Sustainable composite materials based on ethylene-vinylacetate copolymer and organo-modified silica
-
Do Quang Tham
Do Quang Tham received his Bachelor’s degree in Solid State Physics from Hanoi University of Science in 1997 and his Master’s degree in Material Science from Hanoi University of Technology in 2009. In 2014, he obtained his PhD in Polymer and Composite Materials from the Institute for Tropical Technology (ITT), Vietnam Academy of Science and Technology (VAST). Since 2001, he has worked as a researcher at ITT. His present research fields are polymer nanocomposite and polymer composite materials.
Nguyen Thi Thu Trang worked as a researcher atthe Department of Physico-Chemistry of Non-Metallic Materials,ITT, VAST. She received her MSc in Chemistry from the College of Science, Vietnam National University of Hanoi, in 2005. Nearly 30 of her articles and reports are related to conducting polymers and polymer nanocomposites and were published in national and international journals and proceedings of national and international scientific conferences and workshops.
Nguyen Thuy Chinh received her Bachelor’s degree in Chemistry in 2009 and her MSc degree in Physical Chemistry in 2011. Since 2009, she has worked as a researcher at the Department of Physico-Chemistry of Polymers and Non-Metallic Materials, ITT, VAST. Currently, she is working on her PhD thesis at ITT, VAST, where she is investigating drug delivery systems based on poly (lactic acid) and chitosan. Most of her work is related to the properties of nanomaterials and polymer nanocomposites.
Nguyen Vu Giang received his Bachelor’s degree in Physical Engineering in 1994, and his MSc in 2001. He received his PhD degree at the Department of Polymer Science and Engineering, College of Engineering, Sunchon National University, South Korea. Currently, he is working in the fields of polymer nanocomposite and polymer blend materials, degradation and stability of polymers and rubbers, green materials and their applications.
Tran Dai Lam graduated from Belorussian State University (in the former USSR) with a Master’s degree in Solid State Chemistry (1994). He received his PhD in Physical Chemistry (Surface-Interface) from the University of Paris VII, Paris, France, in 2003. He was a lecturer at Hanoi University of Technology from 1998 to 2008. He has been an associate professor at Institute of Materials Science (IMS) since 2009. His research focusses on nanofabrications, characterizations and applications of nanobiomaterials in drug delivery systems and biosensors.
Thai Hoang obtained his PhD in Polymer Chemistry in 1993. Since 2012, he has worked as a professor at the ITT, VAST. He carried out postdoctoral fellowship in polymer blend, polymer composites and plastics technology in South Korea, UK and Japan. He has published 40 papers in international journals and more than 180 papers in national journals. His research fields are polymer blends, nanocomposites, biodegradable polymers, and bio-medical materials.


Abstract
Silica nanoparticles (SNPs) were modified by different amounts of 3-aminopropyltriethoxysilane (APTES). Nanocomposites based on ethylene vinyl acetate copolymer (EVA) and modified SNP (m-SNP) were prepared by the melt mixing method. They were characterized by Haake torque measurement, differential scanning calorimetry (DSC), horizontal burning test, scanning electron microscopy. Accelerated weather testing of the nanocomposites was performed according to ASTM D4329 (cycle A) for 168 h. The Haake torque indicates that the relative melt viscosity of EVA/m-SNPs is slightly higher than that of EVA/SNP nanocomposites. The DSC and burning test results show that m-SNPs decrease crystallinity degree and flammability of EVA. After accelerated weather testing, the relative amount of C=O groups in EVA/m-SNP nanocomposites is lower than that in EVA/SNP nanocomposites. Micro crack of EVA/m-SNP nanocomposite is smaller than that of EVA/SNP nanocomposite. In our study, the EVA/m-SNP nanocomposites with good tensile properties, flame and weather resistance can be used as sustainable materials in some technique fields.
1 Introduction
Recently, silica nanoparticles (SNPs or nanosilica) have been widely used as inorganic additives for polymers because of their interesting advantages. It has been reported in the literature that nanosilica can improve mechanical strength, thermal stability, aging resistance, climate resistance of polymers, etc. [1], [2]. However, the main challenges of using SNPs are their high tendency of agglomeration and incompatibility with most polymers [3]. These problems can be overcome by increasing the affinity between the polymer matrix and nanosilica. Some studies have indicated that surface modification by using a silane coupling agent is an effective way to improve the dispersion of nanosilica in polymer matrix, thus enhancing properties of polymer/silica nanocomposites [4], [5], [6].
In our previous study, SNPs with average particle size of about 80 nm were successfully prepared by the sol-gel method from tetraethoxysilane (TEOS) [7]. By using the compatibilization approach, the nanocomposites based on ethylene vinyl acetate copolymer (EVA) and nanosilica with and without using EVA grafted maleic anhydride (EVAgMA) were prepared by in situ sol-gel methods. The enhancement effects of nanosilica on tensile-, rheological-, thermal properties and the role of EVAgMA as a compatibilizer for enhancing the dispersion of nanosilica into the EVA matrix have been investigated. In the above study, it was strongly suggested that the formations of hydrogen bonds and dipole-dipole interactions between nanosilica and EVA through EVAgMA were the important roles for enhancing the properties and structural morphology of the nanocomposites.
The effects of nanosilica (a commercial product) and EVAgMA on properties and weather resistance of EVA/silica nanocomposites have been investigated [8]. The results have indicated that nanosilica and EVAgMA can improve the tensile properties and thermal stability of EVA and EVA/silica nanocomposites. Under accelerated weather testing, EVA/EVAgMA/silica nanocomposites exhibited higher retentions of tensile properties, smaller micro crack dimensions in the complex testing conditions by heat, high humidity and ultraviolet (UV) irradiation.
In order to enhance the dispersion of SNP in EVA matrix through the surface modification approach, SNP was modified by grafting 3-aminopropyltriethoxysilane (APTES) via the sol-gel method using ecofriendly solvents. Modified SNP was then used for preparation of EVA/modified SNP nanocomposites [9], [10]. The results showed that APTES was grafted on the surface of SNP, and among the three ethoxy groups of APTES, about one to two ethoxy groups reacted with nanosilica in the modification process [9]. The grafted yields of APTES onto the surface of SNP have been evaluated as 2.66–7.25% when increasing amount of APTES from 5% to 20%, respectively [10].
In this study, melt mixing is chosen as a preferred method for preparing EVA filled with modified SNP nanocomposites, because it is economic and environmentally-friendly. The results of melt mixing behavior, thermal properties, flammability, and accelerated weathering characteristics of the nanocomposites will be presented and discussed. Moreover, some comparisons with our previous studies on EVA/silica nanocomposites are also mentioned for more detailed discussion.
