Scalable synthesis and properties of 7-methyl- 4-azaindole
-
Andrii I. Subota


Abstract
An approach to the synthesis of 7-methyl-4-azaindole, which is a valuable building block for drug discovery programs, is described. The method relies on using a bromine atom as a ‘place holding group’ for one of the carbon atoms of the pyridine ring throughout the reaction sequence, and it is removed only upon the final reductive cyclization leading to the azaindole ring. Exhaustive hydrogenation of the target product proceeds in a diastereoselective manner and leads to a bicyclic conformationally restricted diamine derivative.
Introduction
In medicinal chemistry, building an effective structure-activity relationship (SAR) study requires the possibility of introducing substituents into every position of the molecule identified as an initial hit [1]. Even the smallest structural modification, such as a methyl group, can sometimes dramatically influence the biological properties of the compound when placed in a proper site of the molecular scaffold [2]. Pyrrolopyridines (‘azaindoles’) are scaffolds which have been widely used in drug discovery; the recently registered anti-cancer drugs venetoclax (1) and vemurafenib (2) [3], an anti-HIV agent BMS-378806 (3) [4], an anti-mycobacterial compound 4 [5] and a potential agent for the treatment of the cognitive impairments 5 [6] can be mentioned among the most prominent examples (Figure 1).

Some biologically active azaindoles.
While the synthesis of various substituted azaindoles is generally well documented, some of the regioisomeric derivatives remain hardly accessible, in particular, 7-methyl-4-azaindole (6), a building block which was needed for our project on azaindole derivatives in multigram quantities. It was involved in the design of M1 receptor positive allosteric modulators [6], muscarinic acetylcholine receptor modulators [7], decaprenylphosphoryl-β-D-ribose-2′-epimerase (DprE1) inhibitors [8] or HIV-1 attachment inhibitors [9]. However, all preparations of 6 reported so far (Scheme 1) were inefficient for the multigram preparation due to a dramatic yield drop at the key steps of Bartoli cyclization [10] and sp2–sp3 Suzuki coupling [8]) upon scale-up. In this work, we report an efficient method for the multigram synthesis of 6, which in our opinion may also be useful for the preparation of other 7-substituted 4-azaindole derivatives.
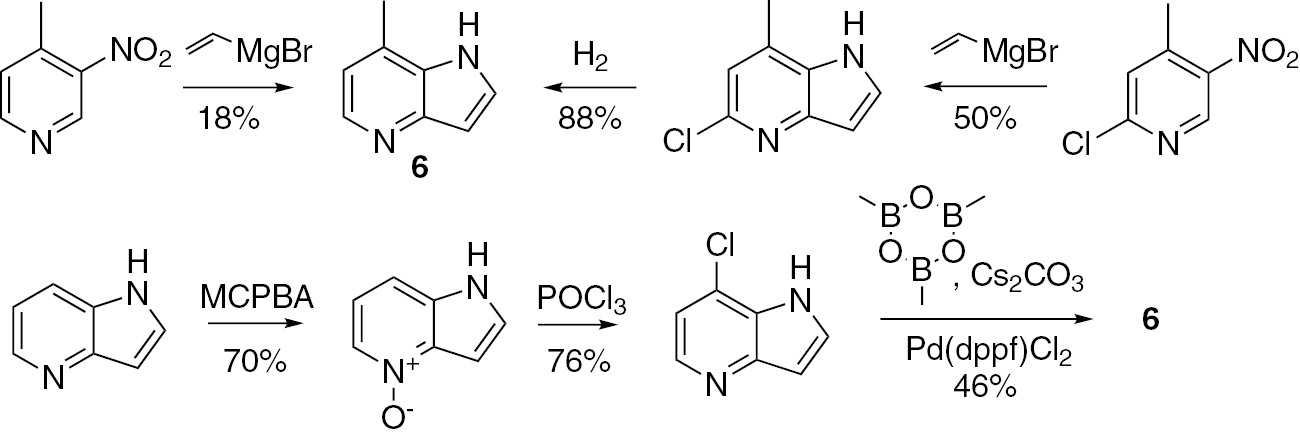
The known syntheses of 7-methyl-4-azaindole (6).
