The first in situ synthesis of 1,3-dioxan-5-one derivatives and their direct use in Claisen-Schmidt reactions
-
M. Javad Poursharifi
 ,
M. Saeed Abaee
,
M. Saeed Abaee

Abstract
A method is developed for in situ generation of 1,3-dioxan-5-one derivatives 2. These compounds are simple precursors for accessing carbohydrate structures and previously had to be produced via stepwise procedures using excessive amounts of reagents. In the present work, three different derivatives of 2 were synthesized via the reaction of trialkoxyalkanes with dihydroxyacetone dimer 1 in the presence of acetic acid as the catalyst. In the same pot, derivatives of 2 were reacted with aromatic aldehydes and 30 mol% of pyrrolidine to obtain high yields of the respective bischalcones 3 within short time periods.
Introduction
Trioses are among the smallest monosaccharide biomolecules playing important roles in cellular respiration [1]. In the course of glycolysis, fructose-1,6-bisphosphate is cleaved to glyceraldehyde-3-phosphate and dihydroxyacetone phosphate (DHAP) [2]. The latter in turn could convert to lactic acid and pyruvic acid [3]. In the nature, DHAP can lead to more complex carbohydrates stereoselectively via enzyme-catalyzed aldol reactions [4]. The chemical equivalent to DHAP is dihydroxyacetone (1, DHA, Scheme 1) which is the only ketotriose and does not exist in enantiomeric forms and is therefore achiral [5]. DHA and its derivatives are successfully employed as C3 building blocks through synthetic manipulations for asymmetric synthesis of various compounds of interest [6].
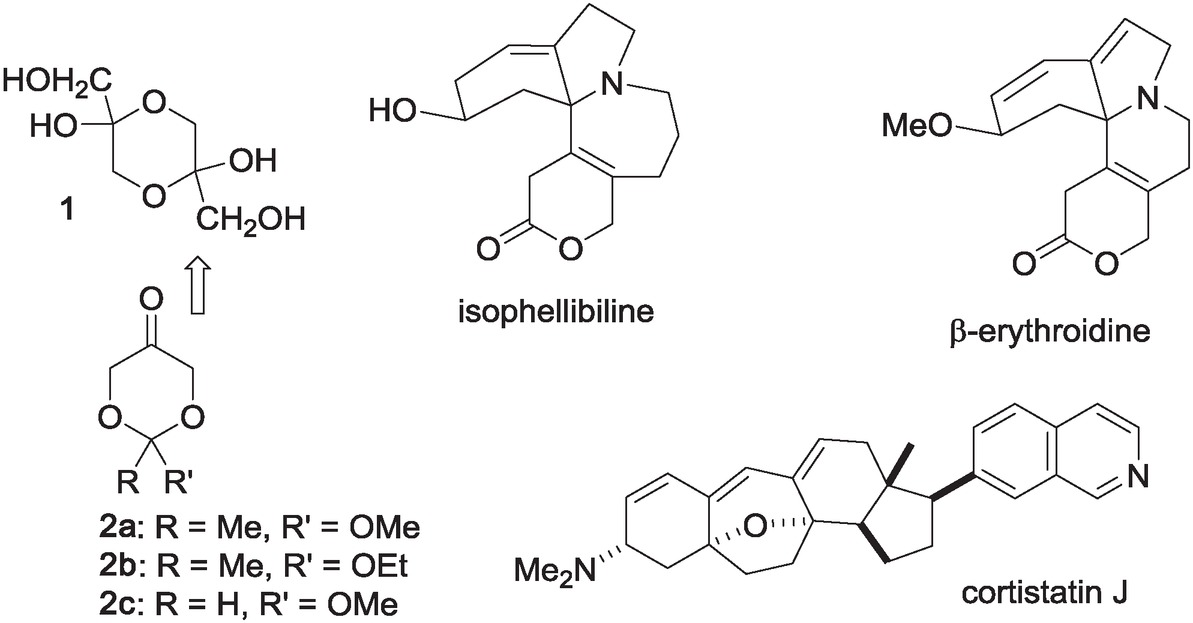
Important structures derived from ketones 2 (a synthon for 1).
The limitation in the chemistry of DHA is that the compound usually exists in relatively inactive dimeric form [7, 8] and researchers have to use its protected heterocyclic synthon, 1,3-dioxan-5-one 2 derivatives instead [9, 10]. In this context, Enders introduced a simple and biomimetic approach for direct proline-catalyzed asymmetric synthesis of several carbohydrate structures and their related compounds through one-step aldol reactions of 2 [11, 12, 13, 14]. Majewski reported stereodivergent synthesis of both enantiomers of glycero-allo-heptose from similar starting ketone 2 [15] and organocatalyzed synthesis of L-deoxymannojirimycin and L-deoxyidonojirimycin via syn-aldol reaction of 2 with (S)-isoserinal hydrate [16]. Interestingly, several applications of this chemistry are reported for the synthesis of natural compounds of interest such as total synthesis of (±)-isophellibiline [17], (±)-cortistatin J [18], and (±)-erythroidines [19].
The difficulties associated with the preparation of derivatives of 2 have persuaded synthetic chemists to design and attempt new methods to obtain 2 via more convenient reactions and by performing less synthetic steps [20, 21, 22, 23]. In the framework of our studies to develop new synthetic procedures in heterocyclic chemistry [24, 25, 26, 27], herein we introduce a new method for in situ preparation of three various derivatives of 2 starting from 1 and trialkoxyalkanes (RCR’3) and acetic acid as catalyst. Then, ketones 2 are reacted in the same pot with aldehydes
to get the respective bischalcone derivatives 3 via Claisen-Schmidt condensation reactions at room temperature. The importance of chalcone functionalities in heterocyclic chemistry from synthetic [28, 29] and biological points of view is well documented [30, 31].
Results and discussion
We first optimized the conditions for the synthesis of 2a by reacting 1 with MeC(OMe)3 and various catalysts (Table 1). Under the conditions reported by Müller et al [32], camphor-10-sulfonic acid (CSA) in dioxane caused 81% formation of 2a after 24 h (entry 1). Use of Lewis acids almost led to minor quantities of the desired product even at a higher temperature or a longer reaction time (entries 2-5). Acetic acid improved the result to give 80% of 2a at 60 °C and after a much shorter time period (entry 6). Also, less amounts of the solvent (entry 7) or the reagent (entry 8) led to comparable results. Other carboxylic acids did not behave better than CH3COOH (entries 9-10). These optimum conditions were applied successfully to prepare two other derivatives of 2 in high yields (entries 11-12).
With these results, we were persuaded to use the optimum conditions to prepare derivatives of 2 and subject them to react with an aromatic aldehyde to evaluate the possibility of the synthesis of bischalcone derivatives 3 in
Optimization of the synthesis of 2.
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Temperature (°C) | Time (h) | Product | Yield (%)a |
| 1 | CSA | 60 | 24 | 2a (R = Me, | 81 |
| R’ = OMe) | |||||
| 2 | MgBr2 | 60 | 48 | 2a | < 5 |
| 3 | MgBr2 | 80 | 72 | 2a | < 5 |
| 4 | LiBr | 60 | 48 | 2a | < 5 |
| 5 | LiBr | 80 | 72 | 2a | < 5 |
| 6 | CH3CO2H | 60 | 8 | 2a | 80 |
| 7 | CH3CO2Hb | 60 | 8 | 2a | 83 |
| 8 | CH3COHb,c2 | 60 | 8 | 2a | 83 |
| 9 | H2C2O4b,c | 80 | 72 | 2a | < 5 |
| 10 | PhCO2Hb,c | 80 | 72 | 2a | < 5 |
| 11 | CH3CO2Hb,c | 60 | 8 | 2b (R = Me, | 85 |
| R’ = OEt) | |||||
| 12 | CH3CO2Hb,c | 60 | 8 | 2c (R = H, | 80d |
| R’ = OMe) | |||||
aIsolated yields. bDioxane (2 mL). cMeC(OMe)3 (2.0 equiv). dGC yield.
the same reaction pot (Table 2). For this purpose, when monitoring of the reaction showed maximum formation of 2a, the mixture was treated with an alkaline hydroxides followed by addition of 4-ClC6H4CHO. As a result, use of NaOH (entries 1-2) or KOH (entries 3-4) in aqueous or solvent-free conditions gave no minor amounts of 3a. However, when organocatalysts were used, pyrrolidine (entry 5) gave 3a in 83% yield after 15 min, while Et2NH (entry 6) or Et3N (entry 7) produced 63% or 41% of the same product after similar time period.
Optimization of the synthesis of 3a.
| Entry | Conditions (30 mol%) | Solvent | Time (min) | Yield (%)a |
|---|---|---|---|---|
| 1 | NaOH | none | 24 | < 5 |
| 2 | NaOH | H2O | 24 | < 5 |
| 3 | KOH | none | 24 | < 5 |
| 4 | KOH | H2O | 24 | < 5 |
| 5 | pyrrolidine | none | 15 | 83 |
| 6 | Et2NH | none | 15 | 63 |
| 7 | Et3N | none | 15 | 41 |
aIsolated yields.
To show the generality of the process, we synthesized various derivatives of 3 by subjecting 2a to react with different aldehydes bearing electron withdrawing groups (Scheme 2). Thus 3a-f were obtained in high yields. Also, the reaction with benzaldehyde itself led to the same observations and 3g was obtained in 80%. Similarly, use of 2b or 2c produced the target products (3h-3n) in 85-95% yields. The condensation step for all reactions occurred within 15-20 min and products precipitated in the mixtures spontaneously.

One-pot synthesis of various derivatives of 3.
Conclusion
In summary, we succeeded to prepare derivatives of 2, which could be either isolated from the reaction mixtures or further subjected to Claisen-Schmidt reactions in the same pot. Thus various derivatives of 2 and 3 could be prepared efficiently. After the prevailing reaction conditions, the products were solidified in the reaction vessels and required no expensive and time consuming chromatographic separations. In addition, synthesis of 2 succeeded by using much less amounts of solvent and the required orthoesters.
Experimental
Melting points are uncorrected. FT-IR spectra were recorded using KBr disks on a Bruker Vector-22 spectrometer. NMR spectra were obtained on a FT-NMR Bruker Ultra ShieldTM (500 MHz for 1H and 125 MHz for 13C) as DMSO-d6 solutions using TMS as internal standard reference. Elemental analyses were performed using a Thermo Finnigan Flash EA 1112 instrument. MS spectra were obtained on a Finnigan Mat 8430 instrument at ionization potential of 70 eV. TLC experiments were carried out on pre-coated silica gel plates using petroleum ether/EtOAc as the eluent. Chemicals and starting materials were purchased from commercial sources. Aldehydes were redistilled or recrystallized before being used. Products 3a, 3f, 3g, and 3k were known [33, 34]. All other products were new and were characterized by analyzing their 1H NMR, 13C NMR, IR, and mass spectra.
General procedure for the synthesis of 2
Acetic acid (125 μl, 20 mol%) was added dropwise to a mixture of dihydroxyacetone dimer 1 (1.01 g, 5.6 mmol) in dioxane (2 mL), while being heated at 60 °C under argon atmosphere. After 10 min, a trialkyl orthoacetate (23 mmol) was added to the mixture and was stirred for another 8 h. The mixture was concentrated under reduced pressure and the residue was distilled to obtain derivatives of 2. Products 2a-b are known [32, 34].The structure of 2c is inferred from the final products (3m and 3n) containing this central ring.
General procedure for one-pot synthesis of 3
Acetic acid (125 μl, 20 mol%) was added dropwise to a mixture of dihydroxyacetone dimer 1 (1.01 g, 5.6 mmol) in dioxane (2 mL), while being heated at 60 °C under argon atmosphere. After 10 min, trialkyl orthoacetate (23 mmol) was added to the mixture and the mixture was stirred for another 8 h. TLC (petroleum ether/EtOAc 4:1) showed complete conversion of the starting materials to 2 after 8 h. The heating source was removed and an aldehyde (18.6 mmol) and pyrrolidine (3.36 mmol, 277 μl, 30 mol%) were added and mixing was continued at room temperature for 10-15 min. The completion of the reaction was monitored with TLC (petroleum ether/EtOAc:10:1). The product precipitated in the mixture. The crude solid product was purified by recrystallization from EtOH and solid products 3 were obtained.
Spectral data of new products
4,6-Bis((Z)-4-bromobenzylidene)-2-methoxy-2-methyl-1,3-dioxan-5-one (3b) Mp: 224-225 ˚C; IR (KBr) ν 2940, 1578, 1464 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 7.79 (d, J = 8.5 Hz, 4H), 7.62 (d, J = 8.5 Hz, 4H), 6.87 (s, 2H), 3.30 (s, 3H), 1.98 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.6, 144.5, 133.0, 132.9, 132.5, 123.4, 114.6, 113.7, 52.7, 21.1; MS (70 eV) m/z 480 (M+), 404, 325, 196, 89; Anal. Calcd for C20H16Br2O4: C, 50.03; H, 3.36. Found: C, 50.16; H, 3.27.
2 -Methoxy- 2 -methyl- 4 , 6 -bis ( ( Z ) - 3 - nitrobenzylidene)-1,3-dioxan-5-one (3c). Mp: > 250 ˚C; IR (KBr) ν 2920, 1701, 1615, 1528, 1345 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 8.71 (s, 2H), 8.26 (d, J = 8.0 Hz, 2H), 8.21 (d, J = 8.0 Hz, 2H), 7.74 (dd, J = 8.0, 8.0 Hz, 2H), 7.07 (s, 2H), 3.35 (s, 3H), 2.05 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.4, 148.6, 145.3, 137.2, 134.6, 130.9, 125.2, 124.2, 114.0, 113.7, 52.9, 21.0; MS (70 eV) m/z 412 (M+), 381, 337, 163, 129; Anal. Calcd for C20H16N2O8: C, 58.26; H, 3.91; N, 6.79. Found: C, 58.33; H, 3.79; N, 6.90.
4,4’-((1Z,1’Z)-(2-Methoxy-2-methyl-5-oxo-1,3-dioxane-4,6-diylidene)bis(methanylylidene))dibenzonitrile (3d). Mp: 248-249 ˚C; IR (KBr) ν 3053, 2228, 1603, 1279 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 8.02 (d, J = 8.0 Hz, 4H), 7.90 (d, J = 8.0 Hz, 4H), 6.97 (s, 2H), 3.34 (s, 3H), 2.03 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.6, 145.6, 137.6, 133.2, 131.7, 119.3, 114.1, 113.9, 111.7, 52.9, 20.9; MS (70 eV) m/z 372 (M+), 341, 297, 173, 143; Anal. Calcd for C22H16N2O4: C, 70.96; H, 4.33; N, 7.52. Found: C, 70.77; H, 4.60; N, 7.59.
4,6-Bis((Z)-2,4-dichlorobenzylidene)-2-methoxy-2-methyl-1,3-dioxan-5-one (3e). Mp: 195-196 ˚C; IR (KBr) ν 1739, 1577, 1466, 1281, 826 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 8.12 (d, J = 8.0 Hz, 2H), 7.73 (s, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.08 (s, 2H), 3.32 (s, 3H), 1.96 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.2, 145.0, 135.3, 134.9, 132.3, 129.9, 129.3, 128.3, 113.9, 109.2, 52.8, 20.7; MS (70 eV) m/z 460 (M+), 427, 349, 186, 123; Anal. Calcd for C20H14Cl4O2: C, 52.21; H, 3.07. Found: C, 52.16; H, 3.28.
4,6-Bis((Z)-4-chlorobenzylidene)-2-ethoxy-2-methyl-1,3-dioxan-5-one (3h). Mp: 176-177 ˚C; IR (KBr) ν 2965, 1595, 1485, 802 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 7.85 (d, J = 8.5 Hz, 4H), 7.50 (d, J = 8.5 Hz, 4H), 6.87 (s, 2H), 3.61 (q, J = 7.0 Hz, 2H), 2.0 (s, 3H), 1.03 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.6, 144.4, 134.3, 132.6, 132.0, 129.3, 114.0, 113.2, 60.9, 21.7, 15.1; MS (70 eV) m/z 404 (M+), 334, 281, 181, 152; Anal. Calcd for C21H18Cl2O4: C, 62.24; H, 4.48. Found: C, 62.03; H, 4.59.
4,6-Bis((Z)-4-bromobenzylidene)-2-ethoxy-2-methyl-1,3-dioxan-5-one (3i). Mp: 234-235 ˚C; IR (KBr) ν 2923, 1597, 1481, 1283, 812 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 7.79 (d, J = 8.5 Hz, 4H), 7.64 (d, J = 8.5 Hz, 4H), 6.85 (s, 2H), 3.60 (q, J = 7.0 Hz, 2H), 1.99 (s, 3H), 1.02 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.6, 144.5, 132.9, 132.3, 123.1, 114.1, 113.2, 112.9, 61.0, 21.6, 15.1; MS (70 eV) m/z 494 (M+), 424, 269, 198, 149; Anal. Calcd for C21H18Br2O4: C, 51.04; H, 3.67. Found: C, 50.92; H, 3.77.
4,6-Bis((Z)-2,4-dichlorobenzylidene)-2-ethoxy-2-methyl-1,3-dioxan-5-one (3j). Mp: 205-206 ˚C; IR (KBr) ν 2923, 1577, 1464, 1126 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 8.11 (d, J = 8.5 Hz, 2H), 7.70 (s, 2H), 7.50 (d, J = 8.5 Hz, 2H), 7.06 (s, 2H), 3.61 (q, J = 7.0 Hz, 2H), 1.96 (s, 3H), 1.04 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.4, 145.2, 135.3, 134.8, 132.3, 129.9, 129.4, 128.4, 113.6, 108.9, 61.3, 21.5, 15.3; MS (70 eV) m/z 474 (M+), 404, 217, 186, 123; Anal. Calcd for C21H16Cl4O4: C, 53.20; H, 3.40. Found: C, 53.36; H, 3.38.
4,6-Bis((Z)-4-(dimethylamino)benzylidene)-2-ethoxy-2-methyl-1,3-dioxan-5-one (3l). Mp: 230-231 ˚C; IR (KBr) ν 1591, 1527, 1127 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 7.69 (d, J = 9.0 Hz, 4H), 6.76 (d, J = 9.0 Hz, 4H), 6.75 (s, 2H), 3.58 (q, J = 7.0 Hz, 2H), 2.98 (s, 12H), 1.94 (s, 3H), 1.02 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 175.7, 151.0, 141.9, 132.6, 121.0, 116.1, 112.5, 112.3, 60.2, 40.1, 22.1, 15.3; MS (70 eV) m/z 422 (M+), 306, 266, 205, 161; Anal. Calcd for C25H30N2O4: C, 71.07; H, 7.16; N, 6.63. Found: C, 71.15; H, 7.27; N, 6.52.
4,6-Bis((Z)-4-chlorobenzylidene)-2-methoxy-1,3-dioxan-5-one (3m). Mp: 155-156 ˚C; IR (KBr) ν 2934, 1606, 1578, 1464, 825 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 7.87 (d, J = 8.5 Hz, 4H), 7.50 (d, J = 8.5 Hz, 4H), 6.93 (s, 2H), 6.55 (s, 1H), 3.43 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.4, 144.1, 134.5, 132.7, 131.8, 129.3, 114.9, 107.2, 54.3; MS (70 eV) m/z 376 (M+), 315, 281, 225, 152; Anal. Calcd for C19H14Cl2O4: C, 60.50; H, 3.74. Found: C, 60.66; H, 3.85.
4,6-Bis((Z)-2,4-dichlorobenzylidene)-2-methoxy-1,3-dioxan-5-one (3n). Mp: 186-187 ˚C; IR (KBr) ν 2897, 1577, 1464, 1268, 824 cm-1; 1H NMR (500 MHz, DMSO-d6) δ 8.12 (d, J = 8.5 Hz, 2H), 7.72 (s, 2H), 7.52 (d, J = 8.5 Hz, 2H), 7.11 (s, 2H), 6.58 (s, 1H), 3.43 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.2, 144.9, 135.3, 135.0, 132.3, 129.9, 129.3, 128.3, 109.9, 107.3, 54.5; MS (70 eV) m/z 446 (M+), 409, 349, 186, 123; Anal. Calcd for C19H12Cl4O4: C, 51.16; H, 2.71. Found: C, 51.27; H, 2.86.
Acknowledgment
The Research Council at CCERCI (Grant # 96-112) is acknowledged for financial support of this work.
References
[1] Jeremy, M. B.; Tymoczko, J. L.; Stryer, L. Biochemistry; 5th ed., Freemanm: New York, 2002.Search in Google Scholar
[2] Robina, I.; Vogel, P. Synthesis of monosaccharides and analogues. part 1. applications of chemoenzymatic methods and organocatalysis. Chim. Oggi 2007, 25 65–71.10.1002/chin.200838246Search in Google Scholar
[3] Lux, S.; Siebenhofer, M. Synthesis of lactic acid from dihydroxyacetone: use of alkaline-earth metal hydroxides. Catal. Sci. Technol 2013, 3 1380–1385.10.1039/c3cy20859aSearch in Google Scholar
[4] Suri, J. T.; Ramachary, D. B.; Barbas, C. F. Mimicking dihydroxyacetone phosphate-utilizing aldolases through organocatalysis: a facile route to carbohydrates and aminosugars. Org. Lett 2005, 7 1383–1385.10.1021/ol0502533Search in Google Scholar
[5] Rao, V. S. R.; Qasba, P. K.; Balaji, P. V.; Chandrasekaran, R. Conformation of carbohydrates; Harwood Academic Publishers: Amsterdam, 1998.Search in Google Scholar
[6] Enders, D.; Voith, M.; Lenzen, A. The dihydroxyacetone unit-a versatile C(3) building block in organic synthesis. Angew. Chem. Int. Ed. Engl 2005, 44 1304–1325.10.1002/anie.200400659Search in Google Scholar
[7] Gardiner, D. The dimeric forms of some α-hydroxycarbonyl compounds. Carbohydr. Res 1966, 2 234–239.10.1016/S0008-6215(00)81217-3Search in Google Scholar
[8] Kobayashi, Y.; Igarashi, T.; Takahashi, H.; Higasi, K. Infrared and raman studies of the dimeric structures of 1,3-dihydroxyacetone, d(+)- and dl-glyceraldehyde. J. Mol. Struct. 1976, 35 85–99.10.1016/0022-2860(76)80104-4Search in Google Scholar
[9] Feng, J.; Liu, B. Formal carbo [3+3] annulation and its application in organic synthesis. Tetrahedron Lett 2015, 56 1474–1485.10.1016/j.tetlet.2015.01.035Search in Google Scholar
[10] Carlsen, P. H. J.; Søerbye, K.; Ulven, T. Aasbøe, K. Synthesis of benzylidene-protected dihydroxyacetone. Acta. Chem. Scand 1996, 50 185–187.10.3891/acta.chem.scand.50-0185Search in Google Scholar
[11] Grondal, C.; Enders, D. Direct asymmetric organocatalytic de novo synthesis of carbohydrates. Tetrahedron 2006, 62 329–337.10.1016/j.tet.2005.09.060Search in Google Scholar
[12] Enders, D.; Grondal, C. Direct organocatalytic de novo synthesis of carbohydrates. Angew. Chem. Int. Ed. Engl 2005, 44 1210–1212.10.1002/anie.200462428Search in Google Scholar PubMed
[13] Enders, D.; Jegelka, U. 1,3-Dioxan-5-one as C3-building block for the diastereo- and enantioselective synthesis of C5- to C9-deoxy sugars using the SAMP-/RAMP-hydrazone method. Tetrahedron Lett 1993, 34 2453–2456.10.1016/S0040-4039(00)60439-2Search in Google Scholar
[14] Enders, D.; Narine, A. A. Lessons from nature: biomimetic organocatalytic carbon−carbon bond formations. J. Org. Chem 2008, 73 7857–7870.10.1021/jo801374jSearch in Google Scholar PubMed
[15] Palyam, N.; Niewczas, I.; Majewski, M. Stereodivergent synthesis of d,d-and l,l-glycero-β-allo-heptopyranoses on a dioxanone scaffold. Synlett 2012, 23 2367–2370.10.1055/s-0032-1290461Search in Google Scholar
[16] Palyam, N.; Majewski, M. Organocatalytic syn-aldol reactions of dioxanones with S-isoserinal hydrate: synthesis of l-deoxymannojirimycin and l-deoxyidonojirimycin. J. Org. Chem. 2009, 74 4390–4392.10.1021/jo900263sSearch in Google Scholar PubMed
[17] Funk, R. L.; Belmar, J. Total synthesis of (±)-isophellibiline. Tetrahedron Lett 2012, 53 176–178.10.1016/j.tetlet.2011.10.161Search in Google Scholar PubMed PubMed Central
[18] Nilson, M. G.; Funk, R. L. Total synthesis of (±)-cortistatin J from furan. J. Am. Chem. Soc 2011, 133 12451–12453.10.1021/ja206138dSearch in Google Scholar PubMed PubMed Central
[19] He Y.; Funk, R. L. Total syntheses of (±)-β-erythroidine and (±)-8-oxo-β-erythroidine by an intramolecular Diels−Alder cycloaddition of a 2-amidoacrolein. Org. Lett 2006, 8 3689–3692.10.1021/ol061267rSearch in Google Scholar PubMed
[20] Enders, D.; Voith, M.; Ince, S. J. Preparation and reactions of 2,2-dimethyl-1,3-dioxan-5-one-SAMP-hydrazone: a versatile chiral dihydroxyacetone equivalent. Synthesis 2002, 34 1775–1779.10.1055/s-2002-33646Search in Google Scholar
[21] Peukert, S.; Giese, B. The pivaloylglycol anchor group: a new platform for a photolabile linker in solid-phase synthesis. J. Org. Chem 1998, 63 9045–9051.10.1021/jo9816055Search in Google Scholar
[22] Majewski, M.; Gleave, D. M.; Nowak, P. 1,3-Dioxan-5-ones: synthesis, deprotonation, and reactions of their lithium enolates. Can. J. Chem 1995, 73 1616–1626.10.1139/v95-201Search in Google Scholar
[23] Smithson, T. L.; Ibrahim, N.; Wieser, H. Cyclohexanones: evidence of chair inversion and estimate for barriers to planarity from the far-infrared spectra. Can. J. Chem 1983, 61 1924–1932.10.1139/v83-330Search in Google Scholar
[24] Abaee, M. S.; Mojtahedi, M. M.; Hamidi, V.; Mesbah, A. W.; Massa, W. The first synthesis of bis (arylmethylidene) dioxan-5-ones: potential scaffolds to access vicinal tricarbonyl derivatives. Synthesis 2008, 40 2122–2126.10.1055/s-2008-1067114Search in Google Scholar
[25] Mojtahedi, M. M.; Mehraban, M.; Darvishi, K.; Abaee, M. S. Ultrasound mediated synthesis of dihydropyrano [3, 2-d] [1, 3] dioxin-7-carbonitrile derivatives in H2O/EtOH medium. Heterocycl. Commun 2017, 23 91–95.10.1515/hc-2017-0014Search in Google Scholar
[26] Abaee, M. S.; Akbarzadeh, E.; Shockravi, A.; Mojtahedi, M. M.; Mehraki, E.; Khavasi, H. R. Three-component anti selective Mannich reactions in a tetrahydro-4-pyranone system by using PDAG-Co catalyst. Heterocycl. Commun 2014, 20 123–128.10.1515/hc-2014-0001Search in Google Scholar
[27] Mojtahedi, M. M.; Pourabdi, L.; Abaee, M. S.; Jami, H.; Dini, M.; Halvagar, M. R. Facile one-pot synthesis of novel ortho-aminocarbonitriles and dicyanoanilines fused to heterocycles via pseudo four-component reactions. Tetrahedron 2016, 72 1699–1705.10.1016/j.tet.2016.02.023Search in Google Scholar
[28] Li, Z.; Song, W.; He, J.; Du, Y.; Yang, J. Synthesis of 4-arylethyl-6-arylpyrimidine-2-thiols through aza-Michael addition/ nucleophilic addition/aromatization tandem reactions. Heterocycl. Commun. 2018, 24 23–26.10.1515/hc-2017-0169Search in Google Scholar
[29] El-Hussieny, M.; Yosef, H. A. A.; Mahran, M. R. H.; Ibrahim, N. M. Reactions of ferrocenyl chalcones with hydrazines and active methylene compounds. Heterocycl. Commun. 2016, 22 69–77.10.1515/hc-2016-0006Search in Google Scholar
[30] Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: a mini review. Eur. J. Med. Chem 2014, 85 758–777.10.1016/j.ejmech.2014.08.033Search in Google Scholar PubMed
[31] Arasavelli, A. M.; Ganapavarapu, V.; S. Vidavalur, S. Design, synthesis, and anticancer activity of novel aryl/ heteroaryl chalcone derivatives. Heterocycl. Commun. 2016, 22 1–5.10.1515/hc-2015-0271Search in Google Scholar
[32] Müeller, S. N.; Batra, R.; Senn, M.; Giese, B.; Kisel, M.; Shadyro, O. Chemistry of C-2 glyceryl radicals: indications for a new mechanism of lipid damage. J. Am. Chem. Soc 1997, 119 2795–2803.10.1021/ja9641416Search in Google Scholar
[33] Mojtahedi, M. M.; Darvishi, K.; Abaee, M. S.; Halvagar, M. R. Synthesis and fluorescence studies of novel bisarylmethylidene derivatives of 2-methoxy-2-methyl-1, 3-dioxan-5-one. Can. J. Chem 2017, 95 785–791.10.1139/cjc-2017-0099Search in Google Scholar
[34] Rudolph, T.; Buehle, P.; Rosskopf, R. US Patent 2013/0309184 A1, 2013.Search in Google Scholar
© 2019 M. Javad Poursharifi et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 Public License.
Articles in the same Issue
- Research Article
- Magnesium porphyrazine with peripheral methyl (3,5-dibromophenylmethyl)amino groups – synthesis and optical properties
- Synthesis and fungicidal activity of novel imidazo[4, 5-b]pyridine derivatives
- Synthesis of indazolo[5,4-b][1,6]naphthyridine and indazolo[6,7-b][1,6]naphthyridine derivatives
- Zinc Chloride Catalyzed Amino Claisen Rearrangement of 1-N-Allylindolines: An Expedient Protocol for the Synthesis of Functionalized 7-Allylindolines
- Synthesis and Biological Evaluation of (E)-N’-Benzylidene-7-methyl-2-propyl-1H-benzo[d] imidazole-5-carbohydrazides as Antioxidant, Anti-inflammatory and Analgesic agents
- Efficient synthesis, reactions and spectral characterization of pyrazolo[4’,3’:4,5]thieno[3,2-d] pyrimidines and related heterocycles
- Asymmetric Mannich Reaction: Synthesis of Novel Chiral 5-(substituted aryl)-1,3,4-Thiadiazole Derivatives with Anti-Plant-Virus Potency
- Synthesis and antitubercular activity of new N-[5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl]-(nitroheteroaryl)carboxamides
- Remarkable electronic effect on the total stereoselectivity of the cycloaddition reaction of arylnitrile oxides with pyrrol-2-one derivatives
- Preliminary Communications
- Crystal structure and molecular docking studies of new pyrazole-4-carboxamides
- Research Article
- Synthesis of polycyclic phosphonates via an intramolecular Diels-Alder reaction of 2-benzoylbenzalaldehyde and alkenyl phosphites
- Asymmetric total synthesis of filamentous fungi related resorcylic acid lactones 7-epi-zeaenol and zeaenol
- The first in situ synthesis of 1,3-dioxan-5-one derivatives and their direct use in Claisen-Schmidt reactions
- Synthesis and fungicidal activities of perfluoropropan-2-yl-based novel quinoline derivatives
- Combined XRD-paramagnetic 13C NMR spectroscopy of 1,2,3-triazoles for revealing copper traces in a Huisgen click-chemistry cycloaddition. A model case
- Cytotoxic and antimicrobial activities of some novel heterocycles employing 6-(1,3-diphenyl-1H-pyrazol-4-yl)-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile
- Substrate-controlled Diastereoselective Michael Addition of Alkylidene Malonates by Grignard Reagents
- Synthesis of 1,2,3 triazole-linked benzimidazole through a copper-catalyzed click reaction