2 Materials and methods
2.1 Materials
EVA containing 10 wt.% vinyl acetate, with density of 0.93 g/cm³ at 23°C and melt flow index of 1.3 g/10 min (at 190°C/2.16 kg) was supplied by Hanwha Co., (South Korea). TEOS (99%) was purchased from Merck KGaA (Germany). APTES was supplied by Sigma Aldrich (USA) with purity of 99%. Absolute ethanol was provided from Duc Giang Chemical Company (Vietnam) with purity of 99.7%.
2.2 Preparation of EVA/nanosilica nanocomposites
2.2.1 Preparation and modification of nanosilica:
SNPs were synthesized from TEOS and ecofriendly solvents by the following process: 50 ml TEOS was dissolved in 300 ml absolute ethanol under continuous stirring for 30 min. Deionized water (10 ml) was then gradually added into this solution and stirring was continued for another 30 min. The solution was adjusted to alkaline pH of 8.5 by dropwise addition of 25% NH4OH solution. After 30 min of magnetic stirring, clear silica sol was formed. SNP powder was obtained by evaporation of this silica sol at room temperature for 48 h. The silica powder was dried in a circulating hot air oven at 150°C for 5 h and in vacuum at 120°C for another 5 h until constant weight was obtained [11]. Several batches were made using this process for enough SNP volume for sample preparation later. The dried SNP was milled, sprayed and kept in a desiccator for later use.
The SNP was modified by APTES with amounts of 5 wt.%, 10 wt.%, 15 wt.%, 20 wt.% and 50 wt.% (compared to original SNP weight) by the following process: APTES was dissolved in absolute ethanol at 70°C for 60 min, then a stoichiometric distilled water and certain weights of SNP were added into this solution under continuous stirring for 2 h. After filtering and washing with absolute ethanol several times, the obtained powder was dried in a vacuum oven at 80°C for 4 h [10]. The final products were denoted as m5-SNP, m10-SNP, m15-SNP, m20-SNP, and m50-SNP for the above APTES contents, respectively. SNPs and m-SNPs mean unmodified and modified SNPs, respectively.
2.2.2 Preparation of EVA/SNP and EVA/m-SNP nanocomposites:
EVA/1.5 wt.% unmodified SNP and EVA/1.5 wt.% modified SNPs nanocomposites (compared to EVA weight) were prepared by melt mixing in a Thermo-Haake intermixer (Germany) at 160°C, with a rotor speed of 50 rpm for 6 min. After melt mixing, the molten products were hot pressed at 160°C under pressure of 5–6 MPa for 5 min to obtain sheets of the nanocomposites with suitable thickness. The EVA/SNP and EVA/m-SNP nanocomposites using m5-SNP, m10-SNP, m15-SNP, m20-SNP, and m50-SNP were coded as EVA/SNP, EVA/m5-SNP, EVA/m10-SNP, EVA/m15-SNP, EVA/m20-SNP, and EVA/m50-SNP, respectively.
2.3 Study methods and equipment
The torque measurements were also carried out in the Thermo-Haake intermixer (the Haake Rheocord 9000 torque rheometer equipped with Rheomix 600p chamber of 69 cm3 and roller rotors) when mixing the nanocomposites. The relative melt viscosity was evaluated by the torque as a function of temperature at steady melt state for each nanocomposite sample.
Differential scanning calorimetry (DSC) measurement was performed on a NETSZCH DSC204 (USA) differential scanning calorimeter from 25°C to 130°C under nitrogen atmosphere with a heating rate of 10°C/min. In order to remove thermal history, DSC curves were obtained from the second heating circle (after the first circle that was carried out from −70°C to 130°C then cooling down to 20°C with a heating rate of 10°C/min). The crystallinity percentage (XC) was evaluated by the ratio between fusion enthalpy (∆Hf) of nanocomposites to that of polyethylene with 100% crystallinity (∆Hf*), which has been widely reported to be about 298 J/g, as following equation [7], [8], [12]:
A horizontal burning test of EVA/SNPs nanocomposites was conducted on a flammability testing instrument (Vietnam) according to the UL-94 HB (USA). The test was carried out at 25°C and relative humidity of 60%. The samples were prepared in a rectangular shape with dimensions of 127 mm×12.7 mm×3 mm.
Nanocomposite specimens for accelerated weather testing were prepared by cutting into a dumbbell-shape using a standard die. The specimens were then fixed into the holders and accelerated weather testing carried out using an Atlas UV-CON instrument (USA). The experiments were performed according to ASTM D4329 (Cycle A): each cycle consists of 8 h in UV irradiation at 60°C followed by 4 h of humidity condensation at 50°C [13]. The total testing time was lasted for 168 h (14 cycles).
Fourier transform infrared (FTIR) spectra of the nanocomposites before and after accelerated weather testing were recorded by using a Nicolet Nexus 670 spectrometer (USA). For preparation of FTIR samples, a piece of every specimen was hot pressed into films (about 30–60 µm in thickness), and its spectrum was obtained with 32 scans from 400 cm−1 to 4000 cm−1 and 2 cm−1 resolution. To obtain a quantitative comparison, the carbonyl index (CI) was evaluated by the ratios of the absorption peak area around 1737 cm−1 (A1737) to the reference peak area around 724 cm−1 (A724) as per the following equation: CI=A1737/A724 [13].
Surface morphology of EVA/SNPs nanocomposites after accelerated weather testing was observed by using a field emission scanning electron microscope (S-4800 Hitachi, Japan).
Mechanical properties (tensile strength, elongation at break) of the nanocomposites were measured by using a Zwick Tensile tester (Germany) at room temperature with a crosshead speed of 100 mm/min, according to ASTM D638.
The weather resistance (UV-thermo-humidity complex stability) of the nanocomposites was assessed in terms of the retentions in tensile properties of the nanocomposites before and after accelerated weather testing.
3 Results and discussion
3.1 Relative melt viscosity
When using a Haake intermixer for preparation of the EVA/SNPs nanocomposites, the torque and temperature were recorded as a function of mixing time. The temperature of the mixture in the chamber is measured by a thermocouple, the tip of which protrudes outside the mixer wall for good contact with the polymer melt. In the beginning of the mixing process, the polymer is in the solid state, then it starts to melt and the measured temperature is affected by the mixer wall temperature (i.e. the set temperature of 160°C). Therefore, the temperature range is taken from 120°C to 160°C, corresponding to the steady melt state of EVA, for estimating the flow behavior of the nanocomposites. Figure 1 represents the mixing torques of the EVA/SNP and EVA/m-SNP nanocomposites as a function of temperature. It is clear that the torques of the nanocomposites decrease as their temperature increases. At a certain temperature, the melt torque of the EVA/m-SNP nanocomposites is slightly higher than that of EVA/SNP nanocomposite and it increases with increasing APTES modifier content. As demonstrated in some studies, the steady melt torque is a measure of relative melt viscosity [14], [15], [16]. This means that the relative melt viscosity of the EVA/m-SNP nanocomposites is higher than that of EVA/SNP nanocomposites [17]. This is explained as follows: during melt mixing m-SNP with EVA, the organic moiety of m-SNP as (3-aminopropyl) triethoxy silane is easier to mix with EVA macromolecules. Therefore, m-SNP adheres with EVA more easily and disperses in EVA matrix more regularly than SNP, leading to increased internal friction of the melt polymer fluid. By contrast, the interaction of m-SNP with EVA matrix is stronger than that of SNP. In the EVA/m-SNPs nanocomposites, there is also existence of the hydrogen bonds between amino (NH2) groups of m-SNPs and C=O or C-O-C groups of EVA matrix. That will be discussed in a later section (3.5.1) of this paper.
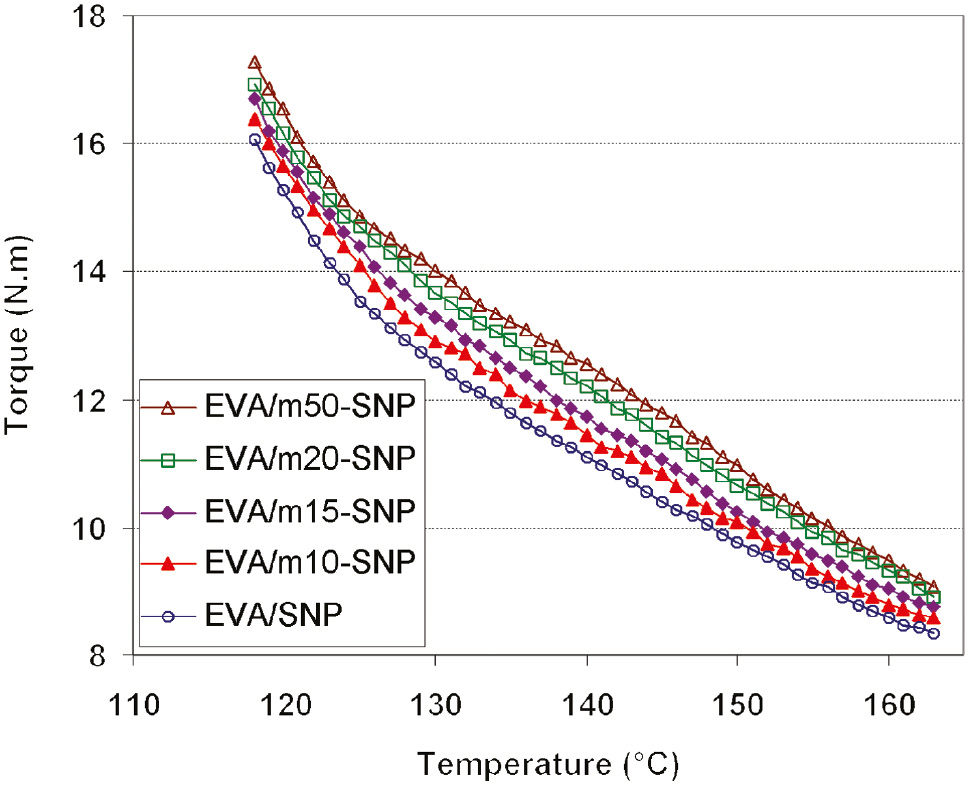
Variation of torque for ethylene vinyl acetate copolymer (EVA)/silica nanoparticle (SNP) and EVA/modified SNP (m-SNP) nanocomposites as function of temperature.
3.2 Thermal properties
The DSC diagrams and thermal characteristics of EVA/SNP and EVA/m-SNP nanocomposites are demonstrated in Figure 2. It can be seen that melting temperature of the nanocomposites has not significantly changed when using 1.5 wt.% of original SNP or m-SNP modified by 10 wt.% and 20 wt.% of APTES (melting point of the nanocomposites around 98.4–99.8°C). However, the areas of melting endotherm of the nanocomposites are slightly reduced, which indicates that the crystallinity of the nanocomposites is reduced by APTES modifier grafted on the surface of SNP. It is worth noting that the crystallinity of neat EVA was 22.7%, as reported in our previous study [8]. As calculated in Table 1, the crystallinity (XC) of the nanocomposites reduces from 22.51% for the EVA/SNP sample to 21.58% and 19.60% for EVA/m10-SNP and EVA/m20-SNP samples, respectively. The major reason is the presence of hydrogen bonds between amino groups (NH2) of APTES and C=O or C-O-C groups of EVA, which restricts the mobility of EVA macromolecules, and as a result, crystallizable segments of EVA are no longer able to be folded into polymer crystals [18]. The melting enthalpys of the nanocomposites using 10 wt.% and 20 wt.% of APTES are also reduced. The melting enthalpy of the nanocomposites reduces from 67.07 J/g for the EVA/SNP sample to 64.31 J/g and 58.40 J/g for the EVA/m10-SNP and EVA/m20-SNP samples, respectively. The melting enthalpy of EVA is also highest with the value of 67.74 J/g [8]. Naturally, the reduction of melting enthalpy of EVA/SNP and EVA/m10-SNP nanocomposites in comparison to EVA relates to the reduction of the part of crystallinity of EVA in the presence of SNP and m-SNP as aforementioned.