Results and discussion
Our approach to the synthesis of compound 6 relied on the known reductive cyclization of 2-(3-nitropyridin-2-yl)acetonitriles of general formula 7 (Scheme 2) [11], [12], [13]. To the best of our knowledge, this method was not used for the preparation of 7-substituted 4-azaindole derivatives previously. The initial retrosynthetic approach to 6, according to the selected strategy, led us to compound 8. As there was no obvious way to assemble this 2,3,4-trisubstituted pyridine or any relevant synthetic precursor in a scalable manner (e.g., the related known compound 9 [14] is obtained via non-chemoselective nitration of amine 10), we switched to the tetrasubstituted derivative 11 as a possible synthetic equivalent of 8. The bromine atom was introduced into the molecule of 11 as a ‘place holding group’ for the sterically accessible C-5 position of the pyridine ring, which might be removed upon reductive conditions of the last step.

Our synthesis of compound 11 commenced from 2-amino-4-methylpyridine (10), which is readily available from commercial sources on a kilogram scale (Scheme 3). Compound 10 was transformed into 2,3,4,5-tetrasubstituted pyridine 12 using reported procedures [15], [16], [17]. Diazotation of 12 in the presence of HBr and Br2 gave dibromide 13 (65%), which was purified by crystallization from hexanes. Reaction of 13 with tert-butyl cyanoacetate proceeded in a regioselective manner and the product 14 was isolated in a 49% yield. It should be noted that 14 existed in equilibrium with its tautomer 14′. Acidic hydrolysis of 14 was accompanied by decarboxylation and led to the target compound 11 in an excellent yield of 92%. The key step of the synthesis – reductive cyclization of 11 – proceeded smoothly using inexpensive and common 10% Pd-C as a catalyst. As expected, heterocyclization was accompanied by debromination and gave the target 4-azaindole 6 in a good yield of 84%. Notably, the reaction sequence described above allowed the preparation of up to 30 g of 6 in a single run. In our opinion, the developed method allows the preparation of even larger quantities of the target compound.

Some transformations were performed with azaindole 6 to demonstrate its chemical properties and potential for functionalization (Scheme 4). A partial reduction of the bicyclic system was achieved by treatment with BH3·Me2S, which furnished 2,3-dihydro-1H-pyrrolo[3,2-b]pyridine derivative 15 in a 71% yield. Further reduction of the pyridine ring was performed by catalytic hydrogenation; it occurred in a diastereoselective manner and gave (3aR*,7R*,7aR*)-isomer 16 in a 76% yield, which was isolated as a sulfate salt. The relative stereochemistry of 16·H2SO4 was established using the nuclear Overhauser effect spectroscopy (NOESY) experiment (Figure 2). It should be noted that direct hydrogenation of 6 to 16 was not successful. Compound 16 is a representative of bicyclic conformationally restricted diamines which are valuable building blocks in medicinal chemistry [18]. Finally, formylation of 6 with urotropine in aqueous AcOH gave 3-formyl derivative 17 in a 62% yield.


Significant correlations in the NOESY spectrum of 16·H2SO4.
Conclusions
A convenient and scalable approach to 7-methyl-4-azaindole was developed. The method relies on using a bromine atom as a ‘place holding group’ for one of the carbon atoms of the pyridine ring throughout the reaction sequence, which is removed only upon the final reductive cyclization. The described reaction sequence can also be used for the synthesis of other substituted 4-azaindoles.
Experimental
The solvents were purified according to the standard procedures [19]. Compound 12 was prepared in a 85% yield from 10 using a reported method [15], [16], [17]. All other starting materials were purchased from commercial sources. Analytical thin-layer chromatography was performed using Polychrom SI F254 plates. Column chromatography was performed using Kieselgel Merck 60 (230–400 mesh) as the stationary phase. 1H and 13C NMR spectra were recorded on a Varian Gemini 2000 spectrometer (at 400MHz for 1H and 101 MHz for 13C). Mass spectra were recorded on an Agilent 1100 LCMSD SL instrument using electrospray ionization (APESI).
2,5-Dibromo-4-methyl-3-nitropyridine (13)
Aqueous HBr (47%, 195 mL) and Br2 (186 g, 1.17 mol, 60 mL) were added to compound 12 (90.0 g, 0.39 mol). A saturated solution of NaNO2 (69.0 g, 1.00 mol) was added slowly to the mixture under mechanical stirring at 0–5°C. The mixture was stirred at 15°C for 2 h and then diluted with H2O (1.5 L). The precipitate was filtered, washed with H2O and dissolved in CHCl3 (500 mL). The solution was washed with 15% aqueous NaHSO3 (3×200 mL) and H2O (200 mL), dried over Na2SO4 and concentrated under reduced pressure. The residue was crystallized from hexane; yield 74.3 g (65%) of orange solid; mp 76–77°C; 1H NMR (CDCl3): δ 8.55 (s, 1H), 2.40 (s, 3H); 13C NMR (CDCl3): δ 151.2, 148.5, 140.8, 130.2, 122.9, 18.2; MS (CI): m/z 295/297/299 (1:2:1, MH+). Anal. Calcd for C6H4Br2N2O2: C, 24.35; H, 1.36; N, 9.47. Found: C, 24.57; H, 1.72; N, 9.45.
tert-Butyl 2-(5-bromo-4-methyl-3-nitropyridin-2-yl)-2-cyanoacetate (14)
To a solution of tert-butyl cyanoacetate (33.8 g, 0.24 mol) in DMF (310 mL), K2CO3 (63.6 g, 0.46 mol) was added, followed by portion-wise addition of compound 13 (50.4 g, 0.17 mol). The mixture was stirred overnight at 95°C, then cooled and poured in H2O (1500 mL). The mixture was neutralized with 15% aqueous HCl and the resultant precipitate was filtered, washed with H2O (2×250 mL) and hexanes (2×100 mL) and dried under reduced pressure. The crude product was taken up in CHCl3/hexane (2:3, 1000 mL). The mixture was filtered and the filtrate was concentrated; yield 30.1 g (49%) of orange solid; mp 182–183°C; the compound exists as a 9:1 mixture of tautomers; 1H NMR (CDCl3), major tautomer: δ 8.86 (s, 1H), 5.15 (s, 1H), 2.52 (s, 3H), 1.50 (s, 9H); 1H NMR (CDCl3), minor tautomer: δ 15.63 (br s, 1H), 7.84 (d, J=6.5 Hz, 1H), 2.38 (s, 3H), 1.54 (s, 9H); 13C NMR (CDCl3), major tautomer: δ 160.6, 152.3, 141.3, 136.0, 124.8, 112.7, 86.1, 43.8, 28.1, 27.5, 18.8; MS (CI): m/z 356/358 (1:1, MH+). Anal. Calcd for C13H14BrN3O4: C, 43.84; H, 3.96; N, 11.80. Found: C, 43.45; H, 3.81; N, 11.86.
2-(5-Bromo-4-methyl-3-nitropyridin-2-yl)acetonitrile (11)
To a solution of compound 14 (25.0 g, 70.2 mmol) in MeCN (500 mL), an aqueous solution of HCl (17%, 250 mL) was added. The mixture was heated under reflux for 2 h, then cooled and concentrated under reduced pressure. The residue was taken up in an aqueous saturated solution of NaHCO3 (250 mL), and the product was extracted with EtOAc (4×300 mL). The extract was dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography eluting with hexanes/EtOAc (1:1) and crystallized from Et2O/hexanes; yield 16.5 g (92%) of beige solid; mp 107–108°C; 1H NMR (CDCl3): δ 8.81 (s, 1H), 3.95 (s, 2H), 2.46 (s, 3H); 13C NMR (CDCl3): δ 152.2, 146.8, 140.8, 140.6, 123.9, 114.0, 23.6, 18.3; Rf 0.57 eluting with hexanes/EtOAc (1:1); MS (CI): m/z 256/258 (1:1, MH+). Anal. Calcd for C8H6BrN3O2: C, 37.53; H, 2.36; N, 16.41. Found: C, 37.48; H, 2.06; N, 16.70.
7-Methyl-1H-pyrrolo[3,2-b]pyridine (6)
A mixture of compound 11 (14.0 g, 54.7 mmol) and 10% Pd/C (8.0 g) in aqueous 96% EtOH (400 mL) was hydrogenated under 50 bar of H2 at room temperature for 30 h. The catalyst was filtered and the filtrate was concentrated under reduced pressure. Aqueous saturated NaHCO3 was added to the residue to adjust to pH 8, and the mixture was concentrated. The residue was treated with hot i-PrOH (400 mL) filtered, and the filtrate was concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography eluting with CHCl3/MeOH (4:1); yield 6.07 g (84%) of beige solid; mp 185–187°C (dec); 1H NMR (DMSO-d6): δ 11.49 (br s, 1H), 8.20 (d, J=4.7 Hz, 1H), 7.62 (s, 1H), 6.91 (d, J=4.6 Hz, 1H), 6.55 (d, J=2.3 Hz, 1H), 2.50 (s, 3H); 13C NMR (DMSO-d6): δ 145.3, 142.2, 129.1, 128.5, 128.4, 117.0, 101.6, 16.3; Rf 0.60 eluting with CHCl3/MeOH (4:1); MS (CI): m/z 133 (MH+). Anal. Calcd for C8H8N2: C, 72.7; H, 6.10; N, 21.2. Found: C, 72.49; H, 6.13; N, 21.37.
7-Methyl-2,3-dihydro-1H-pyrrolo[3,2-b]pyridine (15)
To a stirred solution of compound 6 (1.98 g, 15.0 mmol) in dry THF (100 mL), BH3·Me2S (16.0 g, 12.8 mL, 210 mmol) was added, and the mixture was heated under reflux for 11 h. After cooling, MeOH (25 mL) was added dropwise and the mixture was concentrated. MeOH (150 mL) and concentrated aqueous HCl (20 mL) were added to the residue. The mixture was stirred under reflux for 6 h, then cooled and concentrated under reduced pressure. An aqueous solution of NaOH (20%, 50 mL) was added to the residue, and the product was extracted with Et2O (4×15 mL). The organic layers were combined, dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography eluting with CHCl3/MeOH (9:1); yield 1.43 g (71%) of white solid; mp 97–99°C; 1H NMR (CDCl3): δ 7.76 (d, J=5.1 Hz, 1H), 6.71 (d, J=5.1 Hz, 1H), 3.60 (t, J=8.6 Hz, 2H), 3.60 (br s, 1H, NH), 3.12 (t, J=8.6 Hz, 2H), 2.08 (s, 3H); 13C NMR (CDCl3): δ 151.1, 143.6, 139.0, 125.2, 122.6, 45.1, 31.2, 16.2; Rf 0.28 eluting with CHCl3/MeOH (9:1); MS (CI): m/z 135 (MH+). Anal. Calcd for C8H10N2: C, 71.61; H, 7.51; N, 20.88. Found: C, 71.43; H, 7.80; N, 20.64.
(3aR*,7R*,7aR*)-7-Methyloctahydro-1H-pyrrolo[3,2-b]pyridine sulfate (16·H2SO4)
A mixture of compound 15 (1.00 g, 7.50 mmol), 10% Pd(OH)2-C (1.00 g), MeOH (50 mL) and concentrated aqueous HCl (1.5 mL) was hydrogenated at 35 bar of H2 at 60°C for 24 h. The catalyst was filtered and the filtrate was concentrated under reduced pressure. An aqueous solution of NaOH (10%, 15 mL) was added to the residue, and the product was extracted with Et2O (4×30 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was dissolved in aqueous 96% EtOH (30 mL) and the solution was treated with concentrated H2SO4 to adjust to pH 4. The resultant diamine sulfate was filtered and crystallized from aqueous 96% EtOH (120 mL); yield 1.36 g (76%) of white solid; mp>200°C (dec.); 1H NMR (D2O): δ 4.20 – 4.05 (m, 1H, 3a-CH), 3.98 (t, J=4.7 Hz, 1H, 7a-CH), 3.85 – 3.70 (m, 1H, 1-CHH), 3.62 – 3.43 (m, 2H, 1-CHH and 5-CHH), 3.11 (td, J=13.1, 3.0 Hz, 1H, 5-CHH), 2.67 (ddt, J=15.1, 11.2, 7.5 Hz, 1H, 2-CHH), 2.48 – 2.25 (m, 2H, 7-CH and 2-CHH), 2.00 – 1.88 (m, 1H, 6-CHH), 1.88 – 1.67 (m, 1H, 6-CHH), 1.20 (d, J=7.1 Hz, 3H, CH3); NHs are exchanged with HDO; 13C NMR (DMSO-d6): δ 58.7, 52.7, 40.6, 40.1, 25.2, 24.8, 20.9, 14.3; MS (CI): m/z 141 (MH+). Anal. Calcd for C8H18N2O4S: C, 40.32; H, 7.61; N, 11.76; S, 13.45. Found: C, 40.52; H, 7.81; N, 11.9; S, 13.26.
7-Methyl-1H-pyrrolo[3,2-b]pyridine-3-carbaldehyde (17)
A mixture of compound 6 (3.00 g, 22.7 mmol) and hexamethylenetetramine (4.91 g, 35.0 mmol) in a mixture of acetic acid (20 mL) and H2O (50 mL) was heated under reflux for 6 h, then cooled and concentrated under reduced pressure. A saturated aqueous solution of NaHCO3 (50 mL) was added to the residue, and the product was extracted with EtOAc (4×50 mL). The organic layers were combined, dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography eluting with CHCl3/MeOH (4:1); yield 2.25 g (62%) of light yellow solid; mp 210–212°C; 1H NMR (CD3OD): δ 10.12 (s, 1H), 8.33 (d, J=4.9 Hz, 1H), 8.26 (s, 1H), 7.12 (d, J=4.9 Hz, 1H), 2.57 (s, 3H); NH is exchanged with CD3OD; 13C NMR (CD3OD): δ 187.2, 146.5, 144.4, 138.9, 134.6, 132.6, 121.5, 119.3, 17.4; Rf 0.56 eluting with CHCl3/MeOH (4:1); MS (CI): m/z 161 (MH+). Anal. Calcd for C9H8N2O: C, 67.49; H, 5.03; N, 17.49. Found: C, 67.31; H, 5.43; N, 17.19.
Supplementary material
1H and 13C NMR spectra are available as online-only supplement.
Acknowledgements
This work was supported by Life Chemicals Inc. and Ukrainian Government Funding (state registry No. 0114U003956). The authors are grateful to Oleksiy Ryabitskiy for 2D NMR measurements. O.O.G. thanks Prof. Andrey A. Tolmachev for his encouragement and support.
References
[1] Wermuth, C. G. Application strategies for the primary structure–activity relationship exploration. In The Practice of Medicinal Chemistry; 3rd Edition. Wermuth, C. G., Ed. Academic Press/Elsevier: Amsterdam, 2008; pp 415–427.10.1016/B978-0-12-374194-3.00019-6Suche in Google Scholar
[2] Barreiro, E. J.; Kümmerle, A. E.; Fraga, C. A. M. The methylation effect in medicinal chemistry. Chem. Rev.2011, 111, 5215–5246.10.1021/cr200060gSuche in Google Scholar PubMed
[3] Wishart, D. S.; Knox, C.; Guo, A. C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res.2006, 34, D668–672.10.1093/nar/gkj067Suche in Google Scholar PubMed PubMed Central
[4] Qian, K.; Morris-Natschke, S. L.; Lee, K.-H. HIV entry inhibitors and their potential in HIV therapy. Med. Res. Rev.2009, 29, 369–393.10.1002/med.20138Suche in Google Scholar PubMed PubMed Central
[5] Chatterji, M.; Shandil, R.; Manjunatha, M. R.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; Prabhakar, K. R.; et al. 1,4-Azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob. Agents Chemother.2014, 58, 5325–5331.10.1128/AAC.03233-14Suche in Google Scholar PubMed PubMed Central
[6] Davoren, J. E.; O’Neil, S. V.; Anderson, D. P.; Brodney, M. A.; Chenard, L.; Dlugolenski, K.; Edgerton, J. R.; Green, M.; Garnsey, M.; Grimwood, S.; et al. Design and optimization of selective azaindole amide M1 positive allosteric modulators. Bioorg. Med. Chem. Lett.2016, 26, 650–655.10.1016/j.bmcl.2015.11.053Suche in Google Scholar PubMed
[7] Payne, A.; Castro, P.; Jose; L.; Birch, L. M.; Khan, A.; Braunton, A. J.; Kitulagoda, J. E.; Soejima, M. 4-Azaindole derivatives. U. S. Pat. 2015/0094328, 2015.Suche in Google Scholar
[8] Shirude, P. S.; Shandil, R. K.; Manjunatha, M. R.; Sadler, C.; Panda, M.; Panduga, V.; Reddy, J.; Saralaya, R.; Nanduri, R.; Ambady, A.; et al. Lead optimization of 1,4-azaindoles as antimycobacterial agents. J. Med. Chem.2014, 57, 5728–5737.10.1021/jm500571fSuche in Google Scholar PubMed
[9] Wang, T.; Yang, Z.; Zhang, Z.; Gong, Y. F.; Riccardi, K. A.; Lin, P. F.; Parker, D. D.; Rahematpura, S.; Mathew, M.; Zheng, M.; et al. Inhibitors of HIV-1 attachment. Part 10. The discovery and structure–activity relationships of 4-azaindole cores. Bioorg. Med. Chem. Lett.2013, 23, 213–217.10.1016/j.bmcl.2012.10.120Suche in Google Scholar PubMed
[10] Zhang, Z.; Yang, Z.; Meanwell, N. A.; Kadow, J. F.; Wang, T. A general method for the preparation of 4- and 6-azaindoles. J. Org. Chem.2002, 67, 2345–2347.10.1021/jo0111614Suche in Google Scholar PubMed
[11] Makosza, M.; Danikiewicz, W.; Wojciechowski, K. Reactions of organic anions, 147. Simple and general synthesis of hydroxy- and methoxyindoles via vicarious nucleophilic substitution of hydrogen. Lieb. Ann. Chem.1988, 1988, 203–208.10.1002/jlac.198819880304Suche in Google Scholar
[12] Macor, J. E.; Newman, M. E. The synthesis of a rotationally restricted phenolic analog of 5-methoxy-3-(1,2,5,6-tetrahydropyrid-4-yl)indole (RU-24,969). Heterocycles1990, 31, 805–809.10.1002/chin.199050214Suche in Google Scholar
[13] Choi, I.; Chung, H.; Park, J. W.; Chung, Y. K. Active and recyclable catalytic synthesis of indoles by reductive cyclization of 2-(2-nitroaryl)acetonitriles in the presence of Co–Rh Heterobimetallic nanoparticles with atmospheric hydrogen under mild conditions. Org. Lett.2016, 18, 5508–5511.10.1021/acs.orglett.6b02659Suche in Google Scholar PubMed
