1.0 |
| Growth velocity after rhGH treatment, cm/3 months | 2.3 | 2.6 |
| Treatment time, months | 6 | 18 |
-
rhGH, recombinant human growth hormone; SDS, standard deviation score.
Patient A: A 6-year-old male who visited the clinic in March 2019 due to “Growth delay for over 4 years”. He had a birth weight and length of 3.0 kg and 50 cm and was healthy in the past. In March 2019, his height was 103.7 cm (−3.11SD), weight, 16.2 kg, he was symmetrical, had no bone deformity, no abnormal face, no axillary hair, had long prepuce, G1, and Ph1. The heights of the father, mother, and grandfather were 155 cm (−2.90SD), 155 cm (−1.03SD), and 165 cm (−1.26SD), respectively (shown in Figure 1A). Laboratory examinations: The levels of sex hormones, including the follicle-stimulating hormone, luteinizing hormone, testosterone, estradiol, and prolactin, were within the normal ranges. Furthermore, blood routine, blood gas analysis, urine routine, cortisol, adrenocorticotropic hormone, thyroid function, fasting blood glucose, glycosylated hemoglobin, fasting insulin, liver, and kidney lipid glucose electrolyte, hepatitis series were normal.

(A) The family pedigrees of Patient A. (B) The results of NPR2 gene mutation detection in patient A. (C) The family pedigrees of Patient B. (D) The results of NPR2 gene mutation detection in patient B.
Gene detection: NPR2 NM_003995.4:c.2643G>A; p.(Gln881=) (shown in Figure 1B). This variant occurs at the last base of exon 17 and various splicing prediction programmes suggest that it may affect splicing. Eventually, MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), dbscSNV (https://www.solvebio.com/data/dbscSNV/), and GTAG (www.chigene.org) software packages were applied to predict the functional changes of variants on the splicing sites due to c.2643G>A occurs at the last base of exon 17.
After sequencing, canonical GT-AG dinucleotides, identified as splice site recognition, was formation because of the mutant site. Meanwhile, according to silico analysis of mutated sequences with MaxEntScan showed that variant c.2643G>A was deleterious through affected donor site of exon17 (9.22 → 5.26). Consistent with the above results, the dbscSNV ADA and RF scores were 0.9999 and 0.9840, respectively, thus it was harmful based on the criteria of the ratio value >0.6. However, functional characterization is required to confirm its pathogenicity. Both the father and grandfather had the same gene variant, while the mother had a wild-type gene.
Patient B: A 9-year-old female who visited the clinic in August 2019 for outpatient treatment due to “growth delay for over six years”. The patient had full-term labor without birth asphyxia rescue history. Her birth weight and length were 2.9 kg and 50 cm, respectively, and she was healthy. In August 2019, her height was 123.2 cm (−1.88SD), weight, 27 kg, had breast stage B2, and pubic hair stage Ph1. The heights of the father, mother, grandmother, and aunt were 155 cm (−2.90SD), 157 cm (−0.67SD), 140 cm (−3.81SD), and 148 cm (−2.33SD), respectively (shown in Figure 1C). The growth hormone stimulation tests were normal. Besides, blood routine, blood gas analysis, urine routine, cortisol, adrenocorticotropic hormone, thyroid function, fasting blood glucose, glycosylated hemoglobin, fasting insulin, liver and kidney lipid, glucose and electrolytes, and hepatitis series were normal.
Gene detection: NPR2 NM_003995.4:c.2298_2299delinsTT; p.(Glu766_Arg767delinsAsp) (shown in Figure 1D). The father and grandmother had the same gene variant, while the mother had a wild-type gene.
The height of patient A increased by 5.7 cm (0.36SDS) after half a year of rhGH treatment, and that of patient B increased by 15.8 cm (1.22SDS) after one and a half years of treatment (shown in Figure 2). Both patients showed no serious adverse reactions. Gonadotropin-releasing hormone analog (GnRHa) was added to patient B to inhibit the growth rate due to rapid pubertal progress for one year. Thyroid function, sex hormone level, blood glucose, blood lipid, glycosylated hemoglobin, and other examinations (every 3 months) were normal in both patients during treatments. Bone age examination (every six months) also showed no increased bone age.
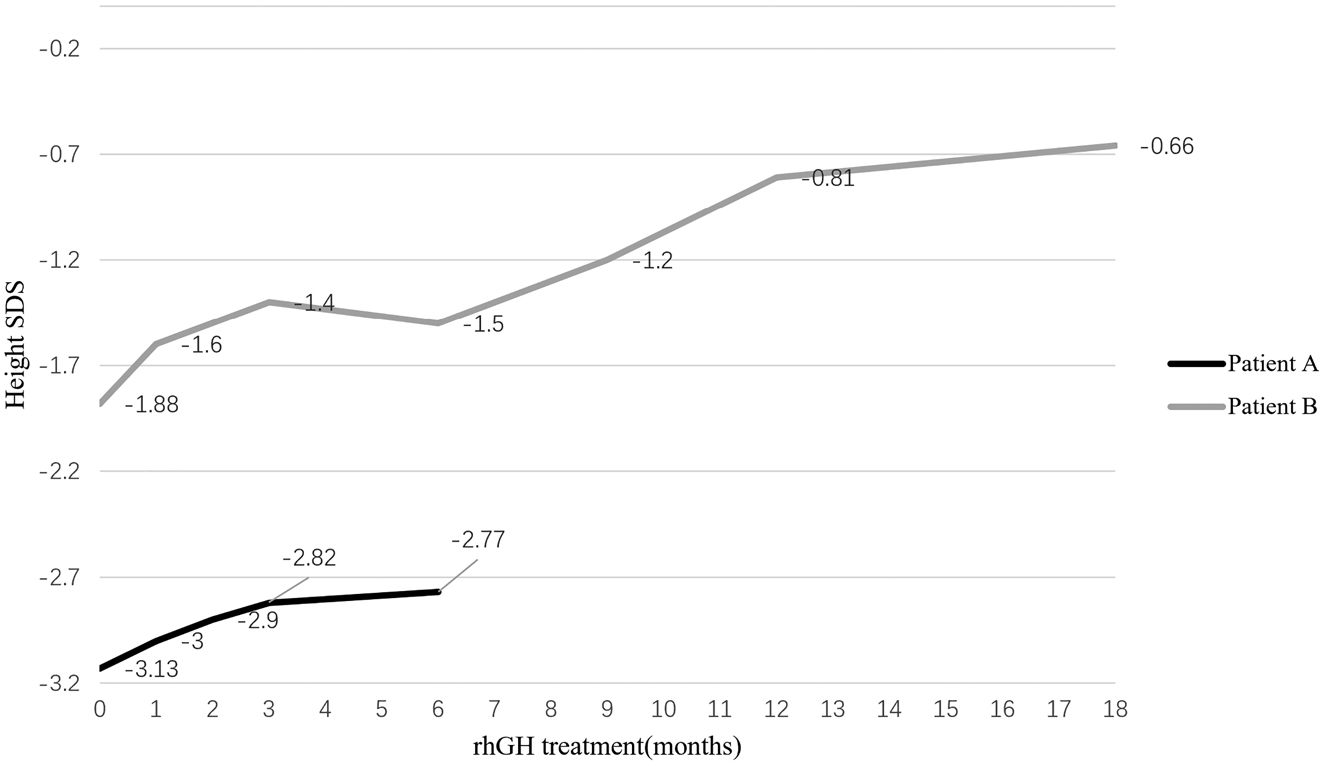
rhGH treatment response in two children with NPR2 gene variants.