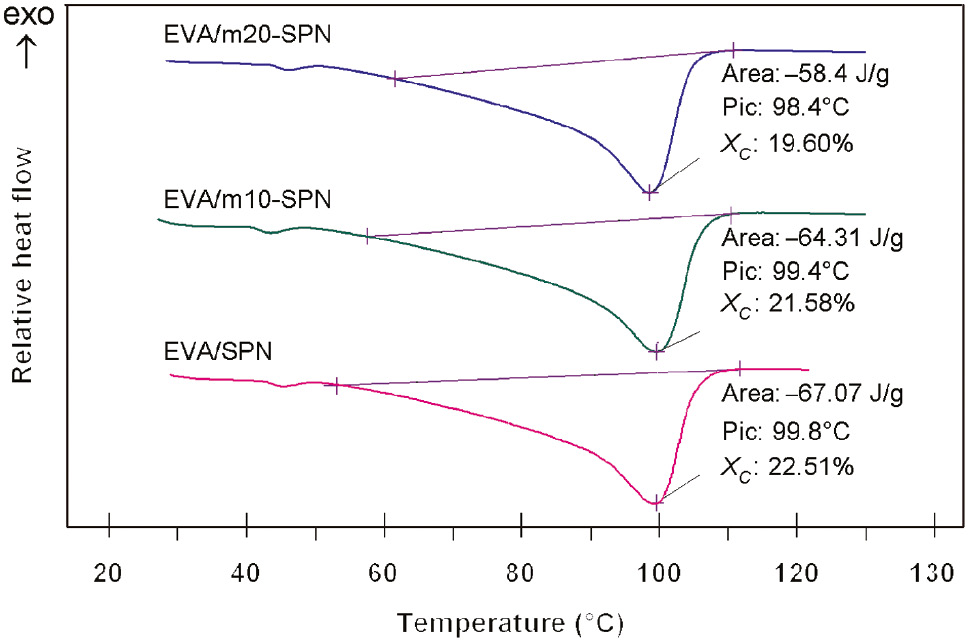
Differential scanning calorimetry (DSC) diagram of ethylene vinyl acetate copolymer (EVA)/silica nanoparticle (SNP) and EVA/modified SNP (m-SNP) nanocomposites.
Thermal characteristics of ethylene vinyl acetate copolymer (EVA), EVA/silica nanoparticle (SNP) and EVA/m-SNP nanocomposites from differential scanning calorimetry (DSC) analysis.
| Sample code | Melting point (°C) | Melting enthalpy (J/g) | Crystallinity (%) |
|---|---|---|---|
| EVA/SNP | 99.8 | 67.07 | 22.51 |
| EVA/m10-SNP | 99.4 | 64.31 | 21.58 |
| EVA/m20-SNP | 98.4 | 58.40 | 19.60 |
EVA, Ethylene vinyl acetate copolymer; SNP, silica nanoparticle.
3.3 Horizontal burning characters
Table 2 represents the horizontal burning time and burning rate of EVA and EVA/SNP nanocomposites. From the results of the burning test, it is realized that all samples were able to catch fire after 30 s of ignition; the flame reached the clamp and the burning drips occurred. All samples have passed the UL94 HB rating and unmodified SNP can also reduce the flammability of EVA matrix. The burning time and burning rate of EVA/m-SNP nanocomposites are higher than those of the EVA/SNP nanocomposite, therefore, m-SNPs are able to reduce the flammability for EVA more effectively than unmodified SNP. When increasing APTES modifier content up to 50 wt.%, the burning time and burning rate of the nanocomposites decrease. This is related to the presence of APTES modifier which improved the dispersion of m-SNP in the EVA matrix, and the adhesion between m-SNP and EVA is enhanced. The structure of EVA/m-SNP nanocomposites becomes tighter than that of EVA/SNP nanocomposite. Therefore, the m-SNPs are able to limit penetration of oxygen into the nanocomposites more strongly than the SNP [19].
Horizontal burning time and burning rate of ethylene vinyl acetate copolymer (EVA), EVA/silica nanoparticle (SNP) and EVA/m-SNP nanocomposites.
| Sample code | Burning time (s) | Burning rate (mm/min) | UL 94 HB rating |
|---|---|---|---|
| EVA | 252 | 17.9 | Passed |
| EVA/SNP | 279 | 16.1 | Passed |
| EVA/m5-SNP | 282 | 16.0 | Passed |
| EVA/m10-SNP | 284 | 15.8 | Passed |
| EVA/m15-SNP | 294 | 15.3 | Passed |
| EVA/m20-SNP | 294 | 15.3 | Passed |
| EVA/m50-SNP | 301 | 15.0 | Passed |
EVA, Ethylene vinyl acetate copolymer; SNP, silica nanoparticle.
In addition, according to Kashiwagi et al. [20], [21], one of the flame retardant mechanisms of silica is the increase of polymer melt viscosity. In this study, the melt viscosity of EVA/m-SNPs nanocomposites is higher than that of EVA/SNP nanocomposite, as demonstrated in Figure 1. Thus, the m-SNPs can reduce the flammability of EVA more effectively than unmodified SNP.
3.4 Accelerated weather resistance
3.4.1 FTIR analysis of EVA, EVA/SNP and EVA/m15-SNP nanocomposites before accelerated weather testing
As discussed in previous studies [7], [19], the hydrogen bonds in EVA/SNP nanocomposites can be characterized by FTIR spectra analysis. As is well known, it is hard to detect the shifts of all peaks of the nanocomposites in comparison to the reference component materials. In fact, only some specific bands with distinguishable changes can be taken into account.
Figure 3 shows the FTIR spectra of EVA and EVA/SNP, EVA/m15-SNP nanocomposites and Table 3 lists some specific bands in spectra of initial EVA, nanosilica, and the nanocomposites. It can be clearly seen that the stretching vibration absorption bands (ν) for the C=O groups at around 1737 cm−1 in EVA and the nanocomposites are not significantly changed. This is due to the numerous C=O groups existing in the nanocomposites and only their small part is available to make interaction with SNPs. Unfortunately, there is no observable peak attributed to the absorption of amino groups in spectra of the nanocomposites. A specific band attributed to ester stretching vibration of C-O-C (ν) at 1135 cm−1 does not appear in the spectra of EVA/SNP and EVA/m-SNP nanocomposites because of the overlap with stretching vibration of Si-O groups at 1100 cm−1 of silica. The wavenumber of the Si-O band is moved to a higher wavenumber at about 1122–1124 cm−1 in spectra of the nanocomposites, corresponding to the shifts of 22–24 cm−1. There are also small changes of bending vibration (δ) of Si-O groups in spectra of the nanocomposites compared to that of pure silica, corresponding to the shifts of 7–10 cm−1. From the above wavenumber shifts, it can be suggested that there are hydrogen bonds between SNP or m-SNP and the EVA matrix. Theoretically, a proposal scheme of hydrogen bonds between EVA and SNP-modified by APTES can be illustrated in Figure 4.
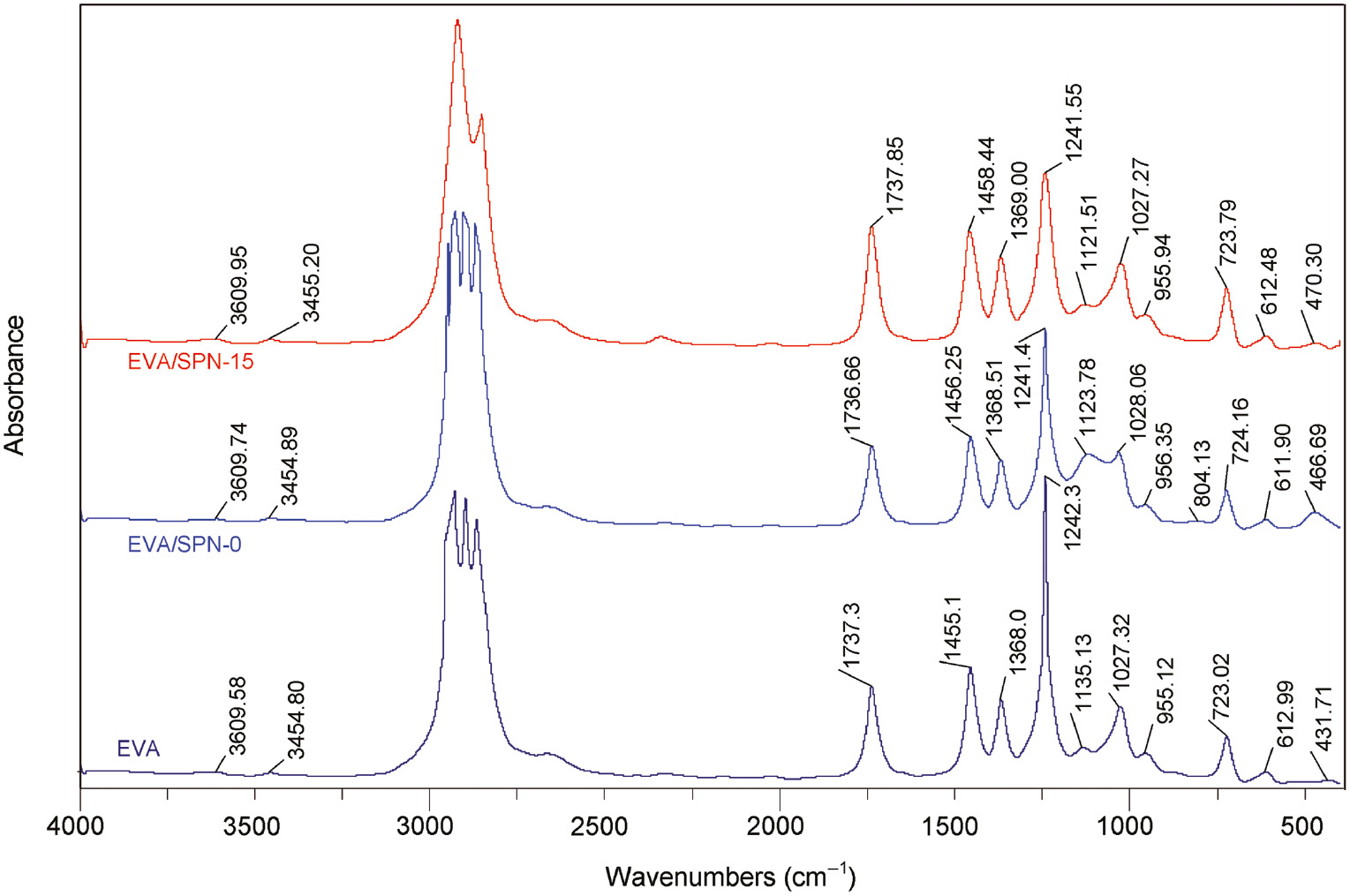
Fourier transform infrared (FTIR) spectra of initial ethylene vinyl acetate copolymer (EVA), EVA/silica nanoparticle (SNP) and EVA/m15-SNP nanocomposites.
Specific peak of some groups in initial ethylene vinyl acetate copolymer (EVA) and EVA/silica nanoparticle (SNP), EVA/m15-SNP nanocomposites.
| Sample code | Wavelength (cm−1) | ||||
|---|---|---|---|---|---|
| νC=O | νC-O | δC-O-C | νSi-O | δSi-O | |
| Nanosilica [8] | – | – | – | 1100 | 460 |
| EVA | 1737 | 1242 1027 | 1135 | – | – |
| EVA/SNP | 1737 | 1241 1028 | – | 1124 | 467 |
| EVA/m15-SNP | 1738 | 1242 1027 | – | 1122 | 470 |
EVA, Ethylene vinyl acetate copolymer; SNP, silica nanoparticle.
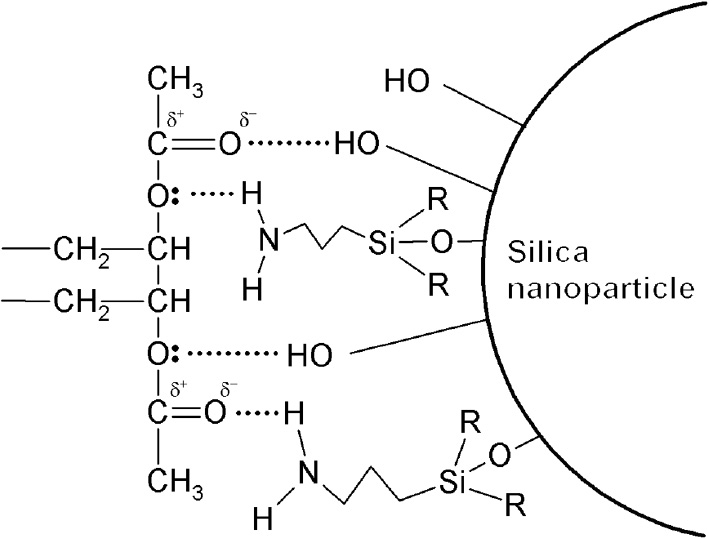
Proposal scheme of hydrogen bonds between ethylene vinyl acetate copolymer (EVA) and silica nanoparticle (SNP) modified by 3-aminopropyltriethoxysilane (APTES).
3.4.2 FTIR analysis of EVA, EVA/SNP and EVA/m15-SNP nanocomposites after accelerated weather testing
Thermo- and photo-oxidation of EVA in accelerated weather testing leads to formation of several photo-oxidation products that can be characterized by the FTIR technique [22]. The FTIR absorbance spectra of EVA, EVA/SNP and EVA/m15-SNP nanocomposites are illustrated in Figure 5, where the bottom and upper curves are spectra of the samples before and after accelerated weather testing, respectively. It is clear that the C=O stretching peak areas of the samples after accelerated weather testing are broadened. An increase of intensity of carbonyl absorption band at around 1739 cm−1 is also observed (the upper curves). This is related to the formation of new carbonyl groups under the effect of UV irradiation, heating and atmosphere oxygen [23].

Fourier transform infrared (FTIR) spectra of: (A) ethylene vinyl acetate copolymer (EVA), (B) EVA/silica nanoparticle (SNP) nanocomposites and (C) EVA/m15-SNP nanocomposite for 0 h, 72 h, and 168 h of accelerated weather testing.
For quantitative comparison, Table 4 represents the results of CI evaluated from the analysis of FTIR spectra of EVA and the nanocomposites. The CI value reflects the relative amount of C=O which existed in the samples. The results show that CI values of the samples before accelerated weather testing are approximately equal to each other. After accelerated weather testing, CI values of investigated samples are increased with increasing testing time. The CI value of the EVA/SNP sample after 72 h and 168 h of testing is lower than that of EVA. In our previous study, it had been demonstrated that nanosilica could limit the photo-degradation of EVA under UV exposure [8]. In this study, a special attention is paid to the effect of SNP modified by APTES on the CI value of the EVA/m-SNP nanocomposites. As expected, the CI value of EVA/m15-SNP nanocomposite after 72 h and 168 h of accelerated weather testing is slightly lower than that of EVA/SNP nanocomposite. This reflects the lower concentration of C=O groups formed by thermo- and photo-oxidation for the EVA/m-SNP nanocomposites, because APTES can enhance the adhesion between SNP and EVA matrix through hydrogen bonds, leading to limitation of the penetration of oxygen into the nanocomposites.
Carbonyl index (CI) value of ethylene vinyl acetate copolymer (EVA), EVA/silica nanoparticle (SNP) and EVA/m-SNP nanocomposites before and after accelerated weather testing.
| Weather testing time (h) | Carbonyl index (CI) | ||
|---|---|---|---|
| EVA | EVA/SNP | EVA/m15-SNP | |
| 0 | 2.83 | 2.82 | 2.82 |
| 72 | 4.24 | 4.05 | 3.93 |
| 168 | 5.60 | 4.57 | 4.50 |
EVA, Ethylene vinyl acetate copolymer; SNP, silica nanoparticle.
3.4.3 Morphology of EVA/SNPs nanocomposites after weather testing
Figure 6 shows the micro cracks that developed on the surface of EVA/SNP and EVA/m-SNP nanocomposites after accelerated weather testing. It is easy to see that the micro cracks of the exposed EVA/SNP nanocomposite are deep, while the micro crack of exposed EVA/m-SNP nanocomposite is shallow. These mean that the APTES modification of nanosilica can reduce the development of the crack tip under the weathering test. It is worth mentioning that in our previous work [8], the long and deep micro cracks had been formed on the surface of EVA after 168 h of accelerated weathering [8]. In the case of polymers with a semicrystalline structure (like EVA), cracking of these polymers is due to internal stress distribution in crystalline and amorphous areas. Since degradation occurs mainly in the amorphous region, the neighboring crystalline areas contribute to stress development around the crack tip, causing orientation of EVA macromolecules. For completely amorphous specimens, a mechanism of the above orientation does not exist; therefore, the samples degrade uniformly [13]. In our study, m-SNP increases the extent of the amorphous phase and this may be a reason for a shallow crack tip developing in EVA/m-SNP nanocomposite. Another reason is that m-SNP can adhere with EVA matrix more strongly than unmodified SNP.

Field emission scanning electron microscopy (FESEM) images of: (A) ethylene vinyl acetate copolymer (EVA)/silica nanoparticle (SNP) and (B) EVA/m15-SNP nanocomposites after 168 h of accelerated weather testing.
3.4.4 Mechanical properties of EVA, EVA/SNP, EVA/m-SNP nanocomposites before and after accelerated weather testing
Table 5 shows initial values of tensile strength and elongation at break of EVA, EVA/SNP, and EVA/m-SNP nanocomposites (SNP and m-SNP were used with 1.5 wt.% compared with EVA) before accelerated weather testing. With introducing unmodified SNP into EVA matrix, the tensile strength of EVA increases slightly from 16.94 MPa to 17.23 MPa. When using m-SNPs, the tensile strength of EVA/m-SNPs nanocomposites is higher than that of EVA/SNP nanocomposite and increases with increasing the APTES modifier content up to 20 wt.%, then it tends to decrease for EVA/m50-SNP nanocomposite (50 wt.% APTES modifier). The enhancement in tensile strength of EVA/m-SNPs nanocomposites can be explained by contribution of APTES modifier grafted on the surface of nanosilica. In our previous studies [9], [10], the grafted APTES weights on the silica surface were increased as 2.66 wt.%, 5.54 wt.%, 6.89 wt.% and 7.52 wt.% when modifying with APTES contents of 5 wt.%, 10 wt.%, 15 wt.%, and 20 wt.%, respectively (all were compared to silica weight). The filler/matrix interaction contributed by APTES coupling agent can be attributed to the formations of hydrogen bond interaction, as illustrated in Figure 4, and molecular interdiffusion interaction between grafted APTES molecules and EVA macromolecules. Therefore, stress transfer at the particle/matrix interfaces is more efficient in the nanocomposites using m-SNP, and tensile strength of nanocomposites using m-SNP is higher than that of nanocomposites using bare SNP. Unfortunately, when modifying with APTES content larger than 15 wt.%, although the grafted weight increased, the higher concentration of silanol that was catalyzed by higher amino group concentration, caused the agglomeration of silica particles, which reduced the effect of APTES on tensile strength of nanocomposites [3]. In contrast, it is observed that elongation at break of EVA decreases with increasing the APTES coupling agent used. This is due to the greater the effective stress transfer, the less elongation at break is gained. As a result, break of nanocomposites occurred sooner. When APTES coupling agent content is larger than 15 wt.%, some defects can be formed in the nanocomposites because of agglomerated m-SNP, and a much higher decrease in elongation at break is observed.
Initial tensile properties of ethylene vinyl acetate copolymer (EVA) and EVA/silica nanoparticle (SNP), EVA/m-SNP nanocomposites and the retentions of tensile properties after accelerated weather testing.
| Sample code | Tensile strength (MPa) | Elongation at break (%) | Retention of tensile strength (%) after testing time | Retention of elongation (%) after testing time | ||
|---|---|---|---|---|---|---|
| 72 h | 168 h | 72 h | 168 h | |||
| EVA | 16.94 | 720 | 46.8 | 32.9 | 38.3 | 7.8 |
| EVA/SNP | 17.23 | 703 | 53.8 | 38.7 | 39.8 | 12.6 |
| EVA/m5-SNP | 17.61 | 677 | 60.3 | 44.3 | 45.8 | 13.4 |
| EVA/m10-SNP | 18.19 | 672 | 61.6 | 45.1 | 56.5 | 13.5 |
| EVA/m15-SNP | 20.47 | 666 | 64.5 | 48.1 | 63.1 | 13.6 |
| EVA/m20-SNP | 18.73 | 657 | 54.4 | 45.8 | 45.7 | 11.0 |
| EVA/m50-SNP | 17.44 | 642 | 48.3 | 41.7 | 43.6 | 10.9 |
EVA, Ethylene vinyl acetate copolymer; SNP, silica nanoparticle.
