Molecular simulations of DEAH-box helicases reveal control of domain flexibility by ligands: RNA, ATP, ADP, and G-patch proteins
-
Robert A. Becker



Abstract
DEAH-box helicases use the energy from ATP hydrolysis to translocate along RNA strands. They are composed of tandem RecA-like domains and a C-terminal domain connected by flexible linkers, and the activity of several DEAH-box helicases is regulated by cofactors called G-patch proteins. We used all-atom molecular dynamics simulations of the helicases Prp43, Prp22, and DHX15 in various liganded states to investigate how RNA, ADP, ATP, or G-patch proteins influence their conformational dynamics. The simulations suggest that apo helicases are highly flexible, whereas binding of RNA renders the helicases more rigid. ATP and ADP control the stability of the RecA1–RecA2 interface, but they have only a smaller effect on domain flexibility in absence of a RecA1–RecA2 interface. Binding of a G-patch protein to DHX15 imposes a more structured conformational ensemble, characterized by more defined relative domain arrangements and by an increased conformational stability of the RNA tunnel. However, the effect of the G-patch protein on domain dynamics is far more subtle as compared to the effects of RNA or ATP/ADP. The simulations characterize DEAH-box helicase as dynamic machines whose conformational ensembles are strongly defined by the presence of RNA, ATP, or ADP and only fine-tuned by the presence of G-patch proteins.
1 Introduction
Helicases are motor enzymes found in all domains of life including viruses, where they play key roles in processes such as DNA transcription, splicing, translation, recombination, DNA repair, proofreading, RNA transport, and several others (Abdel-Monem and Hoffmann-Berling 1976; Abdel-Monem et al. 1976). Helicases convert the chemical energy stored in nucleoside triphosphate (NTP) into mechanical forces with the aim to unwind double-stranded DNA/RNA or to transport single-stranded DNA/RNA (Singleton et al. 2007). Malfunction of helicases is involved in several diseases including cancer (Cantor et al. 2001), Werner syndrome (Gray et al. 1997), Bloom syndrome (Ellis et al. 1995; Selak et al. 2008), and Rothmund–Thomson syndrome (Fuller-Pace 2013; Kitao et al. 1999; Van Brabant et al. 2000). The physiological functions and molecular mechanisms of helicases have been studied and reviewed in detail in recent years (Delagoutte and Von Hippel 2002, 2003; Hall and Matson 1999; Jarmoskaite and Russell 2014; Jeong et al. 2004; Levin et al. 2005, 2003; Lohman 1993; Lohman and Bjornson 1996; Matson and Kaiser-Rogers 1990; Patel and Donmez 2006; Patel and Picha 2000; Singleton et al. 2007; Tuteja and Tuteja 2004; von Hippel and Delagoutte 2001; von Hippel and Delagoutte 2003; West 1996).
Based on structural similarity, helicases have been ordered into six super-families. Super-family 2 (SF2) comprises the largest and most diverse super-family that contains, among others, the DExD/H-box, RecQ, and SnF helicases (Byrd and Raney 2012; Fairman-Williams et al. 2010; Fuller-Pace 2006; Tanner and Linder 2001). Conserved structural motifs of helicases are given by the Walker A/B motifs, which serve as NTP binding domain in various proteins (Walker et al. 1982). In DExD/H-box helicases, which are in the focus of this study, the ATP binding site is formed at the interface between two recombination protein recombinase A-like (RecA-like) domains. The surface of the tandem RecA-like domains together with a C-terminal domain (CTD) form the RNA binding cleft (Figure 1).
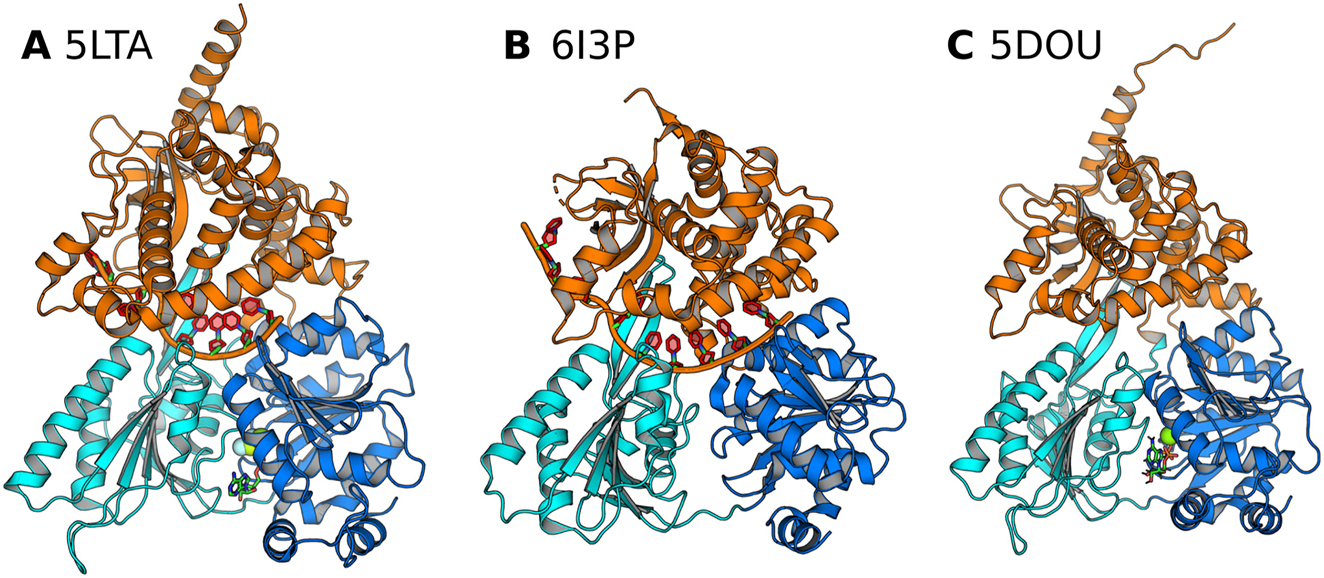
The crystal structures of the DEAH box helicase Prp43 used as starting structures for MD simulations in this study. RecA1, RecA2, and C-terminal domains are colored in blue, cyan, and orange, respectively. RNA (if present) is shown as red/orange cartoon, nucleotide as sticks, and Mg2+ (if present) as green sphere. (A) Prp43 complexed with RNA and an ATP analogue (PDB code 5LTA), (B) with ADP (PDB code 6I3P), and (C) with an ATP analogue (PDB code 5D0U) Tauchert et al. (2016, 2017).
While conventional helicases have been referred to as unwindases as they unwind double-stranded DNA, certain helicases instead serve as translocases as they transport single-stranded DNA or RNA (Singleton et al. 2007). Examples of such translocases include Prp2, Prp22, and Prp43 from yeast as well as the human orthologue of Prp43 called DHX15. Crystal structures of these translocases in various ligand-bound states as well biochemical data revealed that the RNA translocation cycle proceeds via relative domain displacements by several Ångström (Hilbert et al. 2009; Linder and Jankowsky 2011; Studer et al. 2020; Tauchert et al. 2017). Furthermore, structural studies provided the starting point of all-atom molecular dynamics (MD) simulations, which we recently employed to derive a complete and continuous RNA translocation cycle of Prp43 in atomic detail (Becker and Hub 2023). The simulations showed that large-scale domain motions driving RNA translocation are controlled by an interdependent sequence of atomic-scale transitions.
Helicases are regulated by diverse mechanisms including substrate-dependent auto-inhibition (Absmeier et al. 2020; Gowravaram et al. 2018), post-translational modifications (Jacobs et al. 2007; Mathew et al. 2008; Song et al. 2017), recognition of specific RNA features (Kretschmer et al. 2018; Wojtas et al. 2017), and, as studied here, by binding of protein cofactors (Bohnsack et al. 2021; Sloan and Bohnsack 2018). Cofactors may alter the catalytic activity by either creating an electrostatic environment that assists RNA binding or by directly mediating RNA–protein interactions. A variety of cofactors enhance the activity of RNA helicases (Ozgur et al. 2015; Silverman et al. 2003; Sloan and Bohnsack 2018). One family of such binding cofactors are the G-patch proteins characterized by a conserved glycine-rich sequence (Aravind and Koonin 1999). The overall G-patch motif comprises approximately 50 amino acids including the sequence Gx2hhx3Gax2GxGlGx3pxux3sx10–16GhG where ‘a’ denotes an aromatic, ‘h’ a hydrophobic, ‘l’ an aliphatic, ‘s’ a small, ‘u’ a tiny, and ‘x’ a variable amino acid (Bohnsack et al. 2021). The helicases investigated in this study interact with different G-patch regulation proteins. For example, Prp43 in yeast associates with the G-patch cofactors Cmg1, Pxr1, Sqs1, and Ntr1 (also called Spp382), Prp2 binds to the cofactor Spp2, while the human orthologue to Prp43 called DHX15 binds to six different G-patch cofactors including NKRF as shown in Figure 2 (Studer et al. 2020). G-patch proteins are assumed to regulate helicase activity during specific processes; for instance, the G-patch protein Pfa1 regulates ATPase activity and unwinding activity of Prp43 during ribosome biogenesis, whereas Ntr1 and Ntr2 enhance ATPase and helicase activity of Prp43 during pre-mRNA splicing.
