Relativistic effects on the chemistry of heavier elements: why not given proper importance in chemistry education at the undergraduate and postgraduate level?
-
Ankita Das
 ,
Udita Das
,
Ruhi Das
,
Udita Das
,
Ruhi Das


Abstract
Relativistic effects are important to understand the chemistry of heavier elements across the periodic table (PT). Three important relativistic effects are: contraction of s- and p-orbitals (direct relativistic effect), expansion of d- and f-orbitals (indirect relativistic effect) and spin–orbit (SO) coupling to split the p-, d- and f-orbitals. Each of these effects is approximately proportional to Z 2 (Z = atomic number) for the valence shell electrons in many electron atoms and consequently, these relativistic effects dominantly control the properties of the heavier elements (mainly the 6th and 7th period elements). These aspects are not given the proper importance in most of the inorganic chemistry text books and in chemistry education at the university level.
1 Introduction
In terms of Einstein’s relativity theory, the mass (m) of a moving particle increases with the increase of its velocity (v) compared to its rest mass (m 0). This effect is considerable only when the velocity of the moving particle becomes comparable to that of light (c ≈ 3.0 × 108 m s−1). Apparently, we live in a world where the velocities of the moving bodies are negligible with respect to that of light. But for the heavier elements, the velocity of the moving electrons is not negligible with respect to that of light and it influences the properties of such heavier elements. Without introducing this relativistic effect, the conspicuous chemical anomalies observed for the heavier elements across the periodic table (PT) cannot be fully rationalised. These aspects are mainly getting confined in the research journals (Burke & Grant, 1967; Das et al., 2023; Pitzer, 1979; Pyper, 2020; Pyykkö, 1978; Pyykkö, 2012; Pyykkö & Desclaux, 1979; Thayer, 2005; Pyykkö, 1988). Unfortunately, this aspect is grossly overlooked in most of the conventional text books on inorganic chemistry and our present chemistry education at the undergraduate (UG) and postgraduate (PG) chemistry courses. Here it is worth mentioning that the quantitative treatment of relativistic quantum mechanics developed by Dirac in 1929 (Dirac, 1929) is not required at the elementary level to understand qualitatively the relativistic effect on the properties of elements.
2 Background: interaction through the question-answer relationship (QAR) strategy with the students in class rooms and college teachers in refresher course programmes and feedback from them
In teaching the properties of the heavier elements, mainly the 6th and 7th period elements, including the lanthanides and actinides, discussion of relativistic effect appears to be important. In fact, without introducing the relativistic effect controlling the properties of the heavier elements, it becomes difficult to rationalize their properties. But unfortunately, in most of the inorganic chemistry text books, this aspect has been grossly overlooked and very often, properties of the heavier elements have been explained in terms of the effects of lanthanide contraction (in general, f-contraction). For example, by considering the effects of lanthanide contraction, inert pair effect observed for the heavier congeners of the p-block elements has been explained in most of the inorganic chemistry text books. But, it gives the incomplete chemistry. Besides the lanthanide contraction effect on the post-lanthanides, relativistic effect is quite important to understand the reason behind the inert pair effect noted for the heavier p-block congeners. In fact, relativistic effect itself contributes significantly to cause the lanthanide and actinide contractions.
The relativistic effect influencing the properties of the heavier elements across the periodic table (PT) has been discussed in class rooms of different batches of students (each batch of about 50–60 students) and in different forums including the refresher courses (training programmes of college and university teachers) and it has been accepted enthusiastically, by the students and young teachers. These are evident from their spontaneous responses and questions during the open discussions. Being motivated, some of the students and teachers have gone though some selected original papers dealing with the relativistic effects in chemistry. They have pointed out some aspects of these research papers for discussions and clarifications. In the open discussions, different questions raised by the students and other participants have been answered to clear any doubt in the subject. Many questions have been also received from the teachers of different colleges and universities in connection with the online refresher course programmes conducted by different universities. This tutorial review has been developed based on the question-answer relationship (QAR) strategy and feedback from the students and teachers. It addresses to answer all the questions (in different conceptual categories) raised by them on the present topic. The questions asked in the said discussions and class rooms are organized here systematically.
