Synthesis, activity exploration, molecular docking, and affinity assessment of theophylline derivatives as inhibitors targeting IDO1
-
Ziyuan Wu
 und
Jianxue Yang
und
Jianxue Yang

Abstract
Indoleamine 2,3-dioxygenase 1 (IDO1) is an immunomodulatory enzyme associated with tumor immune evasion, making it a promising target for cancer therapy. This study aimed to identify novel holo-IDO1 inhibitors with distinct structural scaffolds. A series of theophylline derivatives containing 1,2,3-triazole groups were designed and synthesized through the condensation of theophylline acetic acid and 4-aminophenylacetylene. Among the synthesized compounds, 3c and 3e exhibited the most potent inhibitory effects in IDO1 enzymatic activity assays. Molecular docking and affinity prediction analyses provided insights into the binding affinities and mechanisms of action of the lead compounds. Our findings suggest that theophylline derivatives are promising holo-IDO1 inhibitors, warranting further development for potential therapeutic applications.
1 Introduction
Theophylline (Figure 1), a member of the xanthine compound class, is known to decrease smooth muscle tension, open air passages, and promote the release of endogenous epinephrine and norepinephrine [1]. Additionally, theophylline inhibits the release of calcium ions (Ca²⁺) from the endoplasmic reticulum of smooth muscle cells, reducing intracellular Ca²⁺ concentrations and producing an airway-dilating effect; thus, it is classified as a potent smooth muscle relaxant [2]. Theophylline is commonly used to treat conditions such as bronchial asthma, acute bronchitis, asthmatic bronchitis, and obstructive emphysema to alleviate wheezing symptoms [3,4]. It is also employed to manage chronic bronchitis and emphysema accompanied by bronchospasm.

Chemical structures of theophylline, caffeine, and pentoxifylline.
Beyond its applications in respiratory disorders, theophylline and its derivatives exhibit a range of biological activities, including antitumor [4,5], anti-inflammatory [6], central nervous system stimulation [7], immunomodulation [8], and hypoglycemic effects [9,10]. For example, the combination of theophylline with carmustine or cyclophosphamide has been shown to produce synergistic antitumor effects. Furthermore, theophylline and caffeine (Figure 1) can enhance the inhibitory effects of Adriamycin on tumor cells [11]. When used in conjunction with gemcitabine or cisplatin, theophylline has been found to induce programmed cell death in various tumors and to promote cancer cell apoptosis by reducing levels of the anti-apoptotic protein Bcl-2 in tumor cells [12,13]. Additionally, theophylline derivative pentoxifylline (Figure 1) and the proteasome inhibitor MG132 exhibit antileukemic activity in experimental models using human leukemia U937 cells, demonstrating an additive effect when used together [14,15].
A research group has synthesized hydrazone compounds with various substituents integrated into the theophylline structure, demonstrating selective inhibitory effects on T lymphoblastic leukemia cells. Notably, compound 34 (Figure 2) exhibited significant selectivity, with an IC50 value of 71.13 µM in suppressing T lymphoblastic leukemia CCRF-CEM cells and 5.75 µM in daunorubicin-resistant CEM-DNR cells. Furthermore, it displayed low toxicity, with IC50 values exceeding 25 µM on normal human skin fibroblast BJ cells and 23.18 µM on human lung fibroblast MRC-5 cells [16]. The compound inhibited RNA and DNA synthesis and mitosis in a dose-dependent manner while inducing apoptosis in CCRF-CEM cells, providing valuable insights for the design and synthesis of novel antineoplastic agents. In another study, a different research group incorporated aryl groups containing a hydroxylamine structure into the theophylline framework. Compound 23 (Figure 2), derived from this modification, demonstrated inhibitory effects on the EGFR target with an IC50 of 0.82 µM. Additionally, it effectively suppressed the human pancreatic cancer cell line Panc-1, exhibiting an IC50 of 1.70 µM. Compound 23 induced mitochondrial apoptosis in Panc-1 cells by increasing caspase-3 levels, downregulating Bcl-2, and upregulating cytochrome c. Cell cycle analysis revealed G1 and G2/M phase arrest in Panc-1 cells upon treatment with compound 23. Molecular docking studies further confirmed its potential as an EGFR inhibitor [17].

Chemical structures of two theophylline derivatives.
Indoleamine 2,3-dioxygenase (IDO1) is a heme-containing oxidoreductase that degrades tryptophan (Trp) to kynurenine, serving as a rate-limiting enzyme in the kynurenine signaling pathway [18]. Kynurenic acid directly inhibits the function of effector T cells and enhances the activities of regulatory T cells (Tregs), while Trp depletion in the microenvironment further suppresses T-cell proliferation, thereby exerting an immunosuppressive effect through multiple mechanisms [19]. Many cancer cells overexpress IDO1, and the IDO1-mediated Trp metabolic pathway is a significant factor in tumor immune evasion [20]. Consequently, inhibiting IDO1 modulates Trp levels in the tumor microenvironment, alleviates T-cell proliferation suppression, and enhances overall immune function, suggesting that IDO1 may serve as a promising target for cancer immunotherapy [21,22]. Additionally, IDO1 is implicated in neurological disorders, as it can reduce levels of 5-hydroxytryptamine, contributing to depression and leading to the accumulation of neurotoxic metabolites such as quinolinic acid in the brain. IDO1 is associated with several major human diseases, including Alzheimer’s disease, cataracts, and cancer [23]. As a result, IDO1 inhibitors are gaining significant attention as potential therapeutic agents.
Currently, multiple IDO1 inhibitors with diverse structures have been reported and have entered clinical research [24]. For instance, researchers at Amgen identified Amg-1 (Figure 3) as a selective IDO1 inhibitor with greater suppressive effects than TDO2 and IDO2, based on high-throughput screening of proprietary compound collections [25]. Its IC50 value was measured at 3.0 μM using the Bridge-IT tryptophan fluorescence assay. Structural analysis of Amg-1 in complex with IDO1 revealed that the hydrophobic 4-methylphenyl group penetrates the hydrophobic pocket (pocket A), while the thiazoltriazol structure coordinates directly with the heme’s ferrous iron (Fe²⁺). The hydrophilic 3,4-methylenedioxyphenyl structure occupies the hydrophilic pocket (pocket B). Thus, Amg-1 competitively inhibits IDO1 activity.

Pocket surface map of the crystal structure of IDO1 complexed with Amg-1 (PDB code: 4PK5). White represents amino acids contained in pocket A, and fuchsia represents amino acids contained in pocket B.
Utilizing the inherent hydrophilicity of the theophylline structure. This advantage facilitated our development of IDO1 inhibitors, leveraging both the hydrophilic properties of the theophylline structure and the specific features of the IDO1 target pockets. The theophylline molecule was strategically used as a reactive group to interact with the hydrophilic pocket (pocket B). We then introduced a 1,2,3-triazole structure that can interact with heme, along with a hydrophobic moiety designed to engage the hydrophobic pocket (pocket A). This innovative approach harnesses the unique characteristics of theophylline to create potent IDO1 inhibitors with a multifaceted mode of action.
2 Chemistry
The specific procedures, detailed in Figure 4, involved a stepwise process. Initially, a condensation reaction occurred between theophylline acetic acid (compound 1) and 4-aminophenylacetylene, leading to the formation of the terminal alkyne (compound 2). This terminal alkyne subsequently reacted with azide compounds containing various hydrophobic substituent groups to yield the desired target compounds. Our selection of hydrophobic groups included seven benzyl compounds (3a–3n) and phenyl compounds with identical substituent groups (3a–3n). The activities of these compounds were systematically compared to evaluate their effectiveness.

The reaction routes to compounds 3a–3n.
3 Results and discussion
3.1 IDO1 inhibition study
We selected HeLa cells, known for their high expression levels of IDO1, as the experimental model for in vitro IDO1 enzyme inhibition studies. Amg-1 served as the positive control, and its IC50 value, measured under our experimental conditions, was 3.97 µM, closely aligning with expected values. Our findings indicated that compounds with benzyl substituents – specifically 3a, 3c, 3e, and 3f – exhibited significant inhibitory effects on IDO1, with corresponding IC50 values of 73.24, 9.91, 19.26, and 80.71 µM, respectively (Table 1). Notably, these benzyl-substituted compounds generally demonstrated superior efficacy compared to their phenyl counterparts with identical substituents. By comparing the structure–activity relationships of compounds 3a and 3c, as well as compounds 3h and 3j, we found that substituents in the para position enhance the insertion into the Packet A active site, improving IDO1 inhibition activity.
IDO1 inhibitory activities of designed derivatives
| Compd no. | IDO1 | Compd no. | IDO1 |
|---|---|---|---|
| IC50 (μM) | IC50 (μM) | ||
| 3a | 73.24 ± 3.08 | 3h | >100 |
| 3b | >100 | 3i | >100 |
| 3c | 9.91 ± 1.74 | 3j | 84.53 ± 3.92 |
| 3d | >100 | 3k | >100 |
| 3e | 19.26 ± 4.93 | 3l | 96.71 ± 4.66 |
| 3f | 80.71 ± 2.78 | 3m | >100 |
| 3g | >100 | 3n | >100 |
IC50 values were fitted from single-point inhibition curves, and two parallel experiments were performed for each compound. IC50 values were calculated using Graph Pad Prism 6.0 software. These results are reported as the averages ± standard deviation.
Cellular-level enzyme activity assays are influenced not only by the inhibitory effects of compounds but also by factors such as cell permeability and substrate and metabolite transport. In contrast, enzyme-level activity assays minimize the impact of these variables by directly assessing the inhibitor’s effects on the enzyme. This method allows for a more accurate reflection of the inhibitor’s potency against IDO1. We conducted enzyme-level activity assays on compounds 3c and 3e, which yielded IC50 values of 29.59 and 23.77 µM, respectively, thereby further confirming the inhibitory effects of the designed compounds on IDO1.
3.2 Inhibitory effect of compound 3e on the transcription level of IDO1
We found that compound 3e maintains consistent inhibitory values against IDO1 enzyme activity at both the cellular and molecular levels, indicating that compound 3e is less affected by other cellular factors. To assess whether 3e also influences the gene expression of IDO1, real-time polymerase chain reaction (PCR) was performed. As shown in Figure 5, IFN-γ exposure caused a robust increase in the mRNA level of IDO1 in Hela cells. However, treatment with 40 μM 3e significantly decreased the IFN-γ-stimulated increase in IDO1 mRNA. This result showed that 3e had the potential to downregulate the transcription of IDO1 under IFN-γ stimulation.

Effect of 3e on IFN-γ-induced mRNA levels of IDO1 in Hela cells. Hela cells were stimulated with IFN-γ (100 ng/mL) for 24 h following treatment with 3e (20, or 40 μM). The mRNA levels of IDO1 were measured by real-time PCR (n = 3). P < 0.001 vs the control group; ***P < 0.001 vs the IFN-γ group.
3.3 Binding modes studies
To provide a more intuitive analysis of the influence of benzyl and phenyl groups on compound activities, we selected four representative compounds for docking studies. Compounds 3a and 3h exhibited docking scores of −7.640 and −5.075, respectively. Compared to compound 3h, the o-chlorobenzyl group of compound 3a demonstrated a greater capacity for insertion into pocket A, which is formed by hydrophobic amino acids. Additionally, the triazole ring attached to the benzyl group was positioned closer to the heme, facilitating enhanced π–π interactions with Phe163. The theophylline moiety of compound 3a engaged in hydrophobic interactions with key amino acids in pocket B and formed a hydrogen bond with Arg231. In contrast, the theophylline moiety of compound 3h formed hydrogen bonds with Gly236 and Gly380 and participated in cation–π interactions with Arg231.
The docking scores for compounds 3c and 3j were −7.381 and −2.996, respectively. Notably, the p-chlorobenzyl group of compound 3c positioned the triazole ring closer to the heme iron than in compound 3j, resulting in stronger interactions with key amino acids in pocket A. The theophylline moiety of compound 3c formed individual hydrogen bonds with Gly236 and Gly261, as well as cation–π interactions with Arg231. Conversely, the theophylline moiety of compound 3j formed hydrogen bonds with Arg231, Gly236, and Thr379.
For compounds 3e and 3l, the docking scores were −7.776 and −4.863, respectively. The presence of a benzyl group in compound 3e caused the triazole ring to tilt toward the heme iron, leading to stronger interactions compared to compound 3l. Both compounds’ theophylline moieties were situated in pocket B, with the imidazole ring of the theophylline moiety in compound 3l forming hydrogen bond interactions with Thr379. For compounds 3f and 3m, the docking scores were −5.952 and −5.274, respectively. In contrast to compound 3m, the o-bromobenzyl group of compound 3f inserted upward into pocket A, while the o-bromophenyl group of compound 3m was positioned slightly farther from pocket A. Additionally, the triazole ring of compound 3f was closer to the heme iron. The theophylline moieties of both compounds were located in pocket B, with the theophylline moiety of compound 3m forming additional hydrogen bonds with Arg231 and Gly261.
3.4 Affinity analysis of compounds with IDO1
To enhance our understanding of the affinities between the compounds and the IDO1 protein, we employed DeepPurpose to predict their interactions. The results are presented in Table 2, revealing that all designed compounds exhibited medium activity. Generally, benzyl compounds with identical substituent groups demonstrated superior affinities compared to their phenyl counterparts. Notably, compound 3c exhibited the highest affinity among them, aligning with the observed trends in the experimental results.
Affinity prediction results of the designed molecules
| Compd no. | Morgan-MLP | Compd no. | Morgan-MLP |
|---|---|---|---|
| 3a | 4.9417 | 3h | 4.4216 |
| 3c | 5.2978 | 3j | 4.8870 |
| 3e | 4.9712 | 3l | 4.9547 |
| 3f | 4.8147 | 3m | 4.7507 |
4 Conclusion
Inspired by the inherent hydrophilicity of theophylline, we designed a series of innovative theophylline derivatives featuring 1,2,3-triazoles and diverse aryl substituents. In our IDO1 inhibitory activity assays, several compounds demonstrated significant inhibitory effects, particularly compound 3c (IC50 = 9.91 µM for cells) and compound 3e (IC50 = 23.77 µM for enzyme), both surpassing the positive control, Amg-1. Docking and affinity prediction analyses indicated that derivatives with benzyl substituents exhibited greater activity than those with phenyl substituents. Overall, our study provides novel insights into the development of innovative holo-IDO1 inhibitors.
5 Experimental protocols
5.1 Materials and chemistry
The theophylline derivatives were synthesized in our laboratory. 1H NMR and 13C NMR spectra were acquired in (dimethyl sulfoxide)-d 6 with a Bruker 400 spectrometer. Hela cell line, DMEM medium, and fetal bovine serum were purchased from ATCC (Virginia, USA). Recombinant human IFN-γ was purchased from R&D systems (Emeryville, CA, USA). Recombinant Human IDO Protein was purchased from R&D systems (Emeryville, CA, USA).
5.2 General synthetic procedure for compound 2
Theophylline acetic acid (compound 1, 5 g), 4-aminophenylacetylene (3.7 g), 2-(7-azobenzotriazole)-N,N,N′,N′-tetramethylurea hexafluorophosphorate (13.0 g), N,N-diisopropyl ethylamine; (8.1 g), and solvent dimethylformamide (DMF) 250 mL were added to a 500 mL reaction flask at room temperature and stirred under nitrogen protection for 24 h. Thin layer chromatography (TLC) was used for monitoring. After 24 h, the reaction was completed and the reaction solution was light brown. DMF was removed by vacuum concentration, dichloromethane was added to extract the reaction solution (150 mL × 3). Combine all organic solutions and wash them with saturated sodium chloride (150 mL × 2) to pH = 7, and the viscous brownish-yellow liquid was obtained by vacuum distillation. Under ultrasonic vibration, methanol was slowly added drop by drop and solid precipitated. After that, it was left to stand, filtered, and dried to obtain the condensation intermediate of theophylline acetic acid and 4-aminophenylacetylene (compound 2, 4.1 g).
5.3 General procedure for preparation of compounds 3a–3n
The reaction reagents were aryl-azide (1.2 mmol) and compound 2 (1.0 mmol), which were added to 20 mL of a mixed solvent (tetrahydrofuran:water:tert-butanol = 1:1:1). The reaction was performed in the presence of copper sulfate pentahydrate (0.1 mmol) and sodium ascorbate (0.2 mmol) at 80°C. The reaction progress was monitored by TLC, and upon completion, the mixture was extracted with dichloromethane (15 mL × 3). The combined organic phase was successively washed with water (10 mL × 3) and brine (10 mL × 3), dried over anhydrous sodium sulfate, and concentrated under reduced pressure [22]. The desired product was then isolated by column chromatography (CH2Cl2/MeOH = 20:1).
Compound 3a. m.p. 277.2–280.4°C. HR-MS(ESI): Calcd. C24H22ClN8O3 [M+H]+ m/z: 505.1503, found: 505.1501. 1H NMR (400 MHz, DMSO-d 6) δ 10.53 (s, 1H, NH-H), 8.53 (s, 1H, CH-H), 8.08 (s, 1H, Ar-H), 7.82 (d, J = 8.0 Hz, 2H, Ar-H), 7.65 (d, J = 8.0 Hz, 2H, CH-H, Ar-H), 7.54 (d, J = 8.0 Hz, 1H, Ar-H), 7.43–7.37 (m, 2H, Ar-H), 7.28 (d, J = 8.0 Hz, 1H, Ar-H), 5.75 (d, J = 8.0, 2H, CH2-H), 5.22 (s, 2H, CH2-H), 3.46 (s, 3H, CH3-H), 3.20 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.4, 154.9, 151.4, 148.4, 146.6, 144.2, 138.7, 133.6, 133.09, 130.9, 130.7, 130.1, 128.2, 126.3, 126.2, 121.8, 119.8, 106.9, 51.2, 49.2, 29.9, 27.9.
Compound 3b. m.p. 271.4–274.3°C. HR-MS(ESI): Calcd. C25H25N8O4 [M+H]+ m/z: 501.1999, found: 501.2004. 1H NMR (400 MHz, DMSO-d 6) δ 10.53 (s, 1H, NH-H), 8.55 (s, 1H, CH-H), 8.08 (s, 1H, Ar-H), 7.81 (d, J = 8.0 Hz, 2H, Ar-H), 7.65 (d, J = 8.0 Hz, 2H, CH-H, Ar-H), 7.30 (t, J = 8.0 Hz, 1H, Ar-H), 6.91 (dd, J 1 = 8.0 Hz, J 2 = 8.0 Hz, 3H, Ar-H), 5.60 (s, 2H, CH2-H), 5.22 (s, 2H, CH2-H), 3.75 (s, 3H, OCH3-H), 3.46 (s, 3H, CH3-H), 3.20 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.4, 159.9, 154.9, 151.4, 148.4, 146.8, 144.2, 138.7, 137.8, 130.4, 126.4, 126.2, 121.5, 120.4, 119.8, 114.2, 113.9, 106.9, 55.6, 53.4, 29.9, 27.9.
Compound 3c. m.p. 260.8–264.8°C. HR-MS(ESI): Calcd. C24H22ClN8O3 [M+H]+ m/z: 505.1503, found: 505.1504. Anal. Calcd for C24H21ClN8O3: C, 57.09; H, 4.19; N, 22.19. Found: C, 57.21; H, 4.13; N, 22.05; Cu, 0. 1H NMR (400 MHz, DMSO-d 6) δ 10.51 (s, 1H, NH-H), 8.54 (s, 1H, CH-H), 8.08 (s, 1H, Ar-H), 7.80 (d, J = 8.0 Hz, 2H, Ar-H), 7.64 (d, J = 8.0 Hz, 2H, CH-H, Ar-H), 7.48–7.43 (m, 2H, Ar-H), 7.37 (d, J = 8.0 Hz, 2H, Ar-H), 5.64 (s, 2H, CH2-H), 5.22 (s, 2H, CH2-H), 3.46 (s, 3H, CH3-H), 3.20 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.4, 155.0, 151.5, 148.5, 146.9, 144.3, 138.7, 135.5, 133.3, 129.3, 126.3, 121.6, 119.8, 106.9, 52.7, 49.2, 29.9, 27.9.
Compound 3d. m.p. 270.0–274.8°C. HR-MS(ESI): Calcd. C25H22F3N8O3 [M+H]+ m/z: 539.1767 found: 539.1766. 1H NMR (400 MHz, DMSO-d 6) δ 10.53 (s, 1H, NH-H), 8.54 (s, 1H, CH-H), 8.08 (s, 1H, Ar-H), 7.82 (d, J = 8.0 Hz, 3H, Ar-H), 7.72–7.57 (m, 4H, CH-H, Ar-H), 7.24 (d, J = 8.0 Hz, 1H, Ar-H), 5.83 (s, 2H, CH2-H), 5.23 (s, 2H, CH2-H), 3.46 (s, 3H, CH3-H), 3.20 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.4, 155.0, 148.4, 146.8, 144.3, 138.8, 133.7, 130.7, 129.4, 126.7, 126.7, 126.3, 122.1, 119.8, 106.9, 52.5, 50.2, 49.2, 39.9.29.9, 27.9, 7.6.
Compound 3e. m.p. 176.2–179.9°C. HR-MS(ESI): Calcd. C24H23N8O3 [M+H]+ m/z: 471.1893, found: 471.1903. 1H NMR (400 MHz, DMSO-d 6) δ 10.53 (s, 1H, NH-H), 8.56 (s, 1H, CH-H), 8.09 (s, 1H, Ar-H), 7.80 (s, 2H, Ar-H), 7.68–7.62 (m, 2H, CH-H, Ar-H), 7.42–7.31 (m, 5H, Ar-H), 5.64 (s, 2H, CH2-H), 5.22 (s, 2H, CH2-H), 3.46 (s, 3H, CH3-H), 3.20 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.4, 146.9, 144.2, 136.5, 129.3, 128.6, 128.4, 126.2, 121.5, 119.8, 53.5, 49.2, 40.2, 29.9, 27.9.
Compound 3f. m.p. 286.1–288.2°C. HR-MS(ESI): Calcd. C24H22BrN8O3 [M+H]+ m/z: 549.0998, found: 549.0992. 1H NMR (400 MHz, DMSO-d 6) δ 10.51 (s, 1H, NH-H), 8.51 (s, 1H, CH-H), 8.08 (s, 1H, Ar-H), 7.82 (d, J = 8.0 Hz, 2H, Ar-H), 7.68 (dd, J 1 = 8.0 Hz, J 2 = 8.0 Hz, 3H, CH-H, Ar-H), 7.45–7.40 (m, 1H, Ar-H), 7.32 (t, J 1 = 12.0 Hz, J 2 = 8.0 Hz, 1H, Ar-H), 7.22 (d, J = 12.0 Hz, 1H, Ar-H), 5.72 (s, 2H, CH2-H), 5.22 (s, 2H, CH2-H), 3.46 (s, 3H, CH3-H), 3.20 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.4, 155.0, 151.5, 148.4, 146.7, 144.3, 138.8, 135.3, 133.4, 128.8, 126.4, 126.3, 123.3, 121.9, 119.8, 106.9, 53.6, 49.2, 29.9, 27.9.
Compound 3g. m.p. 286.5–287.8°C. HR-MS(ESI): Calcd. C25H22F3N8O3 [M+H]+ m/z: 539.1767, found: 539.1776. 1H NMR (400 MHz, DMSO-d 6) δ 10.53 (s, 1H, NH-H), 8.60 (s, 1H, CH-H), 8.08 (s, 1H, Ar-H), 7.79 (dd, J 1 = 8.0 Hz, J 1 = 8.0 Hz, 4H, Ar-H), 7.66 (d, J = 8.0 Hz, 2H, CH-H, Ar-H), 7.54 (d, J = 8.0 Hz, 2H, Ar-H), 5.77 (s, 2H, CH2-H), 5.23 (s, 2H, CH2-H), 3.46 (s, 3H, CH3-H), 3.20 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.4, 155.0, 151.5, 148.4, 147.0, 144.3, 141.1, 138.8, 129.1, 126.3, 126.3, 126.2, 126.2, 121.8, 119.8, 106.9, 52.8, 49.2, 29.9, 27.9.
Compound 3h. m.p. 280.9–282.4°C. HR-MS(ESI): Calcd. C23H20ClN8O3 [M+H]+ m/z: 491.1347, found: 491.1354. 1H NMR (400 MHz, DMSO-d 6) δ 10.58 (s, 1H, NH-H), 9.23 (s, 1H, CH-H), 8.09 (s, 1H, Ar-H), 7.93 (dd, J 1 = 8.0 Hz, J 2 = 8.0 Hz, 4H, Ar-H), 7.71 (d, J = 8.0 Hz, 2H, Ar-H), 7.64 (t, J 1 = 8.0 Hz, J 2 = 8.0 Hz, 2H, CH-H, Ar-H), 7.52 (t, J 1 = 4.0 Hz, J 2 = 8.0 Hz, 1H, Ar-H), 5.24 (s, 2H, CH2-H), 3.47 (s, 3H, CH3-H), 3.21 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.5, 155.0, 151.5, 148.4, 147.5, 144.3, 139.0, 137.1, 129.2, 126.5, 126.0, 120.4, 119.9, 106.9, 49.26, 29.9, 27.9.
Compound 3i. m.p. 266.0–269.7°C. HR-MS(ESI): Calcd. C24H23N8O4 [M+H]+ m/z: 487.1842, found: 487.1771. 1H NMR (400 MHz, DMSO-d 6) δ 10.59 (s, 1H, NH-H), 9.28 (s, 1H, CH-H), 8.09 (s, 1H, Ar-H), 7.87 (dd, J 1 = 12.0 Hz, J 2 = 8.0 Hz, 4H, Ar-H), 7.78–7.64 (m, 3H, CH-H, Ar-H), 7.37 (s, 1H, Ar-H), 5.25 (s, 2H, CH2-H), 3.47 (s, 3H, CH3-H), 3.21 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 155.0, 151.5, 144.3, 132.4, 132.3, 126.5, 119.9, 119.7, 116.3, 55.4, 49.2, 40.2, 29.9, 27.9.
Compound 3j. m.p. 289.2–293.7°C. HR-MS(ESI): Calcd. C23H20ClN8O3 [M+H]+ m/z: 491.1347, found: 491.1351. 1H NMR (400 MHz, DMSO-d 6) δ 10.58 (s, 1H, NH-H), 8.97 (s, 1H, CH-H), 8.09 (s, 1H, Ar-H), 7.91 (d, J = 4.0 Hz, 2H, Ar-H), 7.82–7.76 (m, 2H, Ar-H), 7.71–7.61 (m, 2H, CH-H, Ar-H), 5.24 (s, 2H, CH2-H), 3.47 (s, 3H, CH3-H), 3.21 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 151.5, 144.3, 126.4, 122.8, 122.8, 119.9, 119.9, 117.34, 117.1, 49.2, 40.4, 29.9, 27.9.
Compound 3k. m.p. 299.2–305.1°C. HR-MS(ESI): Calcd. C24H29F3N8O3 [M+H]+ m/z: 525.1610, found: 525.1619. 1H NMR (400 MHz, DMSO-d 6) δ 10.56 (s, 1H, NH-H), 9.25 (s, 1H, CH-H), 8.09 (s, 1H, Ar-H), 7.98 (d, J = 12.0 Hz, 2H, Ar-H), 7.89 (d, J = 12.0 Hz, 2H, Ar-H), 7.71 (d, J = 8.0 Hz, 4H, CH-H, Ar-H), 5.24 (s, 2H, CH2-H), 3.47 (s, 3H, CH3-H), 3.21 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.5, 155.0, 151.5, 148.4, 147.7, 144.3, 139.1, 135.9, 133.4, 130.4, 126.5, 125.8, 122.1, 119.9, 119.7, 106.9, 49.2, 29.9, 27.9.
Compound 3l. m.p. 292.7–296.4°C. HR-MS(ESI): Calcd. C23H21N8O3 [M+H]+ m/z: 457.1737, found: 457.1737. 1H NMR (400 MHz, DMSO-d 6) δ 10.50 (s, 1H, NH-H), 8.54 (s, 1H, CH-H), 8.08 (s, 1H, Ar-H), 7.80 (d, J = 8.0 Hz, 2H, Ar-H), 7.64 (d, J = 8.0 Hz, 2H, CH-H, Ar-H), 7.30 (t, J 1 = 8.0 Hz, J 2 = 8.0 Hz, 1H, Ar-H), 6.91 (dd, J 1 = 8.0 Hz, J 2 = 8.0 Hz, 3H, Ar-H), 5.22 (s, 2H, CH2-H), 3.75 (s, 3H, OCH3-H), 3.46 (s, 3H H, CH3-H), 3.20 (s, 3H H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.4, 155.0, 130.4, 126.2, 121.5, 120.5, 119.8, 114.2, 114.0, 55.6, 53.4, 40.2, 29.9.
Compound 3m. m.p. 267.5–270.2°C. HR-MS(ESI): Calcd. C23H20BrN8O3 [M+H]+ m/z: 535.0842, found: 535.0831. 1H NMR (400 MHz, DMSO-d 6) δ 10.56 (s, 1H, NH-H), 8.94 (s, 1H, CH-H), 8.09 (s, 1H, Ar-H), 7.95–7.90 (m, 3H, Ar-H), 7.77–7.56 (m, 5H, CH-H, Ar-H), 5.24 (s, 2H, CH2-H), 3.47 (s, 3H, CH3-H), 3.21 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.5, 155.0, 151.5, 148.5, 146.7, 144.3, 139.0, 136.7, 134.1, 132.5, 129.5, 129.2, 126.4, 125.9, 123.5, 119.9, 119.4, 49.2, 40.2, 29.9, 27.9.
Compound 3n. m.p. 280.8–285.7°C. HR-MS(ESI): Calcd. C24H20F3N8O3 [M+H]+ m/z: 525.1610, found: 525.1621. 1H NMR (400 MHz, DMSO-d 6) δ 10.57 (s, 1H, NH-H), 9.38 (s, 1H, CH-H), 8.21 (d, J = 8.0 Hz, 2H, Ar-H), 8.09 – 8.02 (m, 3H, Ar-H), 7.92 (d, J = 8.0 Hz, 2H, Ar-H), 7.72 (d, J = 8.0 Hz, 2H, CH-H, Ar-H), 5.24 (s, 2H, CH2-H), 3.47 (s, 3H, CH3-H), 3.21 (s, 3H, CH3-H). 13C NMR (100 MHz, DMSO-d 6) δ 165.5, 155.0, 151.5, 148.4, 144.3, 139.9, 139.2, 127.8, 127.7, 126.5, 125.7,120.8, 119.9, 119.8, 106.9, 49.3, 29.9, 27.9.
5.4 IDO1 enzymatic inhibition assay
Cellular: To demonstrate the inhibitory effect of the designed compounds against IDO1, Hela cells were used to evaluate the enzyme activity inhibition assay in vitro [25]. Hela cells were seeded at 50,000–60,000 cells per well into 96-well plate in 100 μL of modified eagle medium complete growth medium for 12–18 h. On the second day, 100 μL per well of diluted inhibitor was added at a final concentration of 100 ng/mL human IFN-γ and then incubated at 37℃, 5% CO2 for 18 h. On the third day, 140 μL of medium was removed into a new 96-well plate and precipitated the protein with 10 μL of 6.1 N TCA (CCl3COOH) at 50℃ for 30 min. The plate was centrifuged at 2,500 rpm for 10 min. Then, 100 μL of supernatant per well was transferred to another 96-well plate and mixed with 100 μL of p-dimethylaminobenzaldehyde in CH3COOH [2% (w/v)]. The plate was incubated at 25℃ for 10 min, and the yellow color derived from kynurenine was recorded by measuring absorbance at 480 nm using a microplate reader (PE, USA). Graphs of inhibition curves with IC50 values were generated using Prism 6.0.
Enzymatic: Recombinant human IDO1 was expressed and purified in accordance with established protocols. The assay was performed using a standard reaction mixture (100 µL) comprising 100 mM potassium phosphate buffer (pH 6.5), 40 mM ascorbic acid (neutralized with NaOH), 200 µg/mL catalase, 20 µM methylene blue, and 0.05 µM rhIDO1 (65 kD), which was then combined with the substrate l-tryptophan and the test sample at a predetermined concentration. The reaction was conducted at 37°C for 45 min and terminated by adding 20 µL of 30% (w/v) trichloroacetic acid. After heating at 65°C for 15 min, 100 µL of 2% (w/v) p-dimethylaminobenzaldehyde in acetic acid was added to each well. The resulting yellow pigment derived from kynurenine was measured at 492 nm using a microplate reader. IC50 values were calculated using GraphPad Prism 6.0 software.
5.5 Total RNA extraction and real-time PCR
Hela cells were seeded in 12-well plates at a density of 1.5 × 105 cells/mL for 24 h. The cells were stimulated with IFN-γ (100 ng/mL) for 24 h following treatment with 3e (20 or 40 μM). Total RNA was extracted using TRNzol (TIANGEN, Beijing, China) according to the manufacturer’s instructions. cDNA was prepared using HiScript III RT SuperMix for qPCR (Vazyme, Nanjing, China) with 500 ng total RNA. Quantitative real-time PCR was performed using cDNA and ChamQ Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China) on an ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Relative gene expression was calculated using the ΔΔCT method. The primers for IDO-1 were 5′-CACTTTGCTAAAGGCGCTGTTGGA-3′ and 5′-GGTTGCCTTTCCAGCCAGACAAAT-3′ and for β-actin were 5′-CATGTACGTTGCTATCCAGGC-3′ and 5′-CTCCTTAATGTCACGCACGAT-3′.
5.6 Molecular docking
In this study, the complex of IDO1 and Amg-1 (PDB code: 4PK5) was selected as the crystal structure, and the protein was processed by the Protein Preparation Wizard module. The compounds were constructed by ChemDraw and optimized by the LigPrep module. A cubic box sized 20 Å × 20 Å × 20 Å was generated surrounding the cocrystal ligand Amg-1, and the heme iron was used as the center to generate the metal coordination constraints. Docking was performed by utilizing the Extra precision (XP) sampling, and the visualization of docking results was presented by PyMOL.
5.7 Affinity prediction study
We then employed DeepPurpose to analyze the affinities between IDO1 and the portion of compounds with desirable activities. DeepPurpose developed by Huang et al. is a deep-learning library for affinity prediction. In our previous research, we collected the IDO1 dataset containing 1,785 active molecules and verified the structural diversity of the dataset by PCA dimensionality reduction and k-means clustering. We then used the eight encoders in the DeepPurpose library (Morgan, PubChem, Daylight, RDKit2D, CNN, RNN, Transformer, and MPNN) to train the IDO1 dataset. The IDO1 dataset was randomly divided into the training, validation, and test datasets at a ratio of 8:1:1. It was finally demonstrated that the MLP model based on Morgan performed well in the IDO1 dataset (mean squared error = 0.467, P = 0.827, confidence interval = 0.822) (Figure 6). The performance of the IDO1 dataset in different models is shown in Figure 7. Thus, we directly employed the MLP model based on Morgan to predict the affinities of selected compounds in this study.

The prediction performance of the IDO1 dataset on several models.
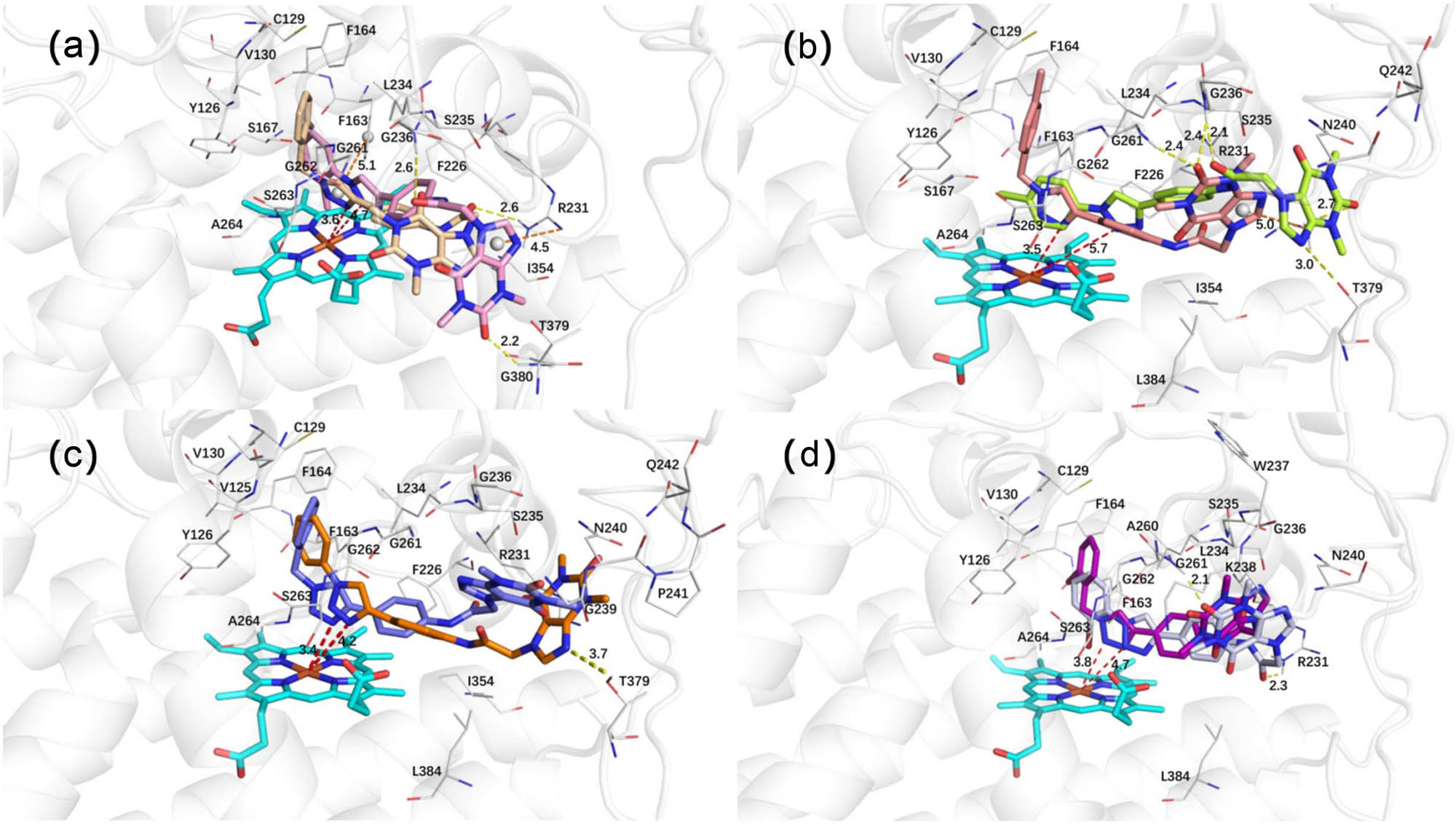
(a) The binding mode of compounds 3a and 3h with IDO1. (b) The binding mode of compounds 3c and 3j with IDO1. (c) The binding mode of compounds 3e and 3l with IDO1. (d) The binding mode of compounds 3f and 3m with IDO1.
-
Funding information: This work was supported by the Innovation Capability Enhancement Project for Technology-based SMEs in Shandong Province (2022TSGC1055), College Students’ Innovative Entrepreneurial Training Plan Program (202310464081), and The Henan Province Medical Science and Technology Research Project (LHGJ20230450).
-
Author contributions: Ziyuan Wu: methodology, data curation, formal analysis, supervision, and writing – original draft. Songmao Liang: conceptualization, validation, and investigation. Shunhua Hu: formal analysis, software, validation, and writing – original draft. Yimian Wang: formal analysis, software, validation, and writing – original draft. Long-Fei Mao: software, validation, and writing – original draft. Xixi Hou: conceptualization, investigation, methodology, formal analysis, and writing – original draft. Jianxue Yang: funding acquisition, project administration, conceptualization, and writing – review and editing.
-
Conflict of interest: Authors state no conflict of interest.
-
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
[1] Schoeder CT, Mahardhika AB, Drabczynska A, Kiec-Kononowicz K, Muller CE. Discovery of tricyclic xanthines as agonists of the cannabinoid-activated orphan G-protein-coupled receptor GPR18. ACS Med Chem Lett. 2020;11:2024–31.10.1021/acsmedchemlett.0c00208Suche in Google Scholar PubMed PubMed Central
[2] Shelke RU, Degani MS, Raju A, Ray MK, Rajan MG. Fragment discovery for the design of nitrogen heterocycles as Mycobacterium tuberculosis dihydrofolate reductase inhibitors. Arch Pharmazie. 2016;349:602–13.10.1002/ardp.201600066Suche in Google Scholar PubMed
[3] van den Berg D, Zoellner KR, Ogunrombi MO, Malan SF, Terre’Blanche G, Castagnoli Jr N, et al. Inhibition of monoamine oxidase B by selected benzimidazole and caffeine analogues. Bioorg Med Chem. 2007;15:3692–702.10.1016/j.bmc.2007.03.046Suche in Google Scholar PubMed
[4] Baraldi PG, Preti D, Tabrizi MA, Romagnoli R, Saponaro G, Baraldi S, et al. Structure-activity relationship studies of a new series of imidazo[2,1-f]purinones as potent and selective A(3) adenosine receptor antagonists. Bioorg Med Chem. 2008;16:10281–94.10.1016/j.bmc.2008.10.049Suche in Google Scholar PubMed
[5] Balo MC, Brea J, Caamano O, Fernandez F, Garcia-Mera X, Lopez C, et al. Synthesis and pharmacological evaluation of novel 1- and 8-substituted-3-furfuryl xanthines as adenosine receptor antagonists. Bioorg Med Chem. 2009;17:6755–60.10.1016/j.bmc.2009.07.034Suche in Google Scholar PubMed
[6] Kim D, Lee H, Jun H, Hong S-S, Hong S. Fluorescent phosphoinositide 3-kinase inhibitors suitable for monitoring of intracellular distribution. Bioorg Med Chem. 2011;19:2508–16.10.1016/j.bmc.2011.03.025Suche in Google Scholar PubMed
[7] Mostert S, Mentz W, Petzer A, Bergh JJ, Petzer JP. Inhibition of monoamine oxidase by 8-[(phenylethyl)sulfanyl]caffeine analogues. Bioorg Med Chem. 2012;20:7040–50.10.1016/j.bmc.2012.10.005Suche in Google Scholar PubMed
[8] Partyka A, Chlon-Rzepa G, Wasik A, Jastrzebska-Wiesek M, Bucki A, Kolaczkowski M, et al. Antidepressant- and anxiolytic-like activity of 7-phenylpiperazinylalkyl-1,3-dimethyl-purine-2,6-dione derivatives with diversified 5-HT(1)A receptor functional profile. Bioorg Med Chem. 2015;23:212–21.10.1016/j.bmc.2014.11.008Suche in Google Scholar PubMed
[9] Szymanska E, Drabczynska A, Karcz T, Muller CE, Kose M, Karolak-Wojciechowska J, et al. Similarities and differences in affinity and binding modes of tricyclic pyrimido- and pyrazinoxanthines at human and rat adenosine receptors. Bioorg Med Chem. 2016;24:4347–62.10.1016/j.bmc.2016.07.028Suche in Google Scholar PubMed
[10] Zaluski M, Schabikowski J, Schlenk M, Olejarz-Maciej A, Kubas B, Karcz T, et al. Novel multi-target directed ligands based on annelated xanthine scaffold with aromatic substituents acting on adenosine receptor and monoamine oxidase B. Synthesis, in vitro and in silico studies. Bioorg Med Chem. 2019;27:1195–210.10.1016/j.bmc.2019.02.004Suche in Google Scholar PubMed
[11] Mo DW, Dong S, Sun H, Chen JS, Pang JX, Xi BM, et al. Synthesis and potent inhibitory activities of carboxybenzyl-substituted 8-(3-(R)-aminopiperidin-1-yl)-7-(2-chloro/cyanobenzyl)-3-methyl-3,7-dihydro-purin e-2,6-diones as dipeptidyl peptidase IV (DPP-IV) inhibitors. Bioorg Med Chem Lett. 2015;25:1872–5.10.1016/j.bmcl.2015.03.048Suche in Google Scholar PubMed
[12] Bansal R, Kumar G, Gandhi D, Young LC, Harvey AL. Synthesis of a series of 8-(substituted-phenyl)xanthines and a study on the effects of substitution pattern of phenyl substituents on affinity for adenosine A(1) and A(2A) receptors. Eur J Med Chem. 2009;44:2122–7.10.1016/j.ejmech.2008.10.017Suche in Google Scholar PubMed