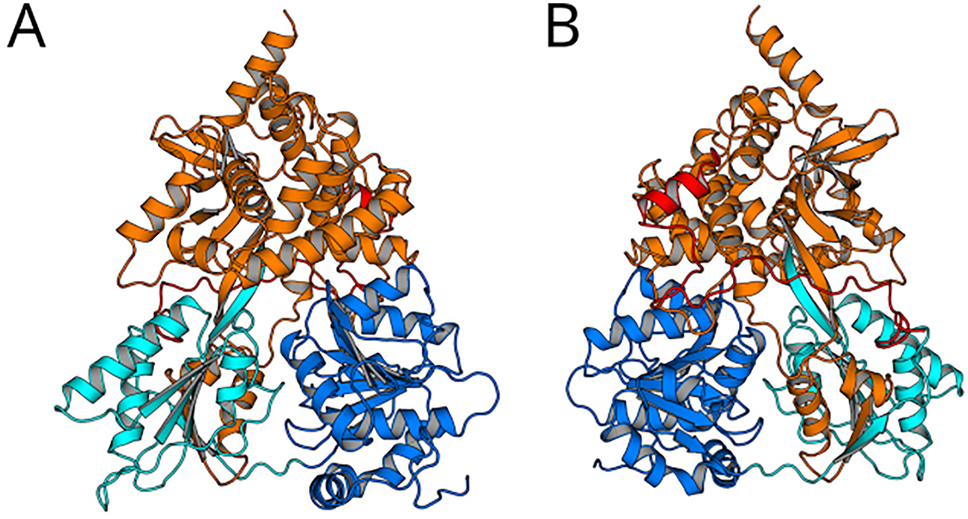
Archtecture of DHX15 complexed with a G-patch protein. DHX15 comprising the RecA1 domain (blue), RecA2 domain (cyan), and the C-terminal domain (orange) in contact with the G-patch protein NKRF (red; PDB code 6SH7; Studer et al. (2020)). NKRF binds to the backside of DHX15 (frontside of panel B) via two defined structures: (i) With a helical motif to the WH motif of the C-terminal domain and (ii) with a loop-like structure to the β-hairpin and to the RecA2 domain.
The function of DEAH-box helicases requires control of concerted domain motions and of domain flexibility. Loading of the RNA into the RNA cleft involves the opening and closing of the RNA tunnel via a hinge-bending motion of CTD relative to the tandem RecA-like domains (Tauchert et al. 2017). RNA translocation involves an inchworm-like stepping of RecA1 and RecA2 along the RNA phosphate backbone (Singleton et al. 2007; Tanner and Linder 2001; Velankar et al. 1999). Recently, we refined the translocation model to an “inchworm-caterpillar model” to emphasize that the RecA-like domains of Prp43 never detach from the RNA during translocation but instead crawl along the RNA by shifting protein–RNA hydrogen bonds one-by-one (Becker and Hub 2023). In this study, we used all-atom MD simulations to reveal how binding of RNA, nucleotides, or G-patch proteins control the conformational domain dynamics of three different DEAH-box helicases: Prp43, Prp22, and DHX15. Starting from six different crystal structures, we devised simulation systems of Prp43, Prp22, and DHX15 in a total of 18 different liganded states including apo conformations as well as conformations complexed with RNA, ATP, ADP, or with a G-patch protein. Using both conventional simulations as well as simulations enhanced with simulated tempering with a total simulation time of 54 µs, we studied how binding of ligands control the conformational fluctuations of DEAH-box helicases.
2 Results
2.1 Effects of ADP, ATP, and RNA on domain fluctuations of Prp43 and Prp22
We carried out extensive MD simulations of Prp43 starting from the crystal structures 5LTA, 5D0U, 5LTK, 5LTJ, as well as well as of Prp22 starting from the crystal structure 6I3P (Hamann et al. 2019; Tauchert et al. 2017, 2016). To reveal how the presence of ADP, ATP, and RNA controls the domain dynamics of DEAH-box helices, we simulated the helicases in the crystallographic conformations and after removal of the RNA (if present) or of a nucleotide (if present), resulting in a total of 18 different complexes. For each complex, multiple simulation replicas were carried out with simulation times between 80 ns and 1000 ns each (Table 1). Although these simulation times are insufficient for obtaining exhaustive sampling of the domain dynamics, they provided insight into the effects of ligands on domain flexibility or on the stability of domain–domain interfaces. We quantified the domain dynamics using two structural features: (i) the RecA1–RecA2 distance, defined as the distance between the centers of mass of the central beta sheets of the RecA-like domains; (ii) the CTD–RecA2 distance, defined as the minimal distance between pairs of amino acids from CTD and RecA2 that frequently formed hydrogen bonds: for Prp43, we considered the distance between Ser614 in the ratchet-like domain and Glu320 in RecA2; for Prp22, we considered Lys468 and Asp233 instead. We analyzed these distances because these pairs of amino acids tend to form or break a hydrogen bond as the RNA cleft closes or opens, hence providing a measure for the CTD–RecA2 arrangement.
Summary of simulations carried out for this study. Name of helicase, PDB ID of starting conformation, ligands, number of independent simulations (Nsim), simulation time per simulation (tsim). Liganded states marked with an asterisk resemble the crystal structure conformation. Sampling of DHX15 simulations was enhanced with simulated tempering (ST).
| Helicase | PDB ID | Ligands | N sim | tsim (ns) |
|---|---|---|---|---|
| Prp43 | 5LTA | RNA, ATP* | 4 | 200 |
| RNA | 10 | 80 | ||
| ATP | 6 | 300 | ||
| ADP | 10 | 800 | ||
| – | 10 | 500 | ||
| 5D0U | ADP* | 6 | 300 | |
| – | 10 | 500 | ||
| 5LTJ | ATP* | 4 | 250 | |
| ADP | 6 | 100 | ||
| 5LTK | ATP* | 4 | 300 | |
| ADP | 4 | 100 | ||
| Prp22 | 6I3P | RNA, ATP | 4 | 200 |
| RNA* | 4 | 200 | ||
| ATP | 4 | 300 | ||
| ADP | 4 | 1000 | ||
| – | 4 | 300 | ||
| DHX15 | 6SH7 | G-patch NKRF | 10 | 1000 (ST) |
| – | 10 | 1000 (ST) |
2.1.1 5LTA structure of Prp43
The crystal structure with the PDB ID 5LTA of Prp43⋅RNA⋅ADP-BeF3 mimics the Prp43 helicase in the RNA- and ATP-bound state. The RNA cleft forms a tunnel for single-stranded RNA (ssRNA), while the two RecA domains, RecA1 and RecA2, are positioned in close proximity, forming a well-defined interface bridged by the presence of ATP. We carried out four 200 ns simulations of the Prp43⋅RNA⋅ATP complex, ten 80 ns simulations of the Prp43⋅RNA complex, six 300 ns simulations of the Prp43⋅ATP complex, ten 800 ns simulations of the Prp43⋅ADP, and ten 500 ns simulations of the Prp43 apo structure (Table 1). Since these simulations cover aggregated simulation times of at most few microseconds, they provide insight into the role of ligands on the conformational stability of the 5LTA conformation; however, it is unlikely that the simulations provide complete domain transitions involved in RNA processing, since such productive domain transitions of helicases likely occur on longer time scales (Becker and Hub 2023; Byrd et al. 2012; Carney et al. 2021; Stano et al. 2005).
Figure 3 analyzes the domain motions during these simulations, revealing that the presence of the nucleotide (ATP or ADP) and of the ssRNA greatly influence the domain dynamics. The distributions of the CTD–RecA2 or RecA1–RecA2 distances are furthermore summarized as violin plots in Figures 4A and 5A, respectively. In the presence of RNA, the CTD–RecA2 and the RecA1–RecA2 interfaces are largely stable on the time scales of 80 ns–180 ns presented here, irrespective of the presence of ATP (Figures 3A/B; 4A/5A, green and yellow). These findings are compatible with our previous study that presented a full conformation cycle of Prp43 (Becker and Hub 2023). In the previous study, we found that, after removal of ATP, the RecA1–RecA2 interfaces opens on much longer time scales of tens of microseconds, suggesting that the stability of the RecA1–RecA2 interface in presence of RNA found here is a consequence of shorter simulation times.
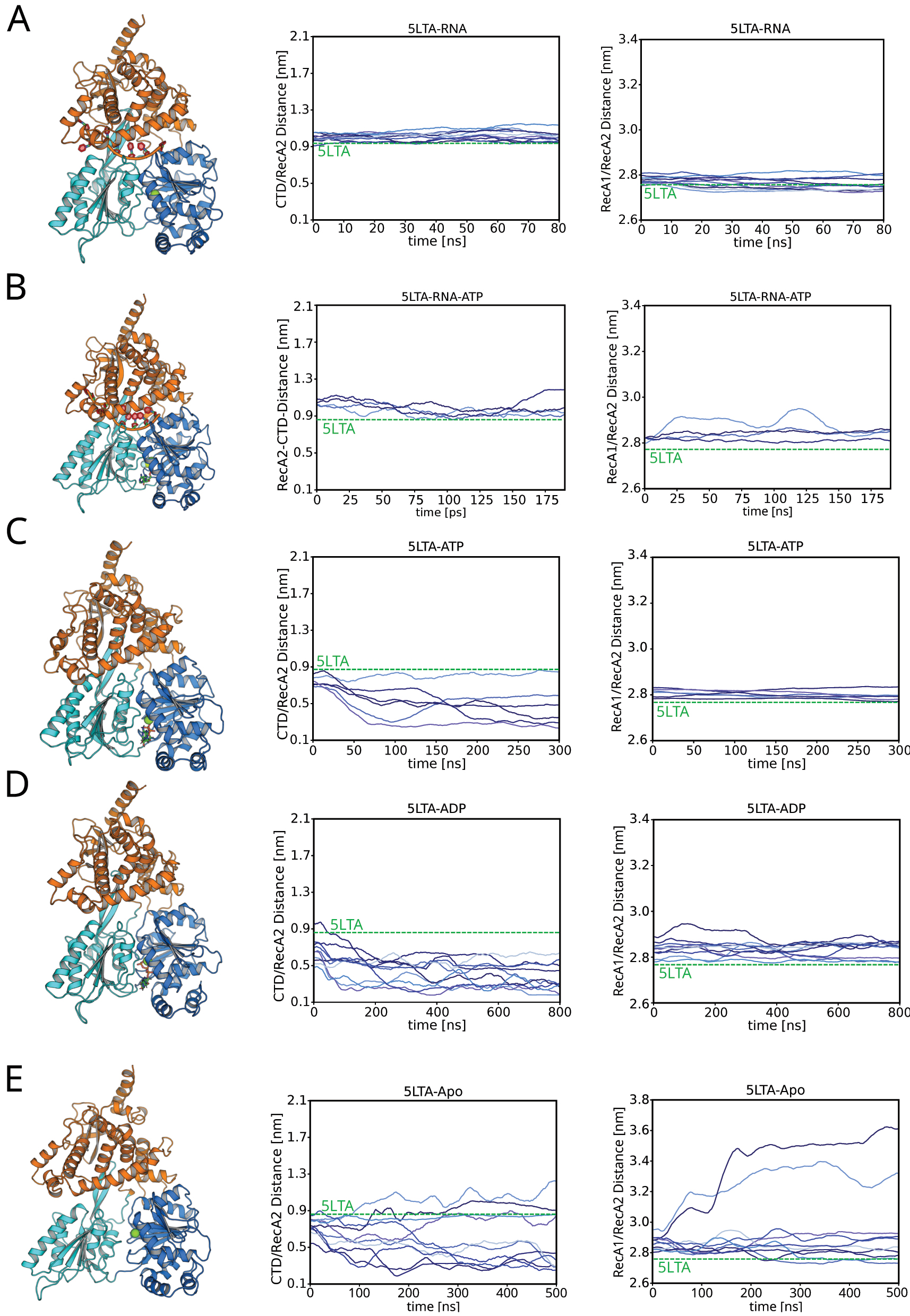
Simulations of Prp43 starting from the 5LTA structure. (A) Simulations of the Prp43⋅RNA complex, (B) Prp43⋅RNA⋅ATP, (C) Prp43⋅ATP, (D) Prp43⋅ADP, and (E) apo Prp43. Left column: Prp43 (coloring scheme as used in Figure 1). Middle column: CTD–RecA2 distance. Right column: RecA1–RecA2 distance.

Violin plots of distributions of the CTD–RecA2 distance. Results were taken from 16 different simulation setups, as indicated in the axis labels.
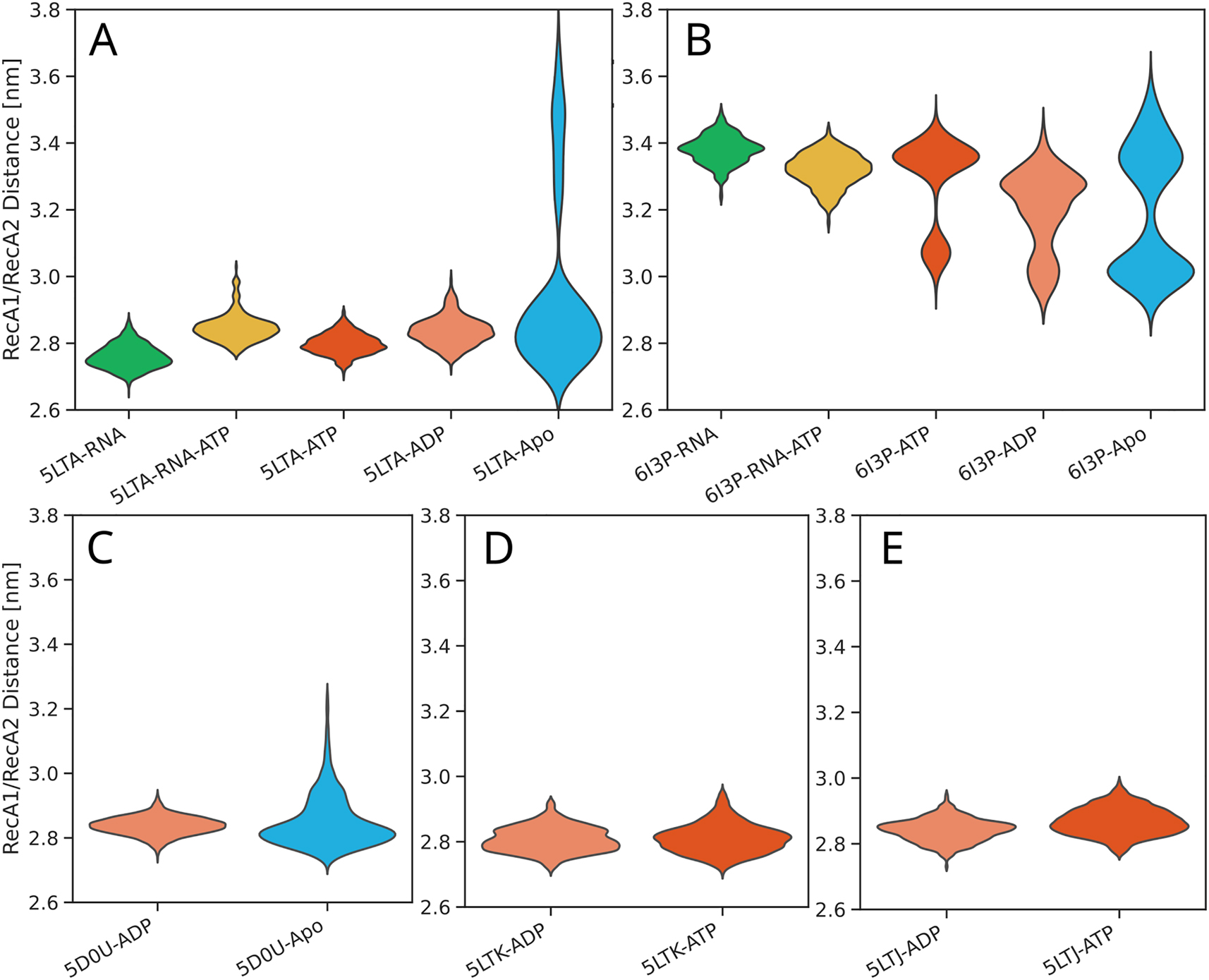
Violin plots of distributions of the RecA1–RecA2 distance. Results were taken from 16 different simulation setups, as indicated in the axis labels.
Upon removal of the RNA, Prp43 becomes far more dynamic (Figure 3C–E; 4A/5A, blue, red, pink, and blue). The structure of the RNA tunnel is perturbed relative to the 5LTA conformation, as revealed by a decrease of the CTD–RecA2 distance by 0.4 nm–0.7 nm. In several simulations, a hydrogen bond forms between Glu320 and Ser614, which furthermore induces a tilt of the CTD towards RecA2, as shown in Figure 3C (left column). Upon replacing ATP with ADP, the RecA1–RecA2 interface is increasingly perturbed, as expected in the light of the RNA translocation cycle that involves the opening after the RecA1–RecA2 interface after ATP hydrolysis (Figure 3C/D, right column).
The simulations of apo Prp43 reveal the largest domain motions among simulations starting from the 5LTA conformation (Figure 3E). Among ten independent simulations, the CTD–RecA2 distance either increased or decreased within 500 ns, reflecting opening or closing of the RNA tunnel. In two among ten simulations, the RecA1–RecA2 distance increases, demonstrating an opening of RecA1–RecA2 interface. However, we did not observe a loop-to-helix transition of the sensor serine loop within our simulations, which was shown to be a critical step of the RNA translocation cycle (Becker and Hub 2023; Hamann et al. 2019), likely because this transition occurs on longer time scales. Hence, the final states of the two simulations exhibiting RecA1–RecA2 opening should not yet be considered as a truly open state as taken by the Prp43⋅RNA complex during the RNA translocation cycle.
2.1.2 5D0U structure of Prp43
The crystal structure 5D0U solved by Tauchert et al. (2016) represents the Prp43⋅ADP complex, adopting a closed state with a small CTD–RecA2 distance and a tight RecA1–RecA2 interface (Figure 1C). The sensor serine takes a helical conformation, as also found in the open Prp43 structure (Becker and Hub 2023; Hamann et al. 2019), suggesting that the loop-to-helix transition of the sensor serine occurs immediately after ATP hydrolysis and phosphate release. The structure shows an open RNA cleft.
Starting from 5D0U, we performed six 300 ns simulations of the native Prp43⋅ADP complex and ten 500 ns simulations of apo Prp43 after removing ADP from the complex (Figures S1 and 4B, 5B). The 5D0U Prp43⋅ADP complex reveals a similar behavior like the Prp43⋅ATP and Prp43⋅ADP complexes derived from 5LTA. Namely, the RecA1–RecA2 interface remains stable, whereas the CTD–RecA2 is more dynamic, reaching a slightly more closed state as compared to the initial 5D0U conformation. Hence, the partly open CTD–RecA2 interface of 5D0U might have been stabilized by crystal contacts (Figure S5), or the slightly more compact conformation may be favored by the applied force field (see Discussion). In contrast to the Prp43⋅ADP complex derived from 5LTA, only one simulation reaches a CTD–RecA2 distance of ∼0.2 nm within 300 ns, suggesting that the RNA tunnel formed between RecA2 and CTD in 5D0U is more stable as compared to the RNA tunnel in the 5LTA crystal structure after RNA removal. The increased flexibility of Prp43 in 5LTA–derived simulations relative to the 5D0U-derived simulations is expected considering that the 5LTA-derived simulations were started from the 5LTA conformation after removal of RNA, which represents a major structural perturbation, whereas the 5D0U-derived simulations were started from the complete 5D0U structure.
The apo Prp43 structure derived from 5D0U reveals similar dynamics like the 5LTA-derived apo structure (Figures S1B; 4C, 5C). The CTD–RecA2 domain distance shows large fluctuation indicating a flexibility of the CTD arrangement relative to the RecA-like domains. The RecA1–RecA2 interface opens in several simulations owing to the loss of the bridging ADP contacts between RecA1 and RecA2.
2.1.3 5LTJ and 5LTK structures of Prp43
The 5LTJ and 5LTK structures solved by Tauchert et al. represent the Prp43⋅ATP complex (Tauchert et al. 2017). In these structures, the RecA2 and CTD do not form an RNA tunnel but instead expose an open RNA cleft (Figure S2). Thus, the conformations represent the pre-catalytic state of Prp43 before RNA binding. To study the stability of the conformations of 5LTJ and 5LTK, we performed four 250 ns simulations of the 5LTJ⋅ATP complex, six 100 ns simulations of the 5LTJ⋅ADP complex, four 300 ns simulations of the 5LTJ⋅ATP complex, and four 300 ns simulations of the 5LTJ⋅ADP complex (Figures S3, 4D/E, 5D/E).
The RecA1–RecA2 distance exhibited only minor fluctuations in the four different simulation setups, indicating that the presence of either ATP or ADP largely stabilizes the RecA1–RecA2 interface. In sharp contrast, the CTD–RecA2 distance is highly dynamic in the four setups, revealing large fluctuations and a partial closure by 0.3 nm–0.9 nm. In most simulations, the CTD formed contact with RecA2, thereby forming an RNA tunnel, as illustrated in Figure S3A/B. Hence, the conformation with a wide-open RNA cleft reported by the 5LTJ and 5LTK structures was not stable in our simulations, but instead approached a more closed CTD–RecA2 arrangement, as also reported by the 5D0U structure of Prp43⋅ADP. These findings may indicate that, in absence of RNA, Prp43 exhibits a heterogeneous ensemble covering various degrees of CTD–RecA2 opening.
2.1.4 6I3P structure of Prp22
The 6I3P crystal structure solved by Hamann et al. represents the helicase Prp22 complexed with RNA and in absence of ATP or ADP (Hamann et al. 2019). The structure exhibits a defined RNA tunnel occupied with ssRNA and an open RecA1–RecA2 interface. The RecA2 loop containing the sensor serine adopts a helical state, enabling the sensor serine to form a hydrogen bond with the RNA. The RecA2 domain has shifted by one RNA nucleotide upstream relative to the conformation of the 5LTA structure, as occurring during the RNA translocation cycle. We performed four 200 ns simulations of Prp22⋅RNA⋅ATP by inserting ATP into the ATP pocket, four 200 ns simulations of the Prp22⋅RNA starting from the 6I3P crystal structure, four 300 ns simulations of Prp22⋅ATP by removing RNA and placing ATP into the ATP pocket, four 1 µs simulations of Prp22⋅ADP by removing RNA and placing ADP into the ATP pocket, as well as four 300 ns simulations of the Prp22 apo structure by removing RNA (Figures S4, 4B, 5B, Table 1).
The Prp22⋅RNA simulations show largely constant CTD–RecA2 and RecA1–RecA2 distances, demonstrating that the conformation of the crystal structure is stable within the simulation time scale. Insertion of ATP into the Prp22⋅RNA complex does not induce any domain motions within 200 ns, as expected from the fact that the closure of the RecA1–RecA2 interface after ATP binding occurs on much longer time scales of tens of microseconds (Becker and Hub 2023) (Figures S4A/B; 4B, 5B, green and yellow).
In contrast, upon removal of the RNA, Prp22 becomes far more dynamic, in line with the increased dynamics of Prp43 in absence of RNA (Figures 4B, 5B, red, pink, blue). In most of the simulations, the CTD–RecA2 distance increases, indicating an opening of the CTD–RecA2 interface. Simultaneously, the arrangement of RecA2 relative to RecA1 becomes more dynamic, enabling RecA2 to from contacts with RecA1. However, we do not observe a complete closing of the RecA1–RecA2 interface, likely because the sensor serine did not carry out the helix-to-loop transition, which is essential for the closing process (Becker and Hub 2023). Notably, the dynamics of apo Prp22 are similar to the dynamics in presence of ADP or ATP on the hundreds of nanosecond time scale (Figure S4E). This finding suggests that the overall flexibility of the enzyme within the simulation time scale is mostly controlled by the presence of RNA but not by the presence of ATP/ADP. Instead, ATP/ADP likely control the stability of the RecA1–RecA2 interface, while the formation or rupture of the RecA1–RecA2 interface occurs on long time scales.
2.2 Effect of G-patch protein NKRF on DHX15 dynamics
Activator or cofactor proteins enhance and regulate the activity of helicases, typically in a function-specific manner (Ozgur et al. 2015; Silverman et al. 2003; Sloan and Bohnsack 2018). DEAH-box helicases interact with a diverse group of cofactors called G-patch proteins (Aravind and Koonin 1999). G-patch proteins contain a glycine-rich G-patch motif embedded in an intrinsically disordered region, and they bind to the outer surface of the helicase (Figure 2). Structural and biochemical data suggested that the G-patch proteins Pfa1 binding to Prp43, Spp2 binding to Prp2, and NKRF binding to DHX15 regulate the helicase by guiding their relative domain arrangements (Enders et al. 2022; Hamann et al. 2020; Studer et al. 2020).
We investigated the apo DHX15 structure solved by Studer et al. (2020) in complex with the NKRF G-patch protein, but in absence of any other additional ligands (PDB ID 6SH7). To shed light on the influence of the NKRF G-patch protein on the DHX15 dynamics, we carried out ten independent 1µs simulations each in presence or in absence of the G-patch protein (Figure 2). Since DEAH-box helicases in apo form (in absence of RNA/ATP/ADP) exhibit high flexibility enabling rapid conformational sampling, as demonstrated above, the apo form of DHX15 provides a useful model system to study the effect of the G-patch protein on the conformational dynamics within accessible simulation time scales. Nevertheless, since sampling is challenging even for the flexible apo DHX15, we here used simulated tempering (ST) on a temperature range between 300 K and 348 K to further enhance the conformational sampling (see Methods) (Marinari and Parisi 1992). ST has been shown to accelerate the conformation sampling by approximately one order of magnitude (Pan et al. 2016), while maintaining the correct Boltzmann ensemble.
Figure 6A and B presents the RecA1–RecA2 distances for the ten independent ST simulations without or with G-patch protein, respectively. Overall, the simulations reveal large conformational fluctuations irrespective of the presence of a G-patch. However, histograms over the RecA1–RecA2 distances for each of the 10 independent simulation replicas reveal differences between the simulations with or without G-patch protein (Figure 6C/D, thin lines). Whereas the distributions from simulations without G-patch protein peak at essentially all possible distances between approx. 2.8 nm and 3.3 nm, the simulations with G-patch protein either peak at lower distances (2.8 nm–2.9 nm, corresponding to a closed RecA1–RecA2 interface) or at larger distances (∼3.2 nm). However, this effect by the G-patch on the RecA1–RecA2 distance histograms is subtle and not visible in the average distribution (Figure 6C/D, thick lines) owing to long tails of the individual distributions. Nevertheless, this subtle difference may indicate that the presence of the G-patch protein leads to more structured RecA1–RecA2 arrangements. This hypothesis is in line with the function of NKRF as a “bracer” that leads to more defined domain arrangements in DHX15/NKRF as compared to DHX15 (Studer et al. 2020).
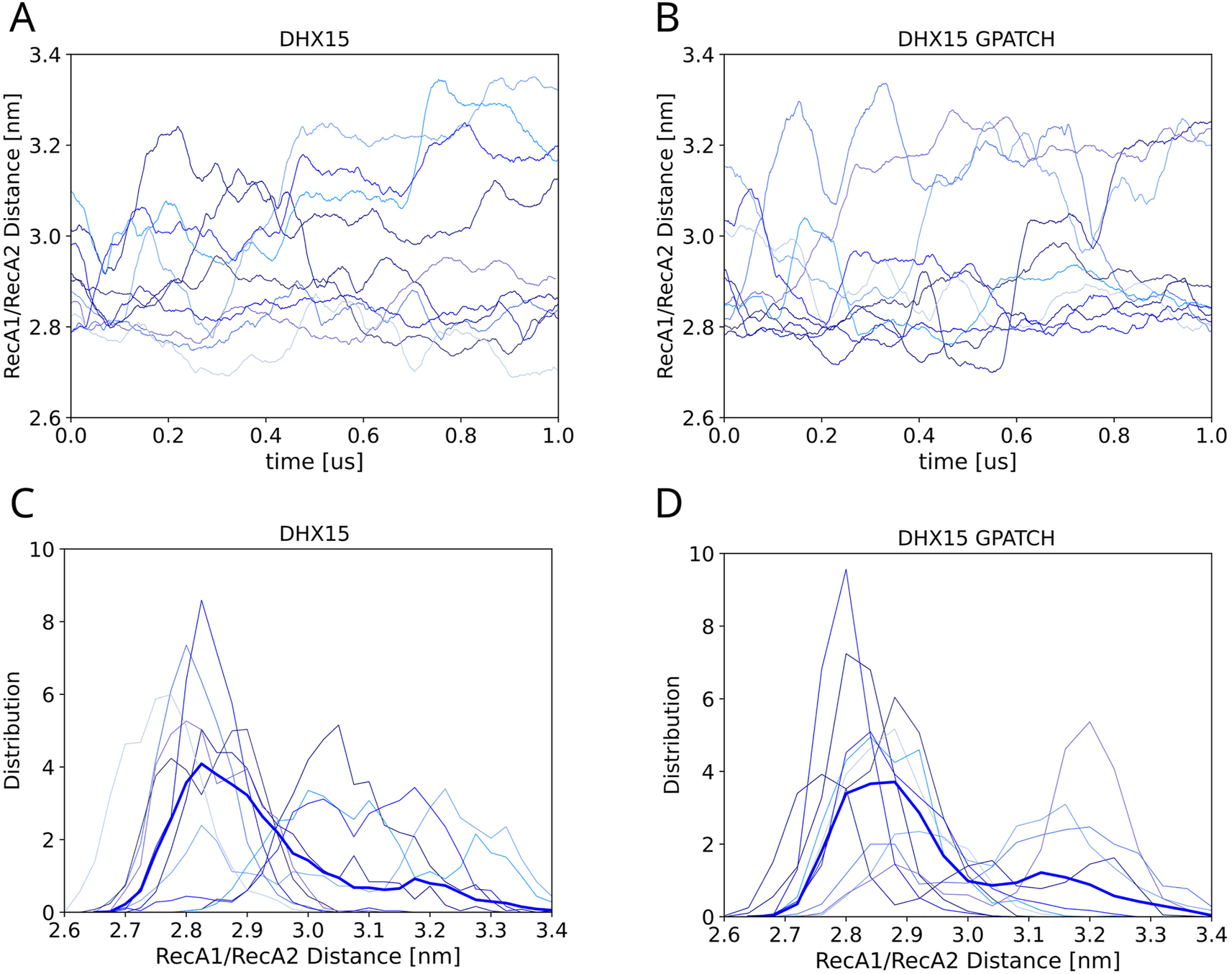
Influence of G-patch protein on the RecA1–RecA2 distance. (A) RecA1–RecA2 domain distance of DHX15 in absence or (B) in presence of the NKRF over 10 independent simulated tempering simulations each. (C) Thin lines show distributions of the RecA1–RecA2 distance, each taken from a 1-µs simulated tempering simulation (thin lines) in absence or (D) in presence of the G-patch. Thick blue lines show the average over ten independent simulations.