Table 5 also displays retentions of tensile strength, and elongation at break of EVA, EVA/SNP, and EVA/m-SNP nanocomposites after accelerated weather testing for two periods (72 h and 186 h). It is clear that the retentions of tensile strength and elongation at break of the nanocomposites after testing periods are higher than those of EVA. The enhancement effect of SNP on the retentions of tensile properties of EVA can be explained by the limitation of degradation of SNP for EVA and heat dissipation under testing conditions. This result is in good agreement with our previous studies, in which commercial SNP (average particle diameter of 12 nm) had been used with SNP content which varied from 2 wt.% to 5 wt.% [8]. In this study, SNP was only used with small content of 1.5 wt.% for studying the effect of APTES modifier on tensile properties retentions of EVA. As expected, the retentions of tensile strength and elongation at break of nanocomposites using m-SNP are improved. The remarkable improvement in tensile properties retentions is observed for the nanocomposite using m15-SNP (1.5 wt.% SNP modified by 15wt.% of APTES). It can be suggested that there is an optimal dosage usage of APTES weight for modifying surface of SNP, because of the limited amount of surface silanol for a certain bare silica weight. Shortage of usage of APTES would cause low hydrophobicity for SNP. Exceeding the amount would cause unwanted effects, such as agglomeration of silica, and hydrogen bonds between amino groups on m-SNP surface. In general, better filler/matrix interaction may provide the better restriction of oxygen and moisture permeation into the nanocomposites, which reduces the thermo-, photo-oxidation and scission reaction of EVA macromolecules. For this reason, adding m-SNP to EVA can limit the reduction in tensile strength and elongation at break of EVA/m-SNP nanocomposites under accelerated weather testing. Remarkably, a nanocomposite using m15-SNP, that was modified by APTES with 15 wt.%, may be an optimal dosage of APTES coupling agent, which possesses the highest weather resistance.
When using m-SNP that was modified with APTES content of 20 wt.% or larger, the agglomeration of m-SNPs and unfavorable intermolecular hydrogen bonds between amino groups would cause irregular dispersion of m-SNP in EVA matrix and the appearance of defects in the nanocomposites [10]. This reduces the effect of coupling agent on prevention of thermo-, photo-oxidation for the nanocomposites. Therefore, a tendency of decreased retention in tensile strength and elongation at break of EVA/m-SNP nanocomposites after accelerated weather testing is observed.
4 Conclusion
In this work, SNP was modified by different amounts of APTES (m-SNP) and using eco-friendly solvents. The EVA/SNP and EVA/m-SNP nanocomposites were prepared by melt mixing EVA and SNP and m-SNP (1.5 wt.% compared to EVA weight) in a Haake intermixer. Relative melt viscosity of EVA/m-SNP nanocomposites is higher than that of EVA/SNP nanocomposite. The DSC results indicate that there is no remarkable change of melting temperature of EVA and the decrease in crystallinity of EVA is due to hydrogen bonds between m-SNP and EVA matrix. The m-SNP enhances tensile strength of EVA and this value reaches the maximum at 15 wt.% of APTES modifier. The m-SNP decreases elongation at break of EVA and reduces flammability of EVA more effectively than SNP. The FTIR spectra analysis of C=O index show that the relative amount of C=O groups formed after accelerated weather testing of the EVA/m-SNP nanocomposites is lower than that of EVA/SNP nanocomposite. The micro crack of exposed EVA/m-SNP sample is not as deep as that of the exposed EVA/SNP sample. The retentions of tensile strength and elongation at break of EVA/m-SNP nanocomposites are higher than those of EVA/SNP nanocomposite. SNP and m-SNP improve weather resistance of EVA. The EVA/m15-SNP nanocomposite with high tensile properties, and flame and weather resistance can be an effective candidate for green fabrication of electric and telecommunication cables.
About the authors

Do Quang Tham received his Bachelor’s degree in Solid State Physics from Hanoi University of Science in 1997 and his Master’s degree in Material Science from Hanoi University of Technology in 2009. In 2014, he obtained his PhD in Polymer and Composite Materials from the Institute for Tropical Technology (ITT), Vietnam Academy of Science and Technology (VAST). Since 2001, he has worked as a researcher at ITT. His present research fields are polymer nanocomposite and polymer composite materials.

Nguyen Thi Thu Trang worked as a researcher atthe Department of Physico-Chemistry of Non-Metallic Materials,ITT, VAST. She received her MSc in Chemistry from the College of Science, Vietnam National University of Hanoi, in 2005. Nearly 30 of her articles and reports are related to conducting polymers and polymer nanocomposites and were published in national and international journals and proceedings of national and international scientific conferences and workshops.

Nguyen Thuy Chinh received her Bachelor’s degree in Chemistry in 2009 and her MSc degree in Physical Chemistry in 2011. Since 2009, she has worked as a researcher at the Department of Physico-Chemistry of Polymers and Non-Metallic Materials, ITT, VAST. Currently, she is working on her PhD thesis at ITT, VAST, where she is investigating drug delivery systems based on poly (lactic acid) and chitosan. Most of her work is related to the properties of nanomaterials and polymer nanocomposites.

Nguyen Vu Giang received his Bachelor’s degree in Physical Engineering in 1994, and his MSc in 2001. He received his PhD degree at the Department of Polymer Science and Engineering, College of Engineering, Sunchon National University, South Korea. Currently, he is working in the fields of polymer nanocomposite and polymer blend materials, degradation and stability of polymers and rubbers, green materials and their applications.

Tran Dai Lam graduated from Belorussian State University (in the former USSR) with a Master’s degree in Solid State Chemistry (1994). He received his PhD in Physical Chemistry (Surface-Interface) from the University of Paris VII, Paris, France, in 2003. He was a lecturer at Hanoi University of Technology from 1998 to 2008. He has been an associate professor at Institute of Materials Science (IMS) since 2009. His research focusses on nanofabrications, characterizations and applications of nanobiomaterials in drug delivery systems and biosensors.

Thai Hoang obtained his PhD in Polymer Chemistry in 1993. Since 2012, he has worked as a professor at the ITT, VAST. He carried out postdoctoral fellowship in polymer blend, polymer composites and plastics technology in South Korea, UK and Japan. He has published 40 papers in international journals and more than 180 papers in national journals. His research fields are polymer blends, nanocomposites, biodegradable polymers, and bio-medical materials.