(1) What is the origin of relativistic effect in chemistry? (2) Why is it important only for the heavier elements in the periodic table (PT)? (3) How can we explain the nonexistence of elements with atomic number (Z) higher than 137 in terms of the theory of relativity? (4) What is the difference between the direct and indirect relativistic effects? (5) What is the relativistic contraction of orbitals? (6) What is the relativistic expansion of orbitals? (7) How can we rationalize the apparent anomalies in electronic configurations in the pairs: Mo (4d 55s 1, 4d-member) but W (5d 46s 2, 5d-member); Pd (4d 105s 0, 4d-member) but Pt (5d 96s 1, 5d-member)? (8) What is spin orbit (SO) coupling? (9) How is it related with the relativistic quantum mechanics? (10) How does it destroy the degeneracy of the p-, d- and f-orbitals? (11) What are the shapes of p 1/2 and p 3/2 orbitals? (12) Why does thallium (Tl) show the unexpectedly high electron affinity? (13) How does bismuth earn the stability of +1 oxidation state, i.e. Bi(I)? (14) What does make the difference in colours of gold (yellow), silver (white) and copper (golden reddish lustre)? (16) What does make gold so noble? (15) Why does gold (Au) show the unexpectedly high electron affinity? (17) Why does gold form auride (Au−)? (18) How can we rationalise the stability of CsAu (cesium auride) and Cs2Pt (cesium platinide)? (19) Very often, the noble metals show the high electron affinity and high electronegativity. How can we explain it? (20) Why is mercury liquid? (21) Why are the nearest neighbours of mercury in PT solid? (22) Why is it so inert? (23) Why is its boiling point relatively low? (24) How can you rationalise the higher stability of the dimeric species Au2 compared to that of Ag2 and Cu2 in Gr. 11 of PT? (25) How can we explain the relationship between Au and Hg with respect to the relationship between H and He in PT? (26) Why is the mercurous ion (Hg2 2+) so stable? (27) Why are the cadmous (Cd2 2+) and zincous (Zn2 2+) ions not stable in the same group of mercury in PT? (28) What is the reason behind the strong tendency for coordination number 2 in Au(I), Ag(I) and Hg(II)? (29) Why does mercury amalgamate sodium, gold and silver easily but not the common transition metals? (30) What is the aurophilic interaction? (31) How do we interpret it? (32) Is relativistic effect responsible for the low melting point of gallium (m.p. 29.8 °C)? (33) Why are the higher oxidation states more stable for the heavier d-block elements? (34) Why is the reverse trend true for the heavier p-block congeners? (35) What are the highest possible oxidation states of metals? (36) How can we rationalize the stability of unusual oxidation state Hg(IV) in HgF4? (37) How can we justify the stability order: Cr(VI) < Mo(VI) < W(VI); Mn(VII) < Tc(VII) < Re(VII); Ni(IV) < Pd(IV) < Pt(IV); Cu(III) < Ag(III) < Au(III)? (38) How can we explain the oxidising power order: CrO4 2− > MoO4 2− > WO4 2− and MnO4 − > TcO4 −> ReO4 −? (39) How can we explain the order of ease of oxidative addition of Ir(I), Rh(I) and Co(I) organometallic complexes? (40) How can we explain the ease of oxidative addition of Ir(I) in the Vaska complex, [Ir(CO)Cl(PPh3)2]? (41) How can we explain the difference in colours of the oxyanions: CrO4 2− (yellow) but MoO4 2− (colourless) and WO4 2− (colourless); MnO4 − (pink) but TcO4 − (colourless)? (42) How can we rationalize the inert pair effect in terms of the relativistic effect? (43) What is the reason behind the so high cell emf (2.0 V) of lead acid battery? (44) Which is the most reactive metal in the earth? (45) Why is cesium (Cs) more reactive than francium (Fr)? (46) How can we explain the difference in colours: tin dioxide (SnO2, white) but lead dioxide (PbO2, dark brown); [PbCl6]2− (yellow) but [SnCl6]2− (colourless), sodium bismuthate (NaBiO3, yellowish brown) but sodium antimonate (NaSbO3, colourless)? (47) What does make the difference between the lanthanides and actinides? (48) How does the relativistic effect contribute to cause the difference in properties of the lanthanides and actinides? (49) How does the relativistic effect contribute to the lanthanide and actinide contractions? (50) Why is actinide contraction more pronounced than lanthanide contraction? (51) Why is crystal field effect more important for the actinides than that for the lanthanides? (52) Why do the actinides but not the lanthanides show some similarities with the transition metals in terms of properties? (53) For the actinide complexes, the absorptions peaks in electronic spectra are broader and more intense compared to those of lanthanides. How can we explain it in terms of the relativistic effect? (54) Why is quenching of orbital contribution to the observed magnetic moment of the actinide complexes more important than that for the lanthanide complexes? (55) Why are the actinides (An3+) relatively softer (HSAB concept) than the lanthanides (Ln3+)? There are so many questions to be answered. This requires the knowledge of relