Novel mutations of the SRD5A2 and AR genes in Thai patients with 46, XY disorders of sex development
-
Chupong Ittiwut
 ,
Sasipa Muensri
,
Sasipa Muensri

Abstract
Background:
Abnormalities of dihydrotestosterone conversion [5α-reductase deficiency: online Mendelian inheritance in man (OMIM) 607306] or actions of androgens [partial androgen insensitivity syndrome (PAIS): OMIM 312300] during the 8th–12th weeks of gestation cause varying degrees of undervirilized external genitalia in 46, XY disorders of sex development (DSD) with increased testosterone production. The objective of the study was to determine clinical and genetic characteristics of Thai patients with 46, XY DSD.
Methods:
A cross-sectional study was conducted in 46, XY DSD with increased testosterone production (n=43) evaluated by a human chorionic gonadotropin (hCG) stimulation test or clinical features consistent with 5α-reductase deficiency or PAIS. PCR sequencing of the entire coding regions of the SRD5A2 and AR genes was performed. Molecular modeling analysis of the androgen receptor-ligand-binding domain (AR-LBD) of a novel mutation was constructed.
Results:
Mutations were found in seven patients (16.3%): five (11.6%) and two (4.7%) patients had mutations in SRD5A2 and AR, respectively. Two novel mutations, SRD5A2 c.383A>G (p.Y128C) and AR c.2176C>T (p.R726C), were identified. Dimensional structural analysis of the novel mutated AR (p.R726C) revealed that it affected the co-activator binding [binding function-3 (BF-3)], not the testosterone binding site. Short phallus length was associated with 5α-reductase deficiency.
Conclusions:
Around 16.3% of our patients with 46, XY DSD had 5α-reductase deficiency or PAIS. Two novel mutations of SRD5A2 and AR were identified. The novel mutated AR (p.R726C) might affect the co-activator binding (BF-3), not the testosterone binding site.
Introduction
The differentiation of male external genitalia requires an adequate amount of testosterone and its 5α-reduced derivative, dihydrotestosterone (DHT), which mediates their actions through androgen receptors [1]. Abnormalities of DHT conversion [5α-reductase deficiency: online Mendelian inheritance in man (OMIM) 607306] or actions of androgens [partial androgen insensitivity syndrome (PAIS): OMIM 312300] during the 8th–12th weeks of gestation, the critical period of external genitalia formation, cause varying degrees of undervirilized external genitalia in 46, XY disorders of sex development (DSD) with normal testosterone production.
The AR gene is located on chromosome Xq11.2–q12 and consists of eight exons that encode the androgen receptor. The androgen receptor is a member of the intracytoplasmic nuclear receptor superfamily [2]. Inactivation mutations of the AR gene are the most common cause of 46, XY DSD and are associated with the variation of phenotypes ranging from complete female external genitalia [complete androgen insensitivity syndrome (CAIS)] to some degree of undervirilization (PAIS). To date, the AR gene mutations database (ARDB) (http://www.mcgill.ca/androgendb/) has reported more than 500 different AR mutations from patients with AIS. Differences in types and locations of AR mutations have been reported without genotype and phenotype correlations [3].
The SRD5A2 gene is located on chromosome 5p15 [4] and consists of five exons that encode the 5α-reductase enzyme. Inactivation mutations of the SRD5A2 gene are rare causes of 46, XY DSD and are frequently associated with clitoromegaly and microphallus with varying degrees of hypospadias [5]. More than 100 mutations in the SRD5A2 gene have been reported to date [Human Gene Mutation Database (HGMD), www.hgmd.cf.ac.uk/].
The prevalence of PAIS and 5α-reductase deficiency among 46, XY DSD has been reported to be 9.8%–28% and 12%–15.5%, respectively [1], [6], [7], [8]. Even with extensive investigations, the genetic defects of at least 50% of 46, XY DSD patients were elusive. Identification of the causative mutations is crucially important due to the differences in the natural history of the disease itself, gender assignment and mode of inheritance. The three-dimensional structure of the androgen receptor-ligand-binding domain (AR-LBD) was first crystalized in the year 2000 [9]. Nowadays, many complex structures with several ligands and co-activators are deposited into the protein databank (PDB). The aim of this study was to determine the prevalence of PAIS and 5α-reductase deficiency, to define features which can differentiate PAIS from 5α-reductase deficiency in Thai patients with 46, XY DSD and to determine the protein structure of AR-LBD of the newly identified mutations by molecular modeling analysis.
Materials and methods
Subjects
All patients with ambiguous genitalia who had a 46, XY karyotype and an increased testosterone production after stimulation with human chorionic gonadotropin (hCG) or clinical features consistent with 5α-reductase deficiency or PAIS were recruited between 2010 and 2014. The hCG stimulation test was done by three daily intramuscular injections of 1500 IU–hCG. Testosterone and DHT were drawn before administration of the first hCG (basal samples) and 1 day after the third hCG (post-stimulation samples). All patients were followed at the Division of Pediatric Endocrinology, Department of Pediatrics, King Chulalongkorn Memorial Hospital. Patients 1 and 2 were previously reported by Sahakitrungruang et al. [10].
Ethical approval
Informed consent and assent was obtained from each parent and individual participant, 7 years and older, respectively. The research protocol was approved by the institutional committees and all procedures performed in this study involving human participants were in accordance with the ethical standards of the Institutional Review Board of Faculty of Medicine, Chulalongkorn University (IRB 332/56) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Study protocol
Clinical and hormonal evaluation data were retrospectively reviewed from the medical records. Phallus length was measured from the tip of the phallus to the mons pubis on the dorsal side of the phallus. The external masculinization score (EMS) was determined by the presence of the following criteria: scrotal fusion, micropenis, urethral meatus and position of gonads [11].
Assays
Serum testosterone and DHT were assayed using a chemiluminescent immunoassay.
Genetic analysis
PCR amplifications using primers designed to amplify the entire coding regions of the SRD5A2 (Transcript SRD5A2-001; ENST00000622030) and AR (Transcript AR-001; ENST00000374690) genes (Supplementary Table 1) were performed on the genomic DNA extracted from ethylenediaminetetraacetic acid (EDTA) peripheral blood of the patients. The amplicons were then electrophoresed using 2% agarose gel and subsequently sent for sequencing by Macrogen Inc. (Beotkkot-ro Geumcheon-gu, Seoul, South Korea) in both forward and reverse directions.
Molecular modeling analyses of human AR-LBD mutants
The models of human AR-LBD mutants described herein were constructed using the resolved X-ray structures of human AR-LBD in complex with testosterone (PDB code 2AM9; 1.64 Å resolution) as starting templates [12]. As a first preparation step, the crystallographic water molecules were removed from the structure. Then harmonic restraint was assigned to the atoms in the protein backbone with a force constant of 50 kcal/mol. A minimization was performed using 3000 steps of the steepest descent method, followed by 3000 steps of the conjugate gradient method for structural convergence toward a local minimum within the CHARMM force field in vacuum. The MODELER program was used to mutate residues to specified types and optimize the conformation of both the mutated residues and any surrounding residues that lie within a 4.5 Å radius. The best mutated model was selected according to the best PDF Total Energy of 10 generated models. The mutant AR-LBD model was then minimized using the same protocol as described for the wild type AR-LBD. All calculations, visualization and analyses were examined using Discovery Studio 3.5 (Accelrys Software Inc)., Discovery Studio Modeling Environment, Release 3.5, San Diego: Accelrys Software Inc., 2013, which was kindly provided by Dr. Jiraporn Chingunpitak, School of Pharmacy, Walailak University, Thailand.
Statistical analysis
Data of each group were expressed as mean and standard deviation (SD). The ANOVA test was used for continuous variables among three different groups. Statistical significance was established at p<0.05 and 80% of power. All statistical analyses were conducted using the SPSS version 17 software (SPSS: An IBM Company, New York, USA).
Results
Forty-three patients with 46, XY DSD with increased testosterone production or clinical features consistent with 5α-reductase deficiency or PAIS were enrolled in the study and divided into three groups according to the mutated genes (Table 1): SRD5A2 (n=5, 11.6%), AR (n=2, 4.7%) and undefined group (n=36, 83.7%).
Clinical, hormonal and genetic data of patients with 5α-reductase deficiency and PAIS.
| Diagnosis | Case no. | Age at hormonal evaluation, year | Genital phenotype | hCG-stimulated | ΔT, ng/dL | T:DHT | Sex of rearing | SRD5A2 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Phallus length, cm | EM S | T, ng/dL | DHT, ng/d L | DNA | Protein | ||||||
| 5α-reductase deficiency | 1 | 2 | 0.5 | 6 | 585 | 8.49 | 582.2 | 68.9 | Female | c.16C>T/c.607G>A | p.Q6X/p.G203S |
| 2 | 2 | 0.5 | 3 | N.D. | N.D. | N.D. | N.D. | Female | c.59T>C/c.547G>A | p.L20P /p.G183S | |
| 3 | 2 | 1.5 | 6 | N.D. | N.D. | N.D. | N.D. | Female | c.383A>G/c.680G>A | p.Y128Ca/p.R227Q | |
| 4 | 4 | 1.5 | 6 | 134 | 8.2 | 131 | 16.34 | Female | c.607G>A/c.680G>A | p.G203S/p.R227Q | |
| 5 | 3 | 1.8 | 7 | 311 | 21.89 | 250.6 | 14 | Male | c.607G>A/c.680G>A | p.G203S/p.R227Q | |
| Diagnosis | Case no. | Age at hormonal evaluation, year | Genital phenotype | hCG stimulated- | ΔT, ng/dL | T:DHT | Sex of rearing | AR | |||
| Phallus length, cm | EMS | T, ng/dL | DHT, ng/dL | DNA | Protein | ||||||
| PAIS | 6 | 1 | 1.3 | 7 | 907.2 | 25 | 898.56 | 36.3 | Male | c.1706G>T | G569V |
| 7 | 4 | 3 | 8 | 233 | 12.56 | 232.28 | 18.6 | Male | c.2176C>T | R726Ca | |
hCG, human chorionic-gonadotropin; EMS, external masculinization score; DHT, dihydrotestosterone; T, testosterone; T/DHT, testosterone/dihydrotestosterone ratio; AR, androgen receptor; SRD5A2, 3-oxo-5 alpha-steroid delta 4-dehydrogenase α 2; N.D., no data. anovel mutation.
Gender of rearing, assigned before mutations were identified, was male in both the patients with PAIS. Of the five patients with 5α-reductase deficiency, four were raised as female. Two of the four were incorrectly diagnosed with PAIS and underwent gonadectomy during childhood before the mutations were found. Unfortunately, the gonadal histology was not available. Estrogen was supplemented at the appropriate ages. However, the gender identity of all these five patients with 5α-reductase deficiency was male. Theoretically, the appropriate gender assignment of patients with 5α-reductase deficiency should be male due to the virilization during the pubertal period by 5α-reductase enzyme type 1.
Two different mutations in the AR gene were identified in two unrelated patients (Table 1). Of these, a novel missense mutation, p.R726C (c.2176C>T) in the LBD, was identified in one patient. The arginine (R-basic amino acid) at residue 127 was substituted by cysteine (C-polar amino acid). The p.R727C was predicted to be damaged with a score of 1.00 and 0.00 by Polyphen 2 and SIFT, respectively (Supplementary Table 2). The AR-LBD consists of 11α-helices and four short β-strands forming two anti-parallel β-sheets. The structure is arranged in a three-layer, antiparallel α-helical sandwich. A ligand-binding pocket (LBP) is surrounded by the N termini of H3, H5 and H11 (Figure 1). The AR-LBD harbors a major co-activator binding surface [activation function-2 (AF-2)], which acts as a docking site for short hydrophobic peptide motifs (NR boxes) featured in AR co-activators and in the AR-NTD and mediates AR functional amino/carboxy (N/C)-terminal interaction [13], [14], [15], [16], [17]. An additional surface cleft of AR-LBD called binding function-3 (BF-3) was found to allosterically regulate AF-2 co-activator binding [17], [18]. Moreover, a series of residues from BF-3, the boundary of BF-3/AF-2 and AF-2 are structurally interconnected and allosterically coupled. In addition, several residues belonging to BF-3 and AF-2 surface pockets are key players of an allosteric network that may influence multiple aspects of AR-LBD function [19]. The new AR p.R727C mutation residue is located at the boundary between AF-2 and BF-3, which are the parts of both pockets (Figure 1). The computational modeling of the wild-type (WT) (R726) and mutant (C726) supports the hypothesis that R726C alters the BF-3 which is an allosteric modulator of the adjacent AF-2 pocket, affecting AR-LBD function. The structural overlay of the WT (R726) and mutant (C726) is illustrated in Figure 1D. The hydrophobic surface analysis clearly indicated that the degree of hydrophobicity was increased when R726 (charged polar basic side chain) was changed to C726 (uncharged polar side chain) (Figure 1E and F).
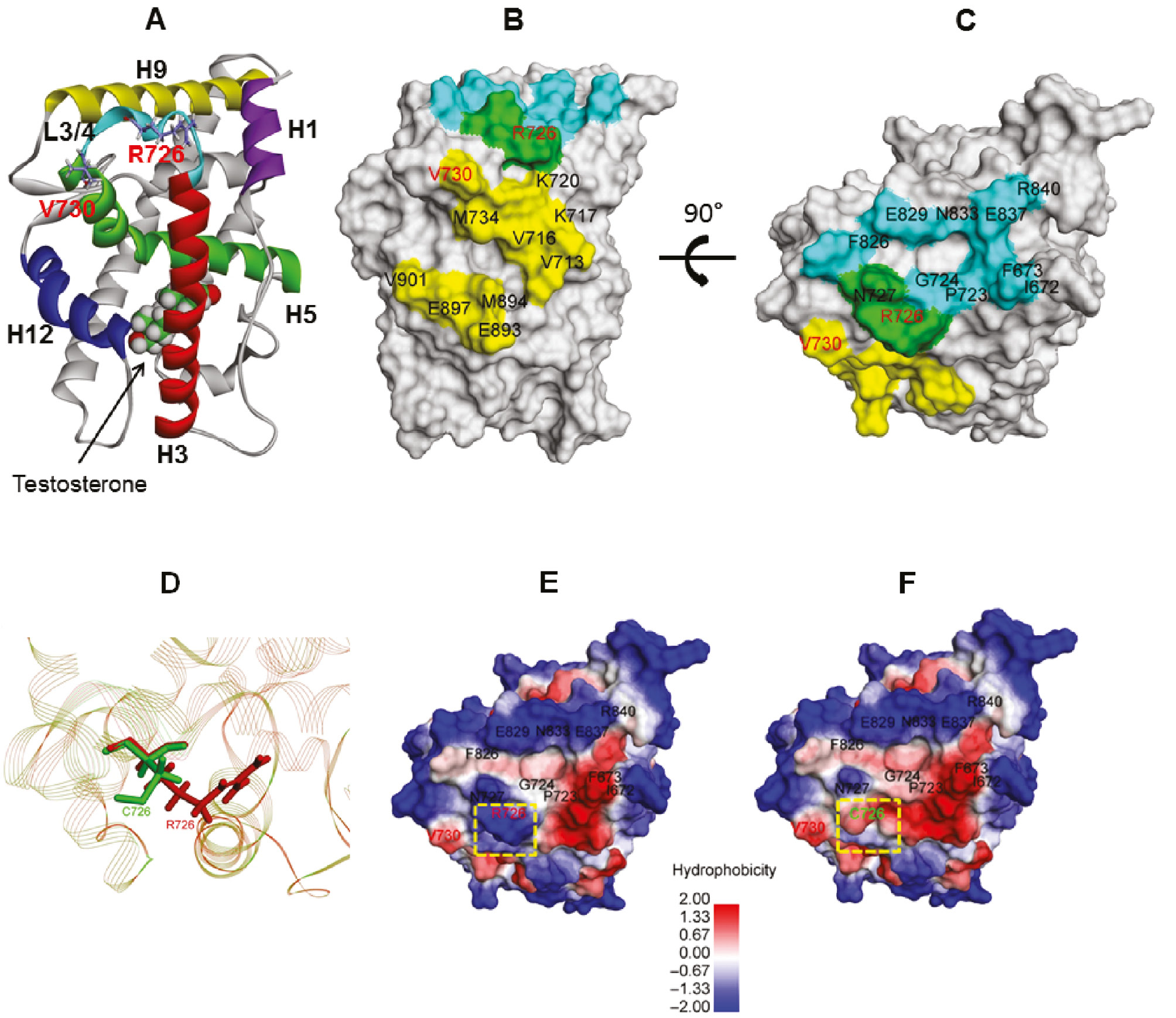
Diagram showing the human AR-LBD crystal structure with a testosterone (PDB accession no. 2am9).
(A) Simplified model representation of the human AR-LBD structure. Helices H1, H3, H4–5, H9 and H12 are depicted as different color helices. Schematic of the AR-LBD showing the location of testosterone. The AF-2 co-activator binding pocket is lined by H3, H5 and H12, whereas the BF-3 pocket is formed by H1, H9 and the loop linking H3 with H4–5 (L3/4). The mutated residues studied herein are shown as sticks. (B) Solid surface representation of the AR-LBD in gray showing the residues lining AF-2 in yellow, the residues lining BF-3 in light blue and the residues R726 and N727 belonging to the boundary between AF-2 and BF-3 pockets shown as a green surface. (C) B rotated 90° to clearly reveal BF-3. Schematic of the human AR-LBD showing the location of the mutated residues. The superposition structure of the WT and R726C mutation (by the RCSB Protein Data Bank system) in the boundary area between AF-2 and BF-3 (D). The WT and mutated residues studied herein are shown as sticks. Hydrophobic surface of the WT (E) and R726C (F), respectively, showing residues lining in the BF-3 pocket.
Six different missense mutations were identified in the SRD5A2 gene in five patients. No patients harbored mutations in both genes. One missense mutation, p.Y128C (c.383A>G), had not been reported previously. It was predicted to be probably damaging (score 0.999) and tolerated (score 0.17) by Polyphen2 and SIFT, respectively (Supplementary Table 2). Unfortunately, his parental DNA was not available. Both AR p.R726C and SRD5A2 p.Y128C mutations were not found in 122 Thai controls and conserved among vertebrate species (Figure 2).
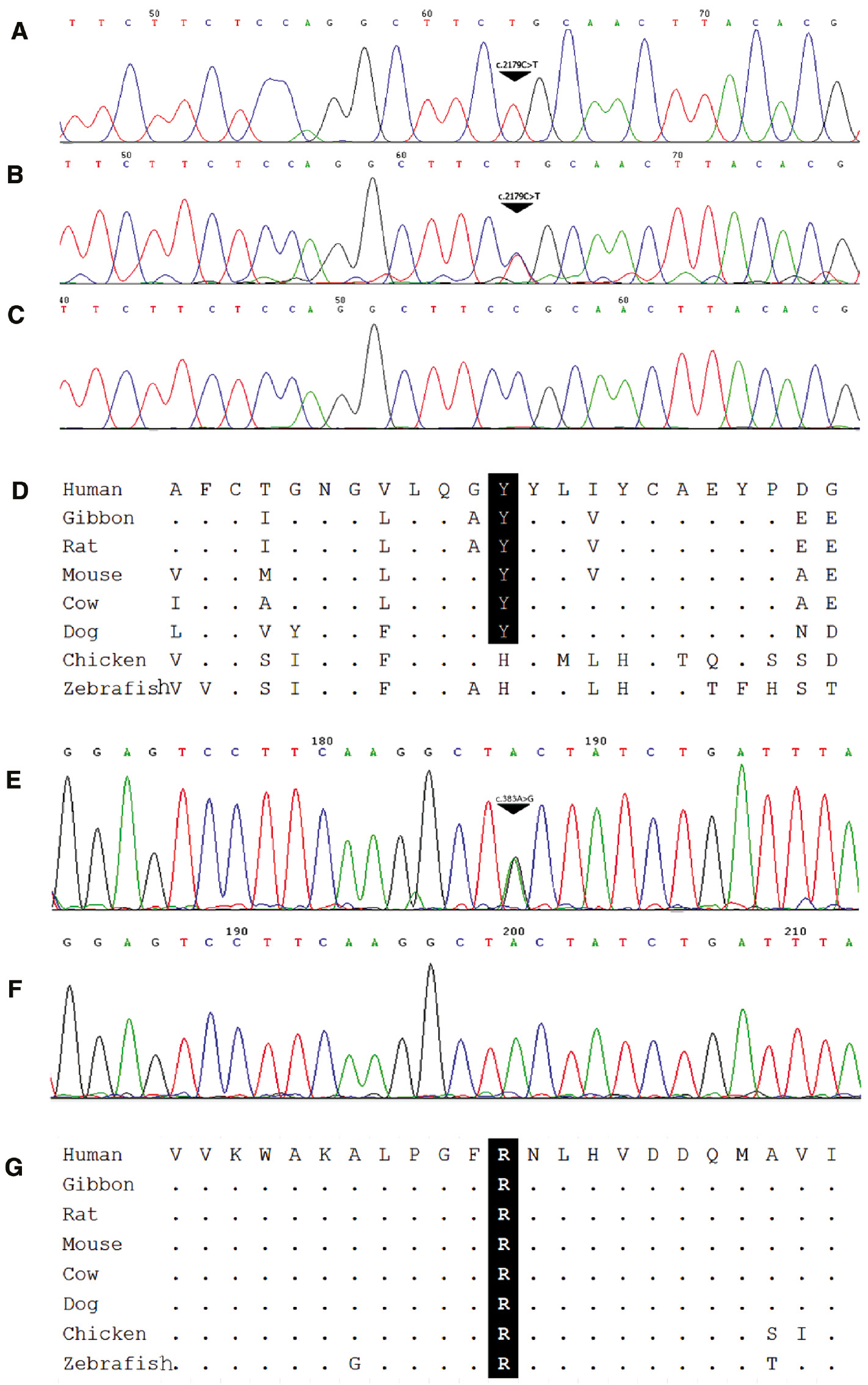
Novel mutations in (A) AR and (B) SRD5A2.
(A) Electropherograms showing hemizygous p.R726C of the AR gene in Patient 7 and his heterozygous mother and normal father (B and C), respectively. Comparative genomic analysis showing that p.R726 is conserved among mammalian species (D). The heterozygous SRD5A2 p.Y128C mutation found in Patient 3 (E) compared to normal control (F) and conserved among species (G).
Discussion
Of our 43 Thai patients with 46, XY DSD, seven (16.3%) were found to have mutations in SRD5A2 or AR. The percentage of PAIS and 5α-reductase deficiency among the 46, XY DSD patients in our study was 4.7 and 11.6%, respectively. The prevalence of PAIS reported in the UK, Spain, Brazil and Turkey was 24%–28%, 9.8%, 17.2% and 22%, respectively [1], [6], [7], [8], [20]. The percentage of 5α-reductase deficiency in this study was consistent with the previous studies in Brazil and Turkey which revealed 15.5% and 12%, respectively [7], [8]. The percentage of both diseases varied from study to study, which might be due to the clinical and hormonal criteria to include patients into the study.
A novel p.Y128C (c.383A>G) mutation in SRD5A2 was found in Patient 3. Previous studies have demonstrated that SRD5A2 mutations in some populations were mainly (60%) homozygous missense [21], [22]. Interestingly, all SRD5A2 mutations in our study were compound heterozygous missense. Patient 3 was compound heterozygous for this newly identified mutation and the other known p.R227Q (c.680G>A) mutation. Patients 1, 2, 4 and 5 were compound heterozygous for two known mutations (Table 1). Patients 4 and 5 were unrelated but harbored the same known mutations, p.G203S (c.607G>A) and p.R227Q (c.680G>A). They had similar genital appearances and hormonal profiles. Notably, gender of rearing was different. Patient 4 was raised as female, while Patient 5 was raised as male. Nonetheless, all the five of our 5α-reductase deficiency patients, including Patient 4, had a strong male identity, assessed by a pediatric psychiatrist during hospital visits (aged between 5 and 7 years). This was reported to be due to the effect of an androgen exposure on postnatal psychological development. p.G203S has been previously reported in Chinese and Koreans (23, 24). Most were compound heterozygous. The clinical and hormonal characteristics and gender of rearing were not different from the other types of mutations of 5α-reductase deficiency. Gender of rearing might be dependent on the age at presentation. Patients were tended to be raised as female if they presented in the early infancy period [23], [24], [25], but as male if presented during pubertal age [26], [27]. Our Patients 2 and 3 were incorrectly diagnosed with PAIS and underwent gonadectomy during childhood. This emphasizes the importance of a definite diagnosis of 5α-reductase deficiency for a proper management and genetic counseling.
A novel mutation, p.R726C (c.2176C>T), in AR was found in Patient 7. Based on the molecular modeling technique, the novel p.R726C mutation within the AR-LBD was shown to alter the propensity of the AF-2 surface to form deep sub-pockets that accommodated bulky side chains of co-regulator motifs. Therefore, the mechanism of AR dysfunction in this mutation was probably mediated through a co-activator binding defect, but not mediated through a testosterone binding site. Interestingly, the same point mutation, but with a different amino acid substitution, p.R726L, was previously reported in a patient with prostate carcinoma due to somatic mutations [28], [29] but not PAIS, which is caused by a germline mutation. It was found that p.R726L marginally reduced the AR-LBD activity in vitro [19]. The mutant p.R727L displayed a moderately higher increase in its interaction with the NTD, but had little effect on corepressor interaction with N-Cor and SMRT. To our knowledge, this is the first time that the novel p.R726C mutation in the human AR-LBD associated with PAIS has been reported.
No mutations in the SRD5A2 and AR genes could be identified in 36 (83.7%) of the 43 patients who were classified as undefined. This is consistent with previous studies showing that mutations in the majority of patients with 46, XY DSD were not identified. Several disorders in 46, XY DSD have a similar PAIS-like phenotype, especially in partial gonadal dysgenesis caused by mutations in many transcription factors such as SRY, SF-1, WT-1 and MAMLD1 [11]. The interesting candidate gene in the backdoor pathway for DHT production, the AKR1C2 gene, has been reported in 46, XY DSD patients with ambiguous genitalia or sex reversal and similar clinical features of PAIS [30], [31].
Conclusions
Two new mutations were found, expanding the mutational spectrum of 46, XY DSD. Dimensional structural analysis of the novel mutated AR (p.R726C) revealed that it affected at the co-activator binding site (BF-3) and not at the testosterone binding site. Other candidate genes should be analyzed for the definite cause of 46, XY DSD, for proper management and genetic counseling.
Strength and limitations of the study
This descriptive analytic study demonstrated novel SRD5A2 and AR mutations, expanded the genetic spectrum and showed an abnormal binding to AR in one patient with PAIS by a dimensional structural analysis. However, this study is limited by the small sample size, and the protein modeling method could structure only defects at the LBD and nearby residues.
Acknowledgments
We would like to thank the patients and their parents for participating in this study.
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
Research funding: This study was funded by the grant from the Ratchadapiseksomphot Endowment Fund of Chulalongkorn University, Bangkok, Thailand. The Faculty of Medicine, RA56/043 and the Chulalongkorn Academic Advancement into It’s 2nd Century Project.
Employment or leadership: None declared.
Honorarium: None declared.
Competing interests: The funding organization(s) played no role in the study design; in the collection, analysis and interpretation of data; in the writing of the report or in the decision to submit the report for publication.
References
1. Audi L, Fernandez-Cancio M, Carrascosa A, Andaluz P, Toran N, et al. Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development. J Clin Endocrinol Metab 2010;95:1876–88.10.1210/jc.2009-2146Search in Google Scholar PubMed
2. Melo CO, Silva DM, da Cruz AD. Challenges in clinical and laboratory diagnosis of androgen insensitivity syndrome: a case report. J Med Case Rep 2011;5:446.10.1186/1752-1947-5-446Search in Google Scholar PubMed PubMed Central
3. Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat 2012;33:887–94.10.1002/humu.22046Search in Google Scholar PubMed
4. Andersson S, Berman DM, Jenkins EP, Russell DW. Deletion of steroid 5 alpha-reductase 2 gene in male pseudohermaphroditism. Nature 1991;354:159–61.10.1038/354159a0Search in Google Scholar PubMed PubMed Central
5. Maimoun L, Philibert P, Cammas B, Audran F, Bouchard P, et al. Phenotypical, biological, and molecular heterogeneity of 5alpha-reductase deficiency: an extensive international experience of 55 patients. J Clin Endocrinol Metab 2011;96:296–307.10.1210/jc.2010-1024Search in Google Scholar PubMed
6. Deeb A, Mason C, Lee YS, Hughes IA. Correlation between genotype, phenotype and sex of rearing in 111 patients with partial androgen insensitivity syndrome. Clin Endocrinol 2005;63:56–62.10.1111/j.1365-2265.2005.02298.xSearch in Google Scholar PubMed
7. Veiga-Junior NN, Medaets PA, Petroli RJ, Calais FL, de Mello MP, et al. Clinical and laboratorial features that may differentiate 46,XY DSD due to partial androgen insensitivity and 5 alpha-Reductase Type 2 Deficiency. Int J Endocrinol 2012;2012:964876.10.1155/2012/964876Search in Google Scholar PubMed PubMed Central
8. Akcay T, Fernandez-Cancio M, Turan S, Guran T, Audi L, et al. AR and SRD5A2 gene mutations in a series of 51 Turkish 46,XY DSD children with a clinical diagnosis of androgen insensitivity. Andrology 2014;2:572–8.10.1111/j.2047-2927.2014.00215.xSearch in Google Scholar PubMed
9. Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem 2000;275:26164–71.10.1074/jbc.M004571200Search in Google Scholar PubMed
10. Sahakitrungruang T, Wacharasindhu S, Yeetong P, Snabboon T, Suphapeetiporn K, et al. Identification of mutations in the SRD5A2 gene in Thai patients with male pseudohermaphroditism. Fertil Steril 2008;90:2015 e11–5.10.1016/j.fertnstert.2008.01.019Search in Google Scholar PubMed
11. Hughes IA, Werner R, Bunch T, Hiort O. Androgen insensitivity syndrome. Semin Reprod Med 2012;30:432–42. PubMed PMID: 23044881.10.1016/S0140-6736(12)60071-3Search in Google Scholar
12. Pereira de Jesus-Tran K, Cote PL, Cantin L, Blanchet J, Labrie F, et al. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci 2006;15:987–99.10.1110/ps.051905906Search in Google Scholar PubMed PubMed Central
13. Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 1997;387:733–6.10.1038/42750Search in Google Scholar PubMed
14. He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem 2000;275:22986–94.10.1074/jbc.M002807200Search in Google Scholar PubMed
15. Christiaens V, Bevan CL, Callewaert L, Haelens A, Verrijdt G, et al. Characterization of the two coactivator-interacting surfaces of the androgen receptor and their relative role in transcriptional control. J Biol Chem 2002;277:49230–7.10.1074/jbc.M209322200Search in Google Scholar PubMed
16. Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, et al. Recognition and accommodation at the androgen receptor coactivator binding interface. PLoS Biol 2004;2:E274.10.1371/journal.pbio.0020274Search in Google Scholar PubMed PubMed Central
17. Estebanez-Perpina E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci USA 2007;104:16074–9.10.1073/pnas.0708036104Search in Google Scholar PubMed PubMed Central
18. Buzon V, Carbo LR, Estruch SB, Fletterick RJ, Estebanez-Perpina E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Mol Cell Endocrinol 2012;348:394–402.10.1016/j.mce.2011.08.012Search in Google Scholar PubMed
19. Grosdidier S, Carbo LR, Buzon V, Brooke G, Nguyen P, et al. Allosteric conversation in the androgen receptor ligand-binding domain surfaces. Mol Endocrinol (Baltimore, MD) 2012;26:1078–90.10.1210/me.2011-1281Search in Google Scholar PubMed PubMed Central
20. Ahmed SF, Cheng A, Dovey L, Hawkins JR, Martin H, et al. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab 2000;85:658–65.10.1210/jc.85.2.658Search in Google Scholar
21. Nordenskjold A, Ivarsson SA. Molecular characterization of 5 alpha-reductase type 2 deficiency and fertility in a Swedish family. J Clin Endocrinol Metabol 1998;83:3236–8.Search in Google Scholar
22. Wilson JD, Griffin JE, Russell DW. Steroid 5 alpha-reductase 2 deficiency. Endocr Rev 1993;14:577–93.10.1210/er.14.5.577Search in Google Scholar
23. Nie M, Zhou Q, Mao J, Lu S, Wu X. Five novel mutations of SRD5A2 found in eight Chinese patients with 46,XY disorders of sex development. Mol Hum Reprod 2011;17:57–62.10.1093/molehr/gaq072Search in Google Scholar PubMed
24. Vilchis F, Ramos L, Mendez JP, Benavides S, Canto P, et al. Molecular analysis of the SRD5A2 in 46,XY subjects with incomplete virilization: the P212R substitution of the steroid 5 alpha-reductase 2 may constitute an ancestral founder mutation in Mexican patients. J Androl 2010;31:358–64.10.2164/jandrol.109.009407Search in Google Scholar PubMed
25. Ko JM, Cheon CK, Kim GH, Kim SH, Kim KS, et al. Clinical characterization and analysis of the SRD5A2 gene in six Korean patients with 5 alpha-reductase type 2 deficiency. Horm Res Paediatr 2010;73:41–8.10.1159/000271915Search in Google Scholar PubMed
26. Zhu H, Liu W, Han B, Fan M, Zhao S, et al. Phenotypic and molecular characteristics in eleven Chinese patients with 5alpha-reductase Type 2 deficiency. Clin Endocrinol (Oxf) 2014;81:711–20.10.1111/cen.12456Search in Google Scholar PubMed
27. Zhang M, Yang J, Zhang H, Ning G, Li X, et al. A novel SRD5A2 mutation with loss of function identified in Chinese patients with hypospadias. Horm Res Paediatr 2011;76:44–9.10.1159/000324692Search in Google Scholar PubMed
28. Hyytinen ER, Haapala K, Thompson J, Lappalainen I, Roiha M, et al. Pattern of somatic androgen receptor gene mutations in patients with hormone-refractory prostate cancer. Lab Invest 2002;82:1591–8.10.1097/01.LAB.0000038924.67707.75Search in Google Scholar
29. Koivisto PA, Hyytinen ER, Matikainen M, Tammela TL, Ikonen T, et al. Germline mutation analysis of the androgen receptor gene in Finnish patients with prostate cancer. J Urol 2004;171:431–3.10.1097/01.ju.0000089774.99728.efSearch in Google Scholar PubMed
30. Fluck CE, Meyer-Boni M, Pandey AV, Kempna P, Miller WL, et al. Why boys will be boys: two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am J Hum Gen 2011;89:201–18.10.1016/j.ajhg.2011.06.009Search in Google Scholar PubMed PubMed Central
31. Zachmann M, Vollmin JA, Hamilton W, Prader A. Steroid 17,20-desmolase deficiency: a new cause of male pseudohermaphroditism. Clin Endocrinol 1972;1:369–85.10.1111/j.1365-2265.1972.tb00407.xSearch in Google Scholar PubMed
Supplemental Material:
The online version of this article (DOI: 10.1515/jpem-2016-0048) offers supplementary material, available to authorized users.
©2017 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Editorial
- Disorders of sex development
- Original Articles
- Fertility and sexual function: a gap in training in pediatric endocrinology
- Disorders of sex development in children in KwaZulu-Natal Durban South Africa: 20-year experience in a tertiary centre
- Novel mutations of the SRD5A2 and AR genes in Thai patients with 46, XY disorders of sex development
- Sensitivity and specificity of different methods for cystic fibrosis-related diabetes screening: is the oral glucose tolerance test still the standard?
- Anti-hyperglycemic activity of Aegle marmelos (L.) corr. is partly mediated by increased insulin secretion, α-amylase inhibition, and retardation of glucose absorption
- Risk factors that affect metabolic health status in obese children
- Nocturnal levels of chemerin and progranulin in adolescents: influence of sex, body mass index, glucose metabolism and sleep
- Elevated endogenous secretory receptor for advanced glycation end products (esRAGE) levels are associated with circulating soluble RAGE levels in diabetic children
- Food exchange estimation by children with type 1 diabetes at summer camp
- Usefulness of non-fasting lipid parameters in children
- Monitoring steroid replacement therapy in children with congenital adrenal hyperplasia
- Clinical and molecular characterization of Beckwith-Wiedemann syndrome in a Chinese population
- An analysis of the sequence of the BAD gene among patients with maturity-onset diabetes of the young (MODY)
- Case Reports
- Hypogonadotropic hypogonadism in a female patient with congenital arhinia
- Transdermal testosterone gel for induction and continuation of puberty in adolescent boys with hepatic dysfunction
- Long-term response to growth hormone therapy in a patient with short stature caused by a novel heterozygous mutation in NPR2
- Three cases of Japanese acromicric/geleophysic dysplasia with FBN1 mutations: a comparison of clinical and radiological features
Articles in the same Issue
- Frontmatter
- Editorial
- Disorders of sex development
- Original Articles
- Fertility and sexual function: a gap in training in pediatric endocrinology
- Disorders of sex development in children in KwaZulu-Natal Durban South Africa: 20-year experience in a tertiary centre
- Novel mutations of the SRD5A2 and AR genes in Thai patients with 46, XY disorders of sex development
- Sensitivity and specificity of different methods for cystic fibrosis-related diabetes screening: is the oral glucose tolerance test still the standard?
- Anti-hyperglycemic activity of Aegle marmelos (L.) corr. is partly mediated by increased insulin secretion, α-amylase inhibition, and retardation of glucose absorption
- Risk factors that affect metabolic health status in obese children
- Nocturnal levels of chemerin and progranulin in adolescents: influence of sex, body mass index, glucose metabolism and sleep
- Elevated endogenous secretory receptor for advanced glycation end products (esRAGE) levels are associated with circulating soluble RAGE levels in diabetic children
- Food exchange estimation by children with type 1 diabetes at summer camp
- Usefulness of non-fasting lipid parameters in children
- Monitoring steroid replacement therapy in children with congenital adrenal hyperplasia
- Clinical and molecular characterization of Beckwith-Wiedemann syndrome in a Chinese population
- An analysis of the sequence of the BAD gene among patients with maturity-onset diabetes of the young (MODY)
- Case Reports
- Hypogonadotropic hypogonadism in a female patient with congenital arhinia
- Transdermal testosterone gel for induction and continuation of puberty in adolescent boys with hepatic dysfunction
- Long-term response to growth hormone therapy in a patient with short stature caused by a novel heterozygous mutation in NPR2
- Three cases of Japanese acromicric/geleophysic dysplasia with FBN1 mutations: a comparison of clinical and radiological features