Additional indications for the structure-forming effect of the G-patch protein is given by the analysis of the CTD dynamics. As a measure for the CTD–RecA2 distance, we tracked the distance between Glu497 of the ratchet-like domain and two cationic Lys259 and Arg263 residues in an outer helix of the RecA2 domain. This analysis was motivated by the observation that Lys259/Arg263 and Glu497 frequently interact. As shown in Figure 7, the CTD–RecA2 distances reveal similar trends as observed for the RecA1–RecA2 distances. Namely, in the absence of the G-patch protein, CTD–RecA2 distances are more uniformly dispersed and frequently adopt large values beyond 0.8 nm (Figure 7A). In contrast, in presence of the G-patch protein, the CTD–RecA2 distances mostly adopt two distinct states at distances of either 0.2 nm or 0.55 nm whereas large distances >0.8 nm are rarely visited (Figure 7B). Here, the state with lower distance (0.2 nm) involves a hydrogen bond between Glu497 with residues Lys259/Arg263. The state with larger distance (0.55 nm) represents a conformation with different contact points between RecA2 and the ratchet-like domain involving, for instance, hydrogen bonds between the residue pairs Lys259–Tyr537 or Arg544–Asp255, leading to long-living conformations.
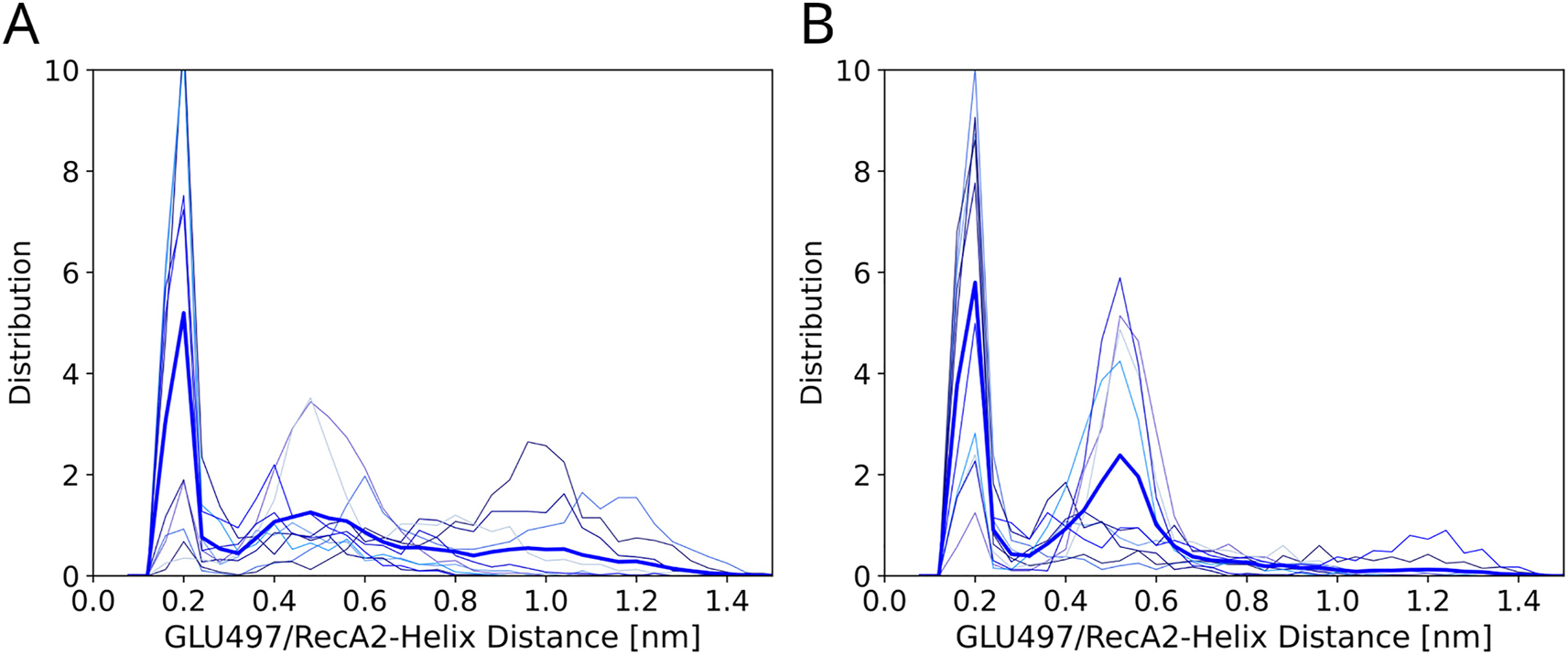
Influence of G-patch protein on the CTD–RecA2 distance. (A) Glu497/RecA2 distance distribution of the DHX15 apo-structure in absence or (B) in presence of the G-patch protein. Thin lines represent distributions from ten individual 1-µs simulated tempering simulations. Thick blue lines show the average over ten independent simulations.
Visual inspection of the simulations revealed that binding of the G-patch protein furthermore influences the section of the RNA tunnel formed between the CTD and the RecA2 domain. We quantified the degree of openness of the RNA tunnel by the distance between Arg748 of the ratchet-like domain and Pro603 (Figure 8C/D). Figure 8A and B presents the average and standard deviation of the tunnel openness as averaged over ten independent simulated tempering simulations in absence or in presence of the G-patch protein, respectively. In absence of the G-patch protein, the Arg748 and Pro603 approach each other during the initial 100 ns of simulations, demonstrating a reproducible tightening of the RNA tunnel (Figure 8A). In contrast, in presence of the G-patch, the openness of the RNA tunnel remains constant and exhibits only reduced conformational fluctuations (Figure 8B, thick red line and orange shaded area). Overall, our simulations suggest that binding of the G-patch protein imposes a more structured confrontational ensemble of DHX15. The G-patch protein stabilizes two distinct arrangements of the CTD relative to RecA2 corresponding to two slightly different conformations of a closed RNA tunnel. In turn, binding of the G-patch protein disfavors large CTD–RecA2 distances corresponding to an open RNA cleft, in agreement with previous observations (Bohnsack et al. 2021; Hamann et al. 2020; Studer et al. 2020). To a lower degree, the G-patch favors more structured relative RecA1–RecA2 arrangements. Furthermore, the G-patch protein stabilizes an open RNA tunnel in our simulations.
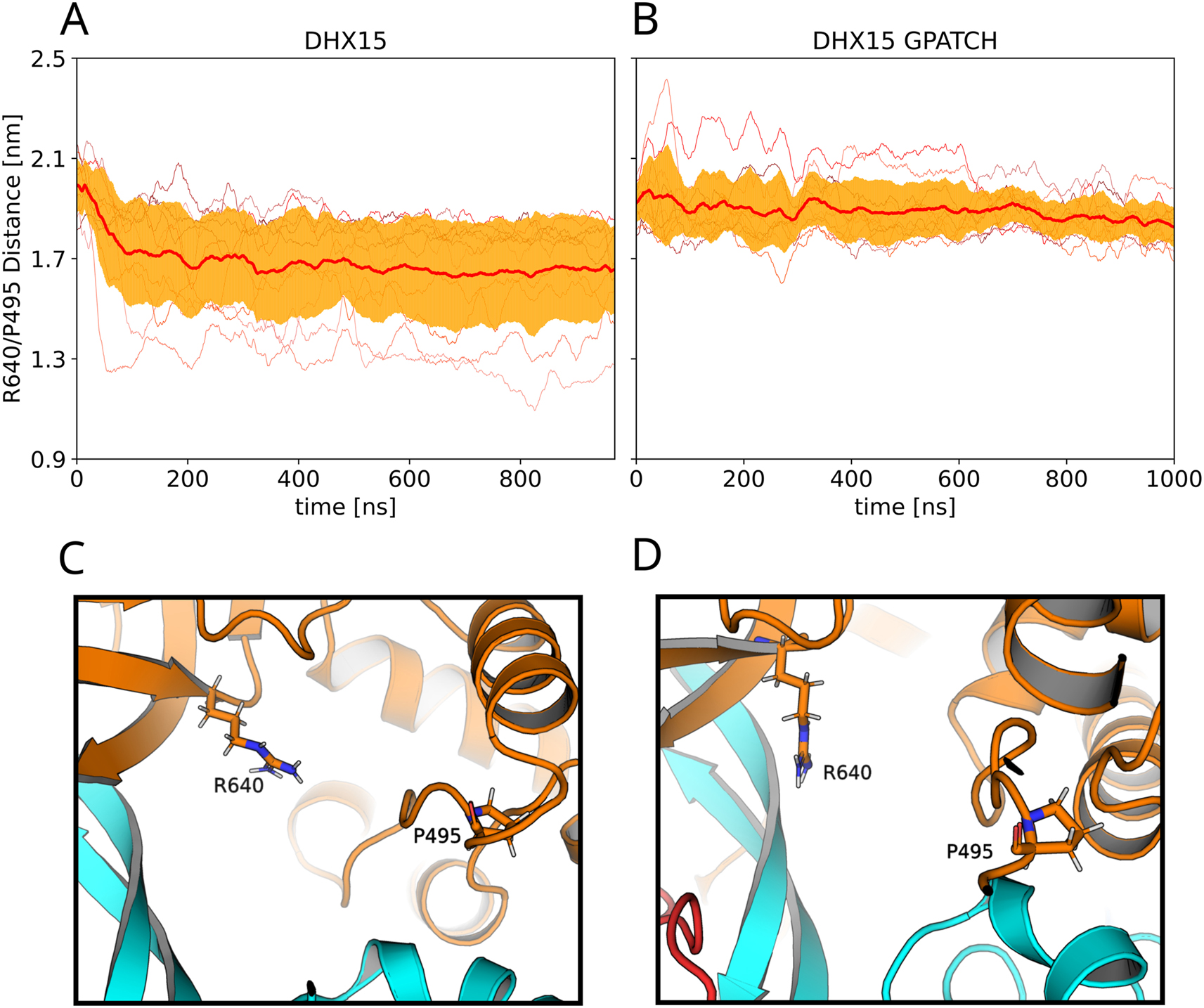
Influence of G-patch protein on the RNA tunnel. (A/B) degree of openness of the RNA tunnel measured as the distance between Arg748 of the ratchet-like domain and Pro603. Thin lines represent ten independent simulated tempering simulations (A) in absence or (B) in presence of the G-path protein. Thick red lines and orange shaded areas represent average and standard deviation among ten independent simulations. (C) Molecular representations of RNA tunnel in a more closed and (D) in a more open conformation.
3 Discussion
We used extensive MD simulations to investigate the effect of ligands on domain fluctuations of DEAH-box helicases. First, starting from five different crystal structure of Prp43 or Prp22, we studied the influence of RNA, ATP, and ADP by either inserting or removing these ligands, followed by multiple simulation replicas of 80 ns to 1 µs each. We found that, in the apo state, Prp43 and Prp22 exhibit large-scale conformational fluctuations of the relative arrangements of CTD, RecA1, and RecA2. These fluctuations occurred on the time scale of tens to hundreds of nanoseconds, reflecting that a shallow free energy landscape underlies the domain dynamics. Upon addition of the RNA, the helicases became far more rigid, demonstrating that the presence of RNA largely defines the relative arrangements of the three domains via the formation of multiple RNA–protein hydrogen bonds. The additional presence of ATP or ADP had only a smaller effect on the domain fluctuations on the hundreds of nanosecond times scales. These findings confirm that ATP and ADP mainly serve to define the stability of the RecA1–RecA2 interface (Bourgeois et al. 2016; Sloan and Bohnsack 2018). In other words, in terms of a free energy landscape, ATP and ADP control the depth of the free energy minimum (if present) of the state with a closed RecA1–RecA2 interface; however, ATP and ADP have only a small effect on the free energy landscape underlying the large-scale fluctuations of more loosely packed domain arrangements.
Our simulations together with previous experimental data (Enders et al. 2022; Studer et al. 2020) suggest that, instead, binding of a G-patch protein modulates the free energy landscape of large-scale domain motions. We found that apo DHX15 exhibits highly diffusive and flexible domain arrangements, in line with the marked flexibility of apo Prp43 or apo Prp22, reflecting rather undefined relative domain arrangements. The addition of the G-patch protein NKRF to DHX15 led to a more structured conformational ensemble with reduced flexibility of the relative domain arrangements. Structurally, the G-patch protein imposed a more stable open RNA tunnel (Figure 8) as well as CTD–RecA2 distances and (to a lower degree) the RecA1–RecA2 distances reflecting mostly two specific relative arrangements, while avoiding large CTD–RecA2 distances. However, the effect of the G-patch protein on domain arrangements was far more subtle as compared to the effects from RNA or APT/ADP, suggesting that the G-patch protein fine-tunes the free energy landscape by only few kilojoules per mole. These conclusions are compatible with a characterization of G-patch proteins as flexible tethers that stabilize the relative domain arrangements (Bohnsack et al. 2021). However, additional studies will be required to clarify why more defined domain arrangements ultimately lead to increased kinetics of the overall conformational cycle as required for increased helicase or ATPase activity.
An unexpected finding in our simulations was the instability of conformations with an open RNA cleft. In absence of RNA, the CTD exhibited increased flexibility, however, the cleft mostly closed in our simulations, which seems to contrast crystallographic studied that revealed conformations with an open RNA cleft under different crystallization conditions (Tauchert et al. 2017). This discrepancy might be a consequence of the applied force field because (i) many force fields overstabilize salt bridges (Ahmed et al. 2018), while (ii) certain Amber force fields in conjunction with the TIP3P water model generally to overstabilize protein–protein interactions (Best et al. 2014). Alternatively, it cannot be fully excluded that crystal contacts favored a wide-open Prp43 conformation (Figure S5). Hence, additional simulations with other force fields, possibly augmented with X-ray solution scattering data (Chen and Hub 2015) may provide a route to unambiguously define the conformational ensemble of DEAH-box helicases in absence of RNA.
4 Materials and methods
MD simulations were set up and carried out with GROMACS 2019.5 (Abraham et al. 2015). Initial conformations of helicases were taken from crystallographic studies (Hamann et al. 2019; Studer et al. 2020; Tauchert et al. 2017). For simulations with ATP, the ATP analogue ADP-BeF3 was replaced with ATP. Initial conformations for different liganded states were generated by removing ligands (RNA, ADP-BeF3, ADP, or the fragment of the G-patch NKRF) from th respective crystal structures (see below). Missing residues were added using Modeller (Webb and Sali 2014) (Supplementary Material). The structures were placed into a simulation box of a dodecahedron keeping a distance of at least 1 nm between the protein and the box boundaries. The boxes were filled with explicit water and neutralized with K+ counter ions. Interactions of protein and RNA were described with the Amber14SB force field (Maier et al. 2015). Water was modeled with the TIP3P model (Jorgensen et al. 1983), and parameters for K+ were taken from Joung and Cheatham III (2008). The energy of the system was minimized with the steepest descent algorithm. Then, the system was equilibrated for 100 ps with position restraints acting on the backbone atoms including RNA and Mg2+ (force constant k = 1000 kJ mol−1 nm−2).
The following MD parameters were used in all simulations. Dispersion interactions and short-range repulsion were described together with a Lennard-Jones potential with a cut-off at 1 nm. Electrostatic interactions were described with the particle-mesh Ewald method (Darden et al. 1993) with a real-space cutoff at 1 nm. The temperature was controlled at 300 K using velocity-scaling (Bussi et al. 2007), thereby coupling protein, RNA, Mg2+, and ATP or ADP (if present) to one heat bath while coupling water and other ions to a second heat bath (τ = 0.5 ps). The pressure was controlled at 1 bar with the Parrinello-Rahman barostat (τ = 5 ps) (Parrinello and Rahman 1981). The velocity Verlet (md-vv) integrator was used for simulated tempering simulations and the Leap-frog (md) integrator was used for all other simulations, both with an integration time step of 2 fs. The geometry of water molecules was constrained with SETTLE (Miyamoto and Kollman 1992), while all other bonds were constrained with P-LINCS (Hess 2008).
Simulated tempering (ST) was used to enhance the conformation sampling of simulations of DHX15 with or without the G-patch protein. The temperature states covered 300 K–348 K in steps of 4 K. Attempts for temperature transitions were carried out every 500 integration steps and accepted or rejected with the Metropolis algorithm. The weights of the states were updated every 500 steps throughout the simulations using the Wang-Landau algorithm (Wang and Landau 2001). The initial weights were chosen using preliminary simulated annealing simulations following Park and Pande (2007).
Funding source: Deutsche Forschungsgemeinschaft
Award Identifier / Grant number: SFB 860/A16 and INST 256/539-1.
Acknowledgment
The authors thank Marieke Enders and Ralf Ficner for helpful discussions.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: This study was supported by the Deutsche Forschungsgemeinschaft (DOI 10.13039/501100001659) via grants SFB 860/A16 and INST 256/539-1.