Acknowledgments
The authors would like to thank the National Foundation for Science and Technology Development of Vietnam (NAFOSTED, 104.04-2010.02) and Vietnam Academy of Science and Technology (VAST) for the financial support of a bilateral international project between ITT, VAST and CIRIMAT, CNRS France during the period 2011-2012.
References
[1] Chen Y, Peng Z, Kong L-X, Huang M-F, Li P-W. Polym. Eng. Sci. 2008, 48, 1674–1677.10.1002/pen.20997Search in Google Scholar
[2] YernChee C, Ching YC, Yaacob I. Adv. Sci. Lett. 2012, 12, 165–169.10.1166/asl.2012.2739Search in Google Scholar
[3] Rahman IA, Jafarzadeh M, Sipaut CS. Ceram. Int. 2009, 35, 1883–1888.10.1016/j.ceramint.2008.10.028Search in Google Scholar
[4] Conradi M. Mater. Technol. 2013, 47, 285–293.10.1002/ciuz.201390060Search in Google Scholar
[5] Wei L-M, Hu N-T, Zhang Y-F. Materials 2010, 3, 4066–4079.10.3390/ma3074066Search in Google Scholar PubMed PubMed Central
[6] Wu Z, Xiang H, Kim T, Chun M-S, Lee K. J. Colloid Interface Sci. 2006, 304, 119–124.10.1016/j.jcis.2006.08.055Search in Google Scholar PubMed
[7] Hoang T, Truc TA, Thanh DTM, Chinh NT, Tham DQ, Trang NTT, Giang NV, Lam VD. J. Compos. Mater. 2014, 48, 505–511.10.1177/0021998313476319Search in Google Scholar
[8] Hoang T, Chinh NT, Trang NTT, Hang TTX, Thanh DTM, Hung DV, Ha C-S, Aufray M. Macromol. Res. 2013, 21, 1210–1217.10.1007/s13233-013-1157-8Search in Google Scholar
[9] Hoang T, Truc TA, Chinh NT, Trang NTT. Viet. J. Chem 2013, 51, 59–63.Search in Google Scholar
[10] Hoang T, Chinh NT, Khu LV, Trang NTT. Viet. J. Chem 2013, 51, 46–0.Search in Google Scholar
[11] Hoang T, Chinh NT, Trang NTT, Manh DQ. Viet. J. Chem 2012, 50, 96–100.Search in Google Scholar
[12] Sadeghi M, Khanbabaei G, Dehaghani AHS, Sadeghi M, Aravand M, Akbarzade M, Khatti S. J. Membrance Sci. 2008, 322, 423–428.10.1016/j.memsci.2008.05.077Search in Google Scholar
[13] Wypych G. Handbook of Material Weathering; Chapter 9, Chapter 13, 2nd ed., ChemTec Publishing: Toronto, 1995.Search in Google Scholar
[14] Freire E, Bianchi O, Monteiro EEC, Nunes RCR, Forte MC. Mater. Sci. Eng., C 2009, 29, 657–661.10.1016/j.msec.2008.12.025Search in Google Scholar
[15] Nouri M, Mehrabzadeh M. Iran. Polym. J. 1996, 5, 237–241.Search in Google Scholar
[16] Zhou J, Yu W, Zhou C-X. Polymer 2009, 50, 4397–4405.10.1016/j.polymer.2009.06.077Search in Google Scholar
[17] Tham DQ, Chinh NT, Hoang T. Viet. J. Sci. Technol. 2015, 53, 18–26.Search in Google Scholar
[18] Bidsorkhi HC, Adelnia H, Ismail AF, Matsuura T. Sep. Purif. Technol. 2015, 146, 351–357.10.1016/j.seppur.2015.03.060Search in Google Scholar
[19] Tham DQ, Tuan VM, Thanh DTM, Chinh NT, Giang NV, Trang NTT, Hang TTX, Huong HT, Dung NTK, Hoang T. J. Nanosci. Nanotechnol. 2015, 15, 2777–2784.10.1166/jnn.2015.9209Search in Google Scholar
[20] Kashiwagi T, Gilman JW, Butler KM, Harris RH, Shields JR, Asano A. Fire Matter. 2000, 24, 277–289.10.1002/1099-1018(200011/12)24:6<277::AID-FAM746>3.0.CO;2-ASearch in Google Scholar
[21] Kashiwagi T, Shields JR, Harris RH, Davis RD. J. Appl. Polym. Sci. 2003, 87, 1541–1553.10.1002/app.11967Search in Google Scholar
[22] Oreski G, Wallner GM, Lang RW. Biosyst. Eng. 2009, 103, 489–496.10.1016/j.biosystemseng.2009.05.003Search in Google Scholar
[23] Jin J, Chen S, Zhang J. Polym. Degrad. Stab. 2010, 95, 725–732.10.1016/j.polymdegradstab.2010.02.020Search in Google Scholar
©2016 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- In this issue
- Green processing of thermosensitive nanocurcumin-encapsulated chitosan hydrogel towards biomedical application
- Supramolecular chemistry at interfaces: host-guest interactions for attaching PEG and 5-fluorouracil to the surface of porous nanosilica
- Study of ATO nanoparticles by the solvothermal method for thermal insulated coated glass: a green energy application
- Synthesis of Cu-BTC, from Cu and benzene-1,3,5-tricarboxylic acid (H3BTC), by a green electrochemical method
- Improving the electrochemical behavior of sustainable polyaniline titanium dioxide composite by intercalation of carbon nanotubes
- Sustainable composite materials based on ethylene-vinylacetate copolymer and organo-modified silica
- Conference announcement
- 7th International Conference of the Flow Chemistry Society (Cambridge, UK, February 7–8, 2017)
Articles in the same Issue
- Frontmatter
- In this issue
- Green processing of thermosensitive nanocurcumin-encapsulated chitosan hydrogel towards biomedical application
- Supramolecular chemistry at interfaces: host-guest interactions for attaching PEG and 5-fluorouracil to the surface of porous nanosilica
- Study of ATO nanoparticles by the solvothermal method for thermal insulated coated glass: a green energy application
- Synthesis of Cu-BTC, from Cu and benzene-1,3,5-tricarboxylic acid (H3BTC), by a green electrochemical method
- Improving the electrochemical behavior of sustainable polyaniline titanium dioxide composite by intercalation of carbon nanotubes
- Sustainable composite materials based on ethylene-vinylacetate copolymer and organo-modified silica
- Conference announcement
- 7th International Conference of the Flow Chemistry Society (Cambridge, UK, February 7–8, 2017)