Discussion
Height can be used as an indicator to describe the growth and development of children. Several factors, such as heredity, environment, endocrine, and nutrition, determine the height. However, human height mainly depends on the growth of long bones via endochondral osteogenesis. The growth hormone, IGF-1, and other external factors regulate proliferation and differentiation of cartilage growth plate in the epiphysis and metaphysis, forming a secondary ossification center, promoting the longitudinal growth of long bone. The growth plate chondrocytes regulate the normal growth of long bone through proliferation, differentiation, and mineralization. The cartilage matrix on the surface of the growing cartilage plate also forms new bone. Besides growth plate chondrocytes, systemic regulatory factors are also essential for longitudinal bone growth. In the past, the GH-IGF-1 axis was known to be the major factor influencing height growth. However, with in-depth research, other genes, such as the NPR2, have been identified to affect height growth. NPR2 is located in chromosome 9p13.3 with 22 exons, 16.5 kb long, and an mRNA length of 3,000 bp. NPR2 encodes natriuretic peptide receptor type B [9], and its extracellular domain binds C-type natriuretic peptide (CNP). CNP is a member of the natriuretic peptide family, located in various body tissues, such as vascular endothelium, nerve, kidney, especially cartilage. CNP has an autocrine and paracrine effect on the hypertrophic bone growth plate region, promoting bone matrix synthesis and stimulating the proliferation and differentiation of cartilage [10]. NPR2 mRNA is expressed in the brain, adrenal gland, kidney, uterus, ovary, lung, and cartilage. Besides, NPR2 is the major brain natriuretic peptide receptor. NPR2 mRNA can also be detected in the cortex, pituitary, cerebellum, and whole nerve axis. NPR2 mRNA is highly expressed in the limbic cortex, hippocampus, neocortex, amygdala, and olfactory bulb [11].
Previous studies have shown that the NPR2 variant causes short stature via several factors. For instance, Bartels et al. identified the NPR2 variant in acromesomelic dysplasia, type maroteaux (AMDM) patients [12]. Olney et al. also showed that the homozygous variant and the compound heterozygotes variant of the NPR2 cause AMDM [13]. Homozygous and compound heterozygotes variants of the NPR2 block the CNP-NPRB signaling pathway, leading to bone dysplasia (AMDM). In the last five years, several in-depth studies have shown that the NPR2 variant causes AMDM [14], [15], [16].
The NPR2 variant is also found in the 3% of individuals with a phenotype similar to LWD, characterized by symmetrical shortness of forearms and limbs. LWD patients have shortened and arched bilateral radii and subluxated distal ulna. Previous studies have shown that SHOX variants or their enhancers can be detected in about 60% of LWD patients. However, the molecular mechanism of the remaining 40% of LWD patients is unknown [17], [18], [19]. Hisako Oliva et al. showed that about 3% of the 173 LWD patients with SHOX variant-negative had NPR2 variant. Therefore, NPR2 detection is necessary for LWD patients with SHOX variant-negative [20].
Some NPR2 heterozygous variants have also been found in ISS. Vasques et al. showed 6% heterozygous variant rate of NPR2 in 47 Brazilian ISS patients. However, further research is needed to determine the exact variant frequency of the NPR2 and its related mechanism in ISS patients [21].
Besides, few FSS-related studies have reported on the NPR2 variant. Currently, Wang et al. have shown that the NPR2 functional deletion variant is found in 13.6% of FSS cases [22]. Lukas et al. also reported five cases (5.7%) of FSS children with NPR2 variant. However, its mechanism is unknown. This study showed that the NPR2 variant causes FSS, similar to the cases reported in this paper. The children had no typical bone dysplasia and responded well to rhGH treatment [23].
To date, no study has reported on the NPR2 variant rate in Chinese FSS. In this study, 12.5% of FSS children had possible pathogenic variants. Treatment of ISS caused by NPR2 variants was also effective. Previous researches have shown that the treatment effect of rhGH varies among different patients, possibly due to different IGF-1 or IGFBP-3 levels [7, 20, 22, 24]. Vaeques et al. showed that rhGH treatment is effective on ISS patients caused by NPR2 variant, with height SDS between −0.3 and +1.8 [21], similar to Ke et al. study. rhGH treatment increased average height SDS by 1.1 ± 0.7. Besides, age at the beginning of treatment was negatively correlated with treatment efficacy [24].