[14] Roe, A.; Seligman, R. B. The preparation of 3-fluoroisonicotinic acid and related compounds. J. Org. Chem.1955, 20, 1729–1732.10.1021/jo01364a020Suche in Google Scholar
[15] Wu, R.; Smidansky, E. D.; Oh, H. S.; Takhampunya, R.; Padmanabhan, R.; Cameron, C. E.; Peterson, B. R. Synthesis of a 6-methyl-7-deaza analogue of adenosine that potently inhibits replication of polio and dengue viruses. J. Med. Chem.2010, 53, 7958–7966.10.1021/jm100593sSuche in Google Scholar PubMed PubMed Central
[16] Graboyes, H.; Day, A. R. Metabolite analogs. VIII. Syntheses of some imidazopyridines and pyridotriazoles. J. Am. Chem. Soc.1957, 79, 6421–6426.10.1021/ja01581a018Suche in Google Scholar
[17] Bhattacharya, A.; Purohit, V. C.; Deshpande, P.; Pullockaran, A.; Grosso, J. A.; DiMarco, J. D.; Gougoutas, J. Z. An alternate route to 2-amino-3-nitro-5-bromo-4-picoline: regioselective pyridine synthesis via 2-nitramino-picoline intermediate. Org. Process Res. Dev.2007, 11, 885–888.10.1021/op700114dSuche in Google Scholar
[18] Grygorenko, O. O.; Radchenko, D. S.; Volochnyuk, D. M.; Tolmachev, A. A.; Komarov, I. V. Bicyclic conformationally restricted diamines. Chem. Rev.2011, 111, 5506–5568.10.1021/cr100352kSuche in Google Scholar PubMed
[19] Armarego, W. L. F.; Chai, C. L. L. Purification of Laboratory Chemicals; Elsevier: Oxford, 2003.10.1016/B978-075067571-0/50008-9Suche in Google Scholar
Supplemental Material:
The online version of this article offers supplementary material (https://doi.org/10.1515//hc-2017-0180).
©2017 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Preliminary Communications
- One-pot synthesis of benzopyrans catalyzed by silica supported dual acidic ionic liquid under solvent-free conditions
- An efficient synthesis of 5-halo-6-trifluoromethylpyridine-3-carbonitriles and carboxylic acids
- Research Articles
- Novel 2H-1,3-benzoxazine ring formation by intramolecular heterocyclization of N-(α-aryloxyalkyl)imidoyl chlorides
- Green synthesis of novel 2-pyrazolyl-1,3-thiazolidine-4-ones using 2-oxoimidazolidine-1,3-disulfonic acid
- A facile synthesis of (E)-2-(aryl/hetaryl)vinyl-4-phenylquinoline-3-carboxylic acids
- Efficient synthesis of new pyrano[3,2-b]pyran derivatives via Fe3O4@SiO2-IL-Fc catalyzed three-component reaction
- An improved and scalable synthesis of zolpidem via a CuI/BINOL-mediated tandem reaction of imine and alkyne
- Scalable synthesis and properties of 7-methyl- 4-azaindole
- Synthesis and insecticidal activities of novel 1H-pyrazole-5-carboxylic acid derivatives
Artikel in diesem Heft
- Frontmatter
- Preliminary Communications
- One-pot synthesis of benzopyrans catalyzed by silica supported dual acidic ionic liquid under solvent-free conditions
- An efficient synthesis of 5-halo-6-trifluoromethylpyridine-3-carbonitriles and carboxylic acids
- Research Articles
- Novel 2H-1,3-benzoxazine ring formation by intramolecular heterocyclization of N-(α-aryloxyalkyl)imidoyl chlorides
- Green synthesis of novel 2-pyrazolyl-1,3-thiazolidine-4-ones using 2-oxoimidazolidine-1,3-disulfonic acid
- A facile synthesis of (E)-2-(aryl/hetaryl)vinyl-4-phenylquinoline-3-carboxylic acids
- Efficient synthesis of new pyrano[3,2-b]pyran derivatives via Fe3O4@SiO2-IL-Fc catalyzed three-component reaction
- An improved and scalable synthesis of zolpidem via a CuI/BINOL-mediated tandem reaction of imine and alkyne
- Scalable synthesis and properties of 7-methyl- 4-azaindole
- Synthesis and insecticidal activities of novel 1H-pyrazole-5-carboxylic acid derivatives