- Synthesis and spectral characteristics of N-(1-([1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-ylamino)-2,2,2-trichloroethyl)carboxamides
- Facile One-pot Protocol of Derivatization Nitropyridines: Access to 3-Acetamidopyridin-2-yl 4-methylbenzenesulfonate Derivatives
- Naphthalene substituted benzo[c]coumarins: Synthesis, characterization and evaluation of antibacterial activity and cytotoxicity
- A Green Synthesis and Antibacterial Activity of N-Arylsulfonylhydrazone Compounds
- Preliminary Communications
- Facile Synthesis of Spiro[cyclohexane-1,3’-indoline]-2,2’-diones
- Research Article
- Synthesis and AChE inhibitory activity of N-glycosyl benzofuran derivatives
- [DMImd-DMP]: A highly efficient and reusable catalyst for the synthesis of 4H-benzo[b]pyran derivatives
Articles in the same Issue
- Research Article
- Magnesium porphyrazine with peripheral methyl (3,5-dibromophenylmethyl)amino groups – synthesis and optical properties
- Synthesis and fungicidal activity of novel imidazo[4, 5-b]pyridine derivatives
- Synthesis of indazolo[5,4-b][1,6]naphthyridine and indazolo[6,7-b][1,6]naphthyridine derivatives
- Zinc Chloride Catalyzed Amino Claisen Rearrangement of 1-N-Allylindolines: An Expedient Protocol for the Synthesis of Functionalized 7-Allylindolines
- Synthesis and Biological Evaluation of (E)-N’-Benzylidene-7-methyl-2-propyl-1H-benzo[d] imidazole-5-carbohydrazides as Antioxidant, Anti-inflammatory and Analgesic agents
- Efficient synthesis, reactions and spectral characterization of pyrazolo[4’,3’:4,5]thieno[3,2-d] pyrimidines and related heterocycles
- Asymmetric Mannich Reaction: Synthesis of Novel Chiral 5-(substituted aryl)-1,3,4-Thiadiazole Derivatives with Anti-Plant-Virus Potency
- Synthesis and antitubercular activity of new N-[5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl]-(nitroheteroaryl)carboxamides
- Remarkable electronic effect on the total stereoselectivity of the cycloaddition reaction of arylnitrile oxides with pyrrol-2-one derivatives
- Preliminary Communications
- Crystal structure and molecular docking studies of new pyrazole-4-carboxamides
- Research Article
- Synthesis of polycyclic phosphonates via an intramolecular Diels-Alder reaction of 2-benzoylbenzalaldehyde and alkenyl phosphites
- Asymmetric total synthesis of filamentous fungi related resorcylic acid lactones 7-epi-zeaenol and zeaenol
- The first in situ synthesis of 1,3-dioxan-5-one derivatives and their direct use in Claisen-Schmidt reactions
- Synthesis and fungicidal activities of perfluoropropan-2-yl-based novel quinoline derivatives
- Combined XRD-paramagnetic 13C NMR spectroscopy of 1,2,3-triazoles for revealing copper traces in a Huisgen click-chemistry cycloaddition. A model case
- Cytotoxic and antimicrobial activities of some novel heterocycles employing 6-(1,3-diphenyl-1H-pyrazol-4-yl)-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile
- Substrate-controlled Diastereoselective Michael Addition of Alkylidene Malonates by Grignard Reagents
- Synthesis of 1,2,3 triazole-linked benzimidazole through a copper-catalyzed click reaction
- Synthesis and spectral characteristics of N-(1-([1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-ylamino)-2,2,2-trichloroethyl)carboxamides
- Facile One-pot Protocol of Derivatization Nitropyridines: Access to 3-Acetamidopyridin-2-yl 4-methylbenzenesulfonate Derivatives
- Naphthalene substituted benzo[c]coumarins: Synthesis, characterization and evaluation of antibacterial activity and cytotoxicity
- A Green Synthesis and Antibacterial Activity of N-Arylsulfonylhydrazone Compounds
- Preliminary Communications
- Facile Synthesis of Spiro[cyclohexane-1,3’-indoline]-2,2’-diones
- Research Article
- Synthesis and AChE inhibitory activity of N-glycosyl benzofuran derivatives
- [DMImd-DMP]: A highly efficient and reusable catalyst for the synthesis of 4H-benzo[b]pyran derivatives