Lukas et al. showed that all FSS patients (NPR2 variant) respond positively to rhGH treatment [23]. Recent studies have shown that rhGH combined with GnRHa is not effective on some patients with NPR2 variant diagnosed as adolescent short stature [25]. However, a further large-scale study is necessary to validate our findings. Notably, the Food and Drug Administration has not approved rhGH to treat FSS patients caused by NPR2 variants. Moreover, patients should be closely monitored to note the changes during treatment.
Patient A had a synonymous gene variant, which is pathogenic since it significantly affects protein expression by splicing mRNA precursor or the stability of mRNA secondary structure [26, 27]. Besides, the impact on translation efficiencies, such as the accuracy or translation speed, causes diseases. In recent years, synonymous variants have been shown to cause several diseases, such as single nucleotide polymorphisms. However, a further in-depth study is necessary to determine the pathogenesis of the study results. According to the classification of variants using ACMG guidelines, we found that c.2643G>A; p.(Gln881=) would be PM1, PM2, and PP3 so would be classified as a VUS although it looks very positive. And c.2298_2299delinsTT; p.(Glu766_Arg767delinsAsp) would be PVS1, PM1, PM2 so would be pathogenic. Other genes variants such as BGN, DSPP, DUOX2, and PCYT1A were also found in the trio-WES examination, but the evidence of pathogenicity was insufficient through ACMG analysis.
Unfortunately, all the studies on FSS caused by the NPR2 variant are case reports with no in-depth research on its mechanisms or factors. Furthermore, the studies have no agreed treatment guidelines, and only a few describe rhGH therapy. Moreover, different case reports have substantial heterogeneity, with no unified standards and programs for the timing, dose, duration, and efficacy evaluation of treatment.
In this study, rhGH therapy was effective in FSS children. The growth rate was significantly improved in the first year, and the effect was similar to that of other children with SHOX deficiency. However, further statistical study is needed. Presently, most children have not reached the age-end height, and further follow-up study is needed to guide the next treatment step.
However, this research has some limitations. a) Only one research center was included b) with small population distribution. The subjects were all Han population in East China, and no other races or nationalities were included. c) The study had a small sample size; only 16 children were included. Therefore, a large sample study is necessary. d) There was no further functional verification or animal experiments. The results were only pathogenic manifestations.
However, several pathogenic genes or loci can be found due to advanced gene research, necessitating increased investment for genetic research.
Funding source: Key Research and Development Program of Zhejiang Province
Award Identifier / Grant number: 2020C03121
Acknowledgments
The authors would like to thank the proband, their family, and the control subjects for providing blood samples and agreeing to participate in this research. The authors thank Zhejiang University for providing a platform.
-
Research funding: This research was supported by grants from the Key Research and Development Program of Zhejiang Province with grant number: 2020C03121.
-
Author contributions: Yuan Ke and Wang Chunlin conceived and designed the study. Yuan Ke and Chen Jiao performed the experiments. Chen Qingqing, Fang Yanlan and Zhu Jianfang provided clinical research. Yuan Ke wrote the article. Yuan Ke, Chen Hong and Wang Chunlin reviewed and edited the manuscript. All authors read and approved the manuscript.
-
Competing interests: The authors declare that they have no conflicts of interest related to this manuscript.
-
Informed consent: Informed consent was obtained from all individuals included in this study.
-
Ethical approval: The Research Ethics Committee of First Affiliated Hospital, College of Medicine, Zhejiang University approved the study. Written informed consent was obtained from the parents. Reference number of the research ethics committee of the First Affiliated Hospital, College of Medicine, Zhejiang University: 2020-727.