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
Abdel-Monem, M., DÜrwald, H., and Hoffmann-Berling, H. (1976). Enzymic unwinding of DNA: 2. Chain separation by an ATP-Dependent DNA unwinding enzyme. Eur. J. Biochem. 65: 441–449, https://doi.org/10.1111/j.1432-1033.1976.tb10359.x.Suche in Google Scholar PubMed
Abdel-Monem, M. and Hoffmann-Berling, H. (1976). Enzymic unwinding of DNA: 1. Purification and characterization of a DNA-Dependent ATPase from Escherichia coli. Eur. J. Biochem. 65: 431–440, https://doi.org/10.1111/j.1432-1033.1976.tb10358.x.Suche in Google Scholar PubMed
Abraham, M.J., Murtola, T., Schulz, R., Páll, S., Smith, J.C., Hess, B., and Lindahl, E. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1: 19–25, https://doi.org/10.1016/j.softx.2015.06.001.Suche in Google Scholar
Absmeier, E., Santos, K.F., and Wahl, M.C. (2020). Molecular mechanism underlying inhibition of intrinsic ATPase activity in a Ski2-like RNA helicase. Structure 28: 236–243, https://doi.org/10.1016/j.str.2019.11.014.Suche in Google Scholar PubMed
Ahmed, M.C., Papaleo, E., and Lindorff-Larsen, K. (2018). How well do force fields capture the strength of salt bridges in proteins?PeerJ 6: e4967, https://doi.org/10.7717/peerj.4967.Suche in Google Scholar PubMed PubMed Central
Aravind, L. and Koonin, E.V. (1999). G-patch: a new conserved domain in eukaryotic RNA-processing proteins and type D retroviral polyproteins. Trends Biochem. Sci. 24: 342–344, https://doi.org/10.1016/s0968-0004(99)01437-1.Suche in Google Scholar PubMed
Becker, R.A. and Hub, J.S. (2023). Continuous millisecond conformational cycle of a DEAH box helicase reveals control of domain motions by atomic-scale transitions. Commun Biol 6: 379, https://doi.org/10.1038/s42003-023-04751-z.Suche in Google Scholar PubMed PubMed Central
Best, R.B., Zheng, W., and Mittal, J. (2014). Balanced protein–water interactions improve properties of disordered proteins and non-specific protein association. J. Chem. Theory Comput. 10: 5113–5124, https://doi.org/10.1021/ct500569b.Suche in Google Scholar PubMed PubMed Central
Bohnsack, K.E., Ficner, R., Bohnsack, M.T., and Jonas, S. (2021). Regulation of DEAH-box RNA helicases by G-patch proteins. Biol. Chem. 402: 561–579, https://doi.org/10.1515/hsz-2020-0338.Suche in Google Scholar PubMed
Bourgeois, C.F., Mortreux, F., and Auboeuf, D. (2016). The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat. Rev. Mol. Cell Biol. 17: 426–438, https://doi.org/10.1038/nrm.2016.50.Suche in Google Scholar PubMed
Bussi, G., Donadio, D., and Parrinello, M. (2007). Canonical sampling through velocity rescaling. J. Chem. Phys. 126: 014101, https://doi.org/10.1063/1.2408420.Suche in Google Scholar PubMed
Byrd, A.K., Matlock, D.L., Bagchi, D., Aarattuthodiyil, S., Harrison, D., Croquette, V., and Raney, K.D. (2012). Dda helicase tightly couples translocation on single-stranded DNA to unwinding of duplex DNA: Dda is an optimally active helicase. J. Mol. Biol. 420: 141–154, https://doi.org/10.1016/j.jmb.2012.04.007.Suche in Google Scholar PubMed PubMed Central
Byrd, A.K. and Raney, K.D. (2012). Superfamily 2 helicases. Front. Biosci. Landmark Ed. 17: 2070, https://doi.org/10.2741/4038.Suche in Google Scholar PubMed PubMed Central
Cantor, S.B., Bell, D.W., Ganesan, S., Kass, E.M., Drapkin, R., Grossman, S., Wahrer, D.C., Sgroi, D.C., Lane, W.S., Haber, D.A., et al.. (2001). BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105: 149–160, https://doi.org/10.1016/s0092-8674(01)00304-x.Suche in Google Scholar PubMed
Carney, S.P., Ma, W., Whitley, K.D., Jia, H., Lohman, T.M., Luthey-Schulten, Z., and Chemla, Y.R. (2021). Kinetic and structural mechanism for DNA unwinding by a non-hexameric helicase. Nat. Commun. 12: 1–14, https://doi.org/10.1038/s41467-021-27304-6.Suche in Google Scholar PubMed PubMed Central
Chen, P.-c. and Hub, J.S. (2015). Interpretation of solution X-ray scattering by explicit-solvent molecular dynamics. Biophys. J. 108: 2573–2584, https://doi.org/10.1016/j.bpj.2015.03.062.Suche in Google Scholar PubMed PubMed Central
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald: an N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 98: 10089–10092, https://doi.org/10.1063/1.464397.Suche in Google Scholar
Delagoutte, E. and Von Hippel, P.H. (2002). Helicase mechanisms and the coupling of helicases within macromolecular machines Part I: structures and properties of isolated helicases. Q. Rev. Biophys. 35: 431–478, https://doi.org/10.1017/s0033583502003852.Suche in Google Scholar PubMed
Delagoutte, E. and Von Hippel, P.H. (2003). Helicase mechanisms and the coupling of helicases within macromolecular machines Part II: integration of helicases into cellular processes. Q. Rev. Biophys. 36: 1–69, https://doi.org/10.1017/s0033583502003864.Suche in Google Scholar PubMed
Ellis, N.A., Groden, J., Ye, T.-Z., Straughen, J., Lennon, D.J., Ciocci, S., Proytcheva, M., and German, J. (1995). The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 83: 655–666, https://doi.org/10.1016/0092-8674(95)90105-1.Suche in Google Scholar PubMed
Enders, M., Ficner, R., and Adio, S. (2022). Regulation of the DEAH/RHA helicase Prp43 by the G-patch factor Pfa1. Proc. Natl. Acad. Sci. U. S. A. 119: e2203567119, https://doi.org/10.1073/pnas.2203567119.Suche in Google Scholar PubMed PubMed Central
Fairman-Williams, M.E., Guenther, U.-P., and Jankowsky, E. (2010). SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 20: 313–324, https://doi.org/10.1016/j.sbi.2010.03.011.Suche in Google Scholar PubMed PubMed Central
Fuller-Pace, F.V. (2006). DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 34: 4206–4215, https://doi.org/10.1093/nar/gkl460.Suche in Google Scholar PubMed PubMed Central
Fuller-Pace, F.V. (2013). DEAD box RNA helicase functions in cancer. RNA Biol. 10: 121–132, https://doi.org/10.4161/rna.23312.Suche in Google Scholar PubMed PubMed Central
Gowravaram, M., Bonneau, F., Kanaan, J., Maciej, V.D., Fiorini, F., Raj, S., Croquette, V., Le Hir, H., and Chakrabarti, S. (2018). A conserved structural element in the RNA helicase UPF1 regulates its catalytic activity in an isoform-specific manner. Nucleic Acids Res. 46: 2648–2659, https://doi.org/10.1093/nar/gky040.Suche in Google Scholar PubMed PubMed Central
Gray, M.D., Shen, J.-C., Kamath-Loeb, A.S., Blank, A., Sopher, B.L., Martin, G.M., Oshima, J., and Loeb, L.A. (1997). The werner syndrome protein is a DNA helicase. Nat. Genet. 17: 100–103, https://doi.org/10.1038/ng0997-100.Suche in Google Scholar PubMed
Hall, M.C. and Matson, S.W. (1999). Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol. 34: 867–877, https://doi.org/10.1046/j.1365-2958.1999.01659.x.Suche in Google Scholar PubMed
Hamann, F., Enders, M., and Ficner, R. (2019). Structural basis for RNA translocation by DEAH-box ATPases. Nucleic Acids Res. 47: 4349–4362, https://doi.org/10.1093/nar/gkz150.Suche in Google Scholar PubMed PubMed Central
Hamann, F., Schmitt, A., Favretto, F., Hofele, R., Neumann, P., Xiang, S., Urlaub, H., Zweckstetter, M., and Ficner, R. (2020). Structural analysis of the intrinsically disordered splicing factor Spp2 and its binding to the DEAH-box ATPase Prp2. Proc. Natl. Acad. Sci. U. S. A.117: 2948–2956, https://doi.org/10.1073/pnas.1907960117.Suche in Google Scholar PubMed PubMed Central
Hess, B. (2008). P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4: 116–122, https://doi.org/10.1021/ct700200b.Suche in Google Scholar PubMed
Hilbert, M., Karow, A.R., and Klostermeier, D. (2009). The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol. Chem. 390: 1237–1250, https://doi.org/10.1515/bc.2009.135.Suche in Google Scholar PubMed
Jacobs, A.-M., Nicol, SM., Hislop, RG., Jaffray, EG., Hay, RT., and Fuller-Pace, FV. (2007). SUMO modification of the DEAD box protein p68 modulates its transcriptional activity and promotes its interaction with HDAC1. Oncogene 26: 5866–5876, https://doi.org/10.1038/sj.onc.1210387.Suche in Google Scholar PubMed
Jarmoskaite, I. and Russell, R. (2014). RNA helicase proteins as chaperones and remodelers. Annu. Rev. Biochem. 83: 697–725, https://doi.org/10.1146/annurev-biochem-060713-035546.Suche in Google Scholar PubMed PubMed Central
Jeong, Y.-J., Levin, M.K., and Patel, S.S. (2004). The DNA-unwinding mechanism of the ring helicase of bacteriophage T7. Proc. Natl. Acad. Sci. U. S. A. 101: 7264–7269, https://doi.org/10.1073/pnas.0400372101.Suche in Google Scholar PubMed PubMed Central
Jorgensen, W.L., Chandrasekhar, J., Madura, J.D., Impey, R.W., and Klein, M.L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79: 926–935, https://doi.org/10.1063/1.445869.Suche in Google Scholar
Joung, I.S. and Cheatham, T.E.III (2008). Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 112: 9020–9041, https://doi.org/10.1021/jp8001614.Suche in Google Scholar PubMed PubMed Central
Kitao, S., Shimamoto, A., Goto, M., Miller, R.W., Smithson, W.A., Lindor, N.M., and Furuichi, Y. (1999). Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nat. Genet. 22: 82–84, https://doi.org/10.1038/8788.Suche in Google Scholar PubMed
Kretschmer, J., Rao, H., Hackert, P., Sloan, K.E., Höbartner, C., and Bohnsack, M.T. (2018). The m6A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5’–3’ exoribonuclease XRN1. RNA 24: 1339–1350, https://doi.org/10.1261/rna.064238.117.Suche in Google Scholar PubMed PubMed Central
Levin, M.K., Gurjar, M.M., and Patel, S.S. (2003). ATP binding modulates the nucleic acid affinity of hepatitis C virus helicase. J. Biol. Chem. 278: 23311–23316, https://doi.org/10.1074/jbc.m301283200.Suche in Google Scholar
Levin, M.K., Gurjar, M., and Patel, S.S. (2005). A Brownian motor mechanism of translocation and strand separation by hepatitis C virus helicase. Nat. Struct. Mol. Biol. 12: 429–435, https://doi.org/10.1038/nsmb920.Suche in Google Scholar PubMed
Linder, P. and Jankowsky, E. (2011). From unwinding to clamping—the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 12: 505–516, https://doi.org/10.1038/nrm3154.Suche in Google Scholar PubMed
Lohman, T.M. (1993). Helicase-catalyzed DNA unwinding. J. Biol. Chem. 268: 2269–2272, https://doi.org/10.1016/s0021-9258(18)53765-0.Suche in Google Scholar
Lohman, T.M. and Bjornson, K.P. (1996). Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem. 65: 169–214, https://doi.org/10.1146/annurev.bi.65.070196.001125.Suche in Google Scholar PubMed
Maier, J.A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K.E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11: 3696–3713, https://doi.org/10.1021/acs.jctc.5b00255.Suche in Google Scholar PubMed PubMed Central
Marinari, E. and Parisi, G. (1992). Simulated tempering: a new Monte Carlo scheme. Europhys. Lett. 19: 451–458, https://doi.org/10.1209/0295-5075/19/6/002.Suche in Google Scholar
Mathew, R., Hartmuth, K., Möhlmann, S., Urlaub, H., Ficner, R., and Lührmann, R. (2008). Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat. Struct. Mol. Biol. 15: 435–443, https://doi.org/10.1038/nsmb.1415.Suche in Google Scholar PubMed
Matson, S.W. and Kaiser-Rogers, K.A. (1990). DNA helicases. Annu. Rev. Biochem. 59: 289–329, https://doi.org/10.1146/annurev.bi.59.070190.001445.Suche in Google Scholar PubMed
Miyamoto, S. and Kollman, P.A. (1992). Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13: 952–962, https://doi.org/10.1002/jcc.540130805.Suche in Google Scholar
Ozgur, S., Buchwald, G., Falk, S., Chakrabarti, S., Prabu, J.R., and Conti, E. (2015). The conformational plasticity of eukaryotic RNA-dependent ATP ases. FEBS J. 282: 850–863, https://doi.org/10.1111/febs.13198.Suche in Google Scholar PubMed
Pan, A.C., Weinreich, T.M., Piana, S., and Shaw, D.E. (2016). Demonstrating an order-of-magnitude sampling enhancement in molecular dynamics simulations of complex protein systems. J. Chem. Theory Comput. 12: 1360–1367, https://doi.org/10.1021/acs.jctc.5b00913.Suche in Google Scholar PubMed
Park, S. and Pande, V.S. (2007). Choosing weights for simulated tempering. Phys. Rev. E 76: 016703, https://doi.org/10.1103/physreve.76.016703.Suche in Google Scholar
Parrinello, M. and Rahman, A. (1981). Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52: 7182–7190, https://doi.org/10.1063/1.328693.Suche in Google Scholar
Patel, S.S. and Donmez, I. (2006). Mechanisms of helicases. J. Biol. Chem. 281: 18265–18268, https://doi.org/10.1074/jbc.r600008200.Suche in Google Scholar PubMed
Patel, S.S. and Picha, K.M. (2000). Structure and function of hexameric helicases. Annu. Rev. Biochem. 69: 651–697, https://doi.org/10.1146/annurev.biochem.69.1.651.Suche in Google Scholar PubMed
Selak, N., Bachrati, C.Z., Shevelev, I., Dietschy, T., van Loon, B., Jacob, A., Hübscher, U., Hoheisel, J.D., Hickson, I.D., and Stagljar, I. (2008). The Bloom’s syndrome helicase (BLM) interacts physically and functionally with p12, the smallest subunit of human DNA polymerase δ. Nucleic Acids Res. 36: 5166, https://doi.org/10.1093/nar/gkn498.Suche in Google Scholar PubMed PubMed Central
Silverman, E., Edwalds-Gilbert, G., and Lin, R.-J. (2003). DExD/H-box proteins and their partners: helping RNA helicases unwind. Gene 312: 1–16, https://doi.org/10.1016/s0378-1119(03)00626-7.Suche in Google Scholar PubMed
Singleton, M.R., Dillingham, M.S., and Wigley, D.B. (2007). Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76: 23–50, https://doi.org/10.1146/annurev.biochem.76.052305.115300.Suche in Google Scholar PubMed
Sloan, K.E. and Bohnsack, M.T. (2018). Unravelling the mechanisms of RNA helicase regulation. Trends Biochem. Sci. 43: 237–250, https://doi.org/10.1016/j.tibs.2018.02.001.Suche in Google Scholar PubMed
Song, C., Hotz-Wagenblatt, A., Voit, R., and Grummt, I. (2017). SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 31: 1370–1381, https://doi.org/10.1101/gad.300624.117.Suche in Google Scholar PubMed PubMed Central
Stano, N.M., Jeong, Y.-J., Donmez, I., Tummalapalli, P., Levin, M.K., and Patel, S.S. (2005). DNA synthesis provides the driving force to accelerate DNA unwinding by a helicase. Nature 435: 370–373, https://doi.org/10.1038/nature03615.Suche in Google Scholar PubMed PubMed Central
Studer, M.K., Ivanović, L., Weber, M.E., Marti, S., and Jonas, S. (2020). Structural basis for DEAH-helicase activation by G-patch proteins. Proc. Natl. Acad. Sci. U. S. A. 117: 7159–7170, https://doi.org/10.1073/pnas.1913880117.Suche in Google Scholar PubMed PubMed Central
Tanner, N.K. and Linder, P. (2001). DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell 8: 251–262, https://doi.org/10.1016/s1097-2765(01)00329-x.Suche in Google Scholar PubMed
Tauchert, M.J., Fourmann, J.-B., Christian, H., Lührmann, R., and Ficner, R. (2016). Structural and functional analysis of the RNA helicase Prp43 from the thermophilic eukaryote Chaetomium thermophilum. Acta Crystallogr. Sect. F Struct. Biol. Commun. 72: 112–120, https://doi.org/10.1107/s2053230x15024498.Suche in Google Scholar
Tauchert, M.J., Fourmann, J.-B., Lührmann, R., and Ficner, R. (2017). Structural insights into the mechanism of the DEAH-box RNA helicase Prp43. Elife 6: e21510, https://doi.org/10.7554/elife.21510.Suche in Google Scholar PubMed PubMed Central
Tuteja, N. and Tuteja, R. (2004). Unraveling DNA helicases: motif, structure, mechanism and function. Eur. J. Biochem. 271: 1849–1863, https://doi.org/10.1111/j.1432-1033.2004.04094.x.Suche in Google Scholar PubMed
Van Brabant, A.J., Stan, R., and Ellis, N.A. (2000). DNA helicases, genomic instability, and human genetic disease. Annu. Rev. Genomics Hum. Genet. 1: 409–459, https://doi.org/10.1146/annurev.genom.1.1.409.Suche in Google Scholar PubMed
Velankar, S.S., Soultanas, P., Dillingham, M.S., Subramanya, H.S., and Wigley, D.B. (1999). Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97: 75–84, https://doi.org/10.1016/s0092-8674(00)80716-3.Suche in Google Scholar PubMed
von Hippel, P.H. and Delagoutte, E. (2001). A general model for nucleic acid helicases and their “coupling” within macromolecular machines. Cell 104: 177–190, https://doi.org/10.1016/s0092-8674(01)00203-3.Suche in Google Scholar PubMed
von Hippel, P.H. and Delagoutte, E. (2003). Macromolecular complexes that unwind nucleic acids. Bioessays 25: 1168–1177, https://doi.org/10.1002/bies.10369.Suche in Google Scholar PubMed
Walker, J.E., Saraste, M., Runswick, M.J., and Gay, N.J. (1982). Distantly related sequences in the alpha-and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1: 945–951, https://doi.org/10.1002/j.1460-2075.1982.tb01276.x.Suche in Google Scholar PubMed PubMed Central
Wang, F. and Landau, D.P. (2001). Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 86: 2050, https://doi.org/10.1103/physrevlett.86.2050.Suche in Google Scholar
Webb, B. and Sali, A. (2014). Protein structure modeling with MODELLER. Prot. Struct. Predict. 1137: 1–15, https://doi.org/10.1007/978-1-4939-0366-5_1.Suche in Google Scholar PubMed
West, S.C. (1996). DNA helicases: new breeds of translocating motors and molecular pumps. Cell 86: 177–180, https://doi.org/10.1016/s0092-8674(00)80088-4.Suche in Google Scholar PubMed
Wojtas, M.N., Pandey, R.R., Mendel, M., Homolka, D., Sachidanandam, R., and Pillai, R.S. (2017). Regulation of m6A transcripts by the 3’ → 5’ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol. Cell 68: 374–387, https://doi.org/10.1016/j.molcel.2017.09.021.Suche in Google Scholar PubMed
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/hsz-2023-0154).
© 2023 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Frontmatter
- Highlight: Integrative Structural Biology of Dynamic Macromolecular Assemblies
- Highlight: integrative structural biology of dynamic macromolecular assemblies
- Bayesian methods in integrative structure modeling
- The many faces of ribosome translocation along the mRNA: reading frame maintenance, ribosome frameshifting and translational bypassing
- Translation termination in human mitochondria – substrate specificity of mitochondrial release factors
- Molecular functions of RNA helicases during ribosomal subunit assembly
- Interaction of nucleoporins with nuclear transport receptors: a structural perspective
- Protein transport along the presequence pathway
- Autophagic and non-autophagic functions of the Saccharomyces cerevisiae PROPPINs Atg18, Atg21 and Hsv2
- Influence of phosphorylation on intermediate filaments
- Mediator structure and function in transcription initiation
- Structure and phase separation of the C-terminal domain of RNA polymerase II
- The DEAD-box RNA helicase Dbp5 is a key protein that couples multiple steps in gene expression
- Structure and function of spliceosomal DEAH-box ATPases
- Molecular simulations of DEAH-box helicases reveal control of domain flexibility by ligands: RNA, ATP, ADP, and G-patch proteins
Artikel in diesem Heft
- Frontmatter
- Highlight: Integrative Structural Biology of Dynamic Macromolecular Assemblies
- Highlight: integrative structural biology of dynamic macromolecular assemblies
- Bayesian methods in integrative structure modeling
- The many faces of ribosome translocation along the mRNA: reading frame maintenance, ribosome frameshifting and translational bypassing
- Translation termination in human mitochondria – substrate specificity of mitochondrial release factors
- Molecular functions of RNA helicases during ribosomal subunit assembly
- Interaction of nucleoporins with nuclear transport receptors: a structural perspective
- Protein transport along the presequence pathway
- Autophagic and non-autophagic functions of the Saccharomyces cerevisiae PROPPINs Atg18, Atg21 and Hsv2
- Influence of phosphorylation on intermediate filaments
- Mediator structure and function in transcription initiation
- Structure and phase separation of the C-terminal domain of RNA polymerase II
- The DEAD-box RNA helicase Dbp5 is a key protein that couples multiple steps in gene expression
- Structure and function of spliceosomal DEAH-box ATPases
- Molecular simulations of DEAH-box helicases reveal control of domain flexibility by ligands: RNA, ATP, ADP, and G-patch proteins