[13] He L, Pei H, Ma L, Pu Y, Chen J, Liu Z, et al. Synthesis and lipid-lowering evaluation of 3-methyl-1H-purine-2,6-dione derivatives as potent and orally available anti-obesity agents. Eur J Med Chem. 2014;87:595–610.10.1016/j.ejmech.2014.09.094Suche in Google Scholar PubMed
[14] Li G, Huan Y, Yuan B, Wang J, Jiang Q, Lin Z, et al. Discovery of novel xanthine compounds targeting DPP-IV and GPR119 as anti-diabetic agents. Eur J Med Chem. 2016;124:103–16.10.1016/j.ejmech.2016.08.023Suche in Google Scholar PubMed
[15] Basu S, Barawkar DA, Ramdas V, Patel M, Waman Y, Panmand A, et al. Design and synthesis of novel xanthine derivatives as potent and selective A2B adenosine receptor antagonists for the treatment of chronic inflammatory airway diseases. Eur J Med Chem. 2017;134:218–29.10.1016/j.ejmech.2017.04.014Suche in Google Scholar PubMed
[16] Kaplanek R, Jakubek M, Rak J, Kejik Z, Havlik M, Dolensky B, et al. Caffeine-hydrazones as anticancer agents with pronounced selectivity toward T-lymphoblastic leukaemia cells. Bioorg Chem. 2015;60:19–29.10.1016/j.bioorg.2015.03.003Suche in Google Scholar PubMed
[17] Abou-Zied HA, Youssif BGM, Mohamed MFA, Hayallah AM, Abdel-Aziz M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg Chem. 2019;89:102997.10.1016/j.bioorg.2019.102997Suche in Google Scholar PubMed
[18] Serafini M, Torre E, Aprile S, Grosso ED, Gesù A, Griglio A, et al. Discovery of highly potent benzimidazole derivatives as indoleamine 2,3-dioxygenase-1 (IDO1) inhibitors: From structure-based virtual screening to in vivo pharmacodynamic activity. J Med Chem. 2020;63:3047–65.10.1021/acs.jmedchem.9b01809Suche in Google Scholar PubMed
[19] Röhrig UF, Majjigapu SR, Grosdidier A, Bron S, Stroobant V, Pilotte L, et al. Rational design of 4-Aryl-1,2,3-triazoles for indoleamine 2,3-dioxygenase 1 inhibition. J Med Chem. 2012;55:5270–90.10.1021/jm300260vSuche in Google Scholar PubMed
[20] Pan S, Zhou Y, Wang Q, Wang Y, Tian C, Wang T, et al. Discovery and structure-activity relationship studies of 1-aryl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione derivatives as potent dual inhibitors of indoleamine 2,3-dioxygenase 1 (IDO1) and trytophan 2,3-dioxygenase (TDO). Eur J Med Chem. 2020;207:112703.10.1016/j.ejmech.2020.112703Suche in Google Scholar PubMed
[21] Nelp MT, Kates PA, Hunt JT, Newitt JA, Balog A, Maley D, et al. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc Natl Acad Sci. 2018;115:3249–54.10.1073/pnas.1719190115Suche in Google Scholar PubMed PubMed Central
[22] Ye JH, Mao LF, Xie LYJ, Zhang RJ, Liu YL, Peng LZ, et al. Discovery of a series of theophylline derivatives containing 1,2,3-triazole for treatment of non-small cell lung cancer. Front Pharmacol. 2021;12:753676.10.3389/fphar.2021.753676Suche in Google Scholar PubMed PubMed Central
[23] Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–9.10.1038/nm1196Suche in Google Scholar PubMed
[24] Kumar S, Waldo JP, Jaipuri FA, Marcinowicz A, Van Allen C, Adams J, et al. Discovery of clinical candidate (1R,4r)-4-((R)-2-((S)-6-fluoro-5H-imidazo[5,1-a]isoindol-5-yl)-1-hydroxyethyl)cyclohexan-1-ol (Navoximod), a potent and selective inhibitor of indoleamine 2,3-dioxygenase 1. J Med Chem. 2019;62:6705–33.10.1021/acs.jmedchem.9b00662Suche in Google Scholar PubMed
[25] Mao LF, Wang YW, Zhao J, Xu GQ, Yao XJ, Li YM. Discovery of icotinib-1,2,3-triazole derivatives as IDO1 inhibitors. Front Pharmacol. 2020;11:579024.10.3389/fphar.2020.579024Suche in Google Scholar PubMed PubMed Central
© 2025 the author(s), published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Artikel in diesem Heft
- Research Articles
- Synthesis, activity exploration, molecular docking, and affinity assessment of theophylline derivatives as inhibitors targeting IDO1
- Synthesis of novel triazole hybrids with pyrene, fluorene, and biphenyl groups and evaluation of their antimycobacterial activity
- Synthesis and antiproliferative evaluation of novel triazolothiadiazine compounds
- Review Article
- Development of heterocyclic-based anticancer agents: A comprehensive review
Artikel in diesem Heft
- Research Articles
- Synthesis, activity exploration, molecular docking, and affinity assessment of theophylline derivatives as inhibitors targeting IDO1
- Synthesis of novel triazole hybrids with pyrene, fluorene, and biphenyl groups and evaluation of their antimycobacterial activity
- Synthesis and antiproliferative evaluation of novel triazolothiadiazine compounds
- Review Article
- Development of heterocyclic-based anticancer agents: A comprehensive review