References
1. Ismail, H, Ness, K. Evaluation of short stature in children. Pediatr Ann 2013;42:217–22. https://doi.org/10.3928/00904481-20131022-08.Suche in Google Scholar PubMed
2. Cohen, P, Rogol, AD, Deal, CL, Saenger, P, Reiter, EO, Ross, JL, et al.. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab 2008;93:4210–7. https://doi.org/10.1210/jc.2008-0509.Suche in Google Scholar PubMed
3. Poyrazoglu, S, Darendeliler, F, Bas, F, Bundak, R, Saka, N, Darcan, S, et al.. Target height estimation in children with idiopathic short stature who are referred to the growth clinic. Horm Res 2009;72:178–83. https://doi.org/10.1159/000232494.Suche in Google Scholar PubMed
4. Song, KC, Jin, SL, Kwon, AR, Chae, HW, Ahn, JM, Kim, DH, et al.. Etiologies and characteristics of children with chief complaint of short stature. Ann Pediatr Endocrinol Metab 2015;20:34–9. https://doi.org/10.6065/apem.2015.20.1.34.Suche in Google Scholar PubMed PubMed Central
5. Blum, WF, Alherbish, A, Alsagheir, A, El, AA, Kaplan, W, Koledova, E, et al.. The growth hormone-insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocr Connect 2018;7:R212–22. https://doi.org/10.1530/ec-18-0099.Suche in Google Scholar PubMed PubMed Central
6. Teran, E, Chesner, J, Rapaport, R. Growth and growth hormone: an overview. Growth Horm Igf Res 2016;28:3–5. https://doi.org/10.1016/j.ghir.2016.02.004.Suche in Google Scholar PubMed
7. Vasques, GA, Arnhold, IJ, Jorge, AA. Role of the natriuretic peptide system in normal growth and growth disorders. Horm Res Paediatr 2014;82:222–9. https://doi.org/10.1159/000365049.Suche in Google Scholar PubMed
8. Poyrazoglu, S, Darendeliler, F, Bas, F, Bundak, R, Saka, N, Darcan, S, et al.. Target height estimation in children with idiopathic short stature who are referred to the growth clinic. Horm Res 2009;72:178–83. https://doi.org/10.1159/000232494.Suche in Google Scholar
9. Potter, LR, Abbey-Hosch, S, Dickey, DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev 2006;27:47–72. https://doi.org/10.1210/er.2005-0014.Suche in Google Scholar PubMed
10. Khan, S, Basit, S, Khan, MA, Muhammad, N, Ahmad, W. Genetics of human isolated acromesomelic dysplasia. Eur J Med Genet 2016;59:198–203. https://doi.org/10.1016/j.ejmg.2016.02.011.Suche in Google Scholar PubMed
11. Herman, JP, Dolgas, CM, Rucker, D, Langub, MJ. Localization of natriuretic peptide-activated guanylate cyclase mRNAs in the rat brain. J Comp Neurol 1996;369:165–87. https://doi.org/10.1002/(sici)1096-9861(19960527)369:2<165::aid-cne1>3.0.co;2-1.10.1002/(SICI)1096-9861(19960527)369:2<165::AID-CNE1>3.0.CO;2-1Suche in Google Scholar
12. Bartels, CF, Bukulmez, H, Padayatti, P, Rhee, DK, van Ravenswaaij-Arts, C, Pauli, RM, et al.. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet 2004;75:27–34. https://doi.org/10.1086/422013.Suche in Google Scholar
13. Olney, RC. C-type natriuretic peptide in growth: a new paradigm. Growth Horm Igf Res 2006;16(A Suppl):S6–14. https://doi.org/10.1016/j.ghir.2006.03.016.Suche in Google Scholar
14. Srivastava, P, Tuteja, M, Dalal, A, Mandal, K, PS, R. Novel mutations in the transmembrane natriuretic peptide receptor NPR-B gene in four Indian families with acromesomelic dysplasia, type Maroteaux. J Genet 2016;95:905–9. https://doi.org/10.1007/s12041-016-0715-1.Suche in Google Scholar
15. Lin, WD, Wang, CH, Tsai, FJ. Identification of one novel homozygous mutation in the NPR2 gene in a patient from Taiwan with acromesomelic dysplasia Maroteaux type. Pediatr Neonatol 2018;59:322–3. https://doi.org/10.1016/j.pedneo.2017.11.017.Suche in Google Scholar
16. Ain, NU, Iqbal, M, Valta, H, Emerling, CA, Ahmed, S, Makitie, O, et al.. Novel variants in natriuretic peptide receptor 2 in unrelated patients with acromesomelic dysplasia type Maroteaux. Eur J Med Genet 2019;62:103554. https://doi.org/10.1016/j.ejmg.2018.10.006.Suche in Google Scholar
17. Huber, C, Rosilio, M, Munnich, A, Cormier-Daire, V. High incidence of SHOX anomalies in individuals with short stature. J Med Genet 2006;43:735–9. https://doi.org/10.1136/jmg.2006.040998.Suche in Google Scholar
18. Benito-Sanz, S, Del, BD, Aza-Carmona, M, Magano, LF, Lapunzina, P, Argente, J, et al.. PAR1 deletions downstream of SHOX are the most frequent defect in a Spanish cohort of Leri-Weill dyschondrosteosis (LWD) probands. Hum Mutat 2006;27:1062. https://doi.org/10.1002/humu.9456.Suche in Google Scholar
19. Chen, J, Wildhardt, G, Zhong, Z, Roth, R, Weiss, B, Steinberger, D, et al.. Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250 kb downstream regulatory domain. J Med Genet 2009;46:834–9. https://doi.org/10.1136/jmg.2009.067785.Suche in Google Scholar
20. Hisado-Oliva, A, Garre-Vazquez, AI, Santaolalla-Caballero, F, Belinchon, A, Barreda-Bonis, AC, Vasques, GA, et al.. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Leri-Weill dyschondrosteosis. J Clin Endocrinol Metab 2015;100:E1133–42. https://doi.org/10.1210/jc.2015-1612.Suche in Google Scholar
21. Vasques, GA, Amano, N, Docko, AJ, Funari, MF, Quedas, EP, Nishi, MY, et al.. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. J Clin Endocrinol Metab 2013;98:E1636–44. https://doi.org/10.1210/jc.2013-2142.Suche in Google Scholar PubMed
22. Wang, SR, Jacobsen, CM, Carmichael, H, Edmund, AB, Robinson, JW, Olney, RC, et al.. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum Mutat 2015;36:474–81. https://doi.org/10.1002/humu.22773.Suche in Google Scholar PubMed PubMed Central
23. Plachy, L, Dusatkova, P, Maratova, K, Petruzelkova, L, Zemkova, D, Elblova, L, et al.. NPR2 variants are frequent among children with familiar short stature and respond well to growth hormone therapy. J Clin Endocrinol Metab 2020;105. https://doi.org/10.1210/clinem/dgaa037.Suche in Google Scholar PubMed
24. Ke, X, Liang, H, Miao, H, Yang, H, Wang, L, Gong, F, et al.. Clinical characteristics of short-stature patients with an NPR2 mutation and the therapeutic response to rhGH. J Clin Endocrinol Metab 2021;106:431–41. https://doi.org/10.1210/clinem/dgaa842.Suche in Google Scholar PubMed
25. Hanley, PC, Kanwar, HS, Martineau, C, Levine, MA. Short stature is progressive in patients with heterozygous NPR2 mutations. J Clin Endocrinol Metab 2020;105. https://doi.org/10.1210/clinem/dgaa491.Suche in Google Scholar PubMed PubMed Central
26. Parmley, JL, Chamary, JV, Hurst, LD. Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol Biol Evol 2006;23:301–9. https://doi.org/10.1093/molbev/msj035.Suche in Google Scholar PubMed
27. Duan, J, Wainwright, MS, Comeron, JM, Saitou, N, Sanders, AR, Gelernter, J, et al.. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet 2003;12:205–16. https://doi.org/10.1093/hmg/ddg055.Suche in Google Scholar PubMed
© 2021 Ke Yuan et al., published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- Editorial
- Obesity after the Covid-19 pandemic and beyond
- Review Article
- Clinical profile and management challenges of disorders of sex development in Africa: a systematic review
- Original Articles
- Development and validation of a mobile application for point of care evaluation of growth failure
- Children-Dietary Inflammatory Index (C-DII), cardiometabolic risk, and inflammation in adolescents: a cross-sectional study
- Accelerated pubertal onset in short children with delayed bone age
- Screening for hypophosphatasia: does biochemistry lead the way?
- Subcutaneous regular insulin use for the management of diabetic ketoacidosis in resource limited setting
- NPR2 gene variants in familial short stature: a single-center study
- The effect of the COVID-19 pandemic on metabolic control in children with type 1 diabetes: a single-center experience
- Evaluating a standardized protocol for the management of diabetes insipidus in pediatric neurosurgical patients
- Development and assessment of a low-health-literacy, pictographic adrenal insufficiency action plan
- Effect of insulin resistance on lung function in asthmatic children
- A major health problem facing immigrant children: nutritional rickets
- Clinical profile, etiology, and diagnostic challenges of primary adrenal insufficiency in Sudanese children: 14-years’ experience from a resource limited setting
- Non-inferiority of liquid thyroxine in comparison to tablets formulation in the treatment of children with congenital hypothyroidism
- Short Communication
- Increased frequency of idiopathic central precocious puberty in girls during the COVID-19 pandemic: preliminary results of a tertiary center study
- Case Reports
- Gordon syndrome caused by a CUL3 mutation in a patient with short stature in Korea: a case report
- Nitisinone treatment during two pregnancies and breastfeeding in a woman with tyrosinemia type 1 – a case report
- Myxedema crisis and ovarian hyperstimulation in a child with Down syndrome
- First successful concomitant therapy of immune tolerance induction therapy and desensitization in a CRIM-negative infantile Pompe patient
Artikel in diesem Heft
- Frontmatter
- Editorial
- Obesity after the Covid-19 pandemic and beyond
- Review Article
- Clinical profile and management challenges of disorders of sex development in Africa: a systematic review
- Original Articles
- Development and validation of a mobile application for point of care evaluation of growth failure
- Children-Dietary Inflammatory Index (C-DII), cardiometabolic risk, and inflammation in adolescents: a cross-sectional study
- Accelerated pubertal onset in short children with delayed bone age
- Screening for hypophosphatasia: does biochemistry lead the way?
- Subcutaneous regular insulin use for the management of diabetic ketoacidosis in resource limited setting
- NPR2 gene variants in familial short stature: a single-center study
- The effect of the COVID-19 pandemic on metabolic control in children with type 1 diabetes: a single-center experience
- Evaluating a standardized protocol for the management of diabetes insipidus in pediatric neurosurgical patients
- Development and assessment of a low-health-literacy, pictographic adrenal insufficiency action plan
- Effect of insulin resistance on lung function in asthmatic children
- A major health problem facing immigrant children: nutritional rickets
- Clinical profile, etiology, and diagnostic challenges of primary adrenal insufficiency in Sudanese children: 14-years’ experience from a resource limited setting
- Non-inferiority of liquid thyroxine in comparison to tablets formulation in the treatment of children with congenital hypothyroidism
- Short Communication
- Increased frequency of idiopathic central precocious puberty in girls during the COVID-19 pandemic: preliminary results of a tertiary center study
- Case Reports
- Gordon syndrome caused by a CUL3 mutation in a patient with short stature in Korea: a case report
- Nitisinone treatment during two pregnancies and breastfeeding in a woman with tyrosinemia type 1 – a case report
- Myxedema crisis and ovarian hyperstimulation in a child with Down syndrome
- First successful concomitant therapy of immune tolerance induction therapy and desensitization in a CRIM-negative infantile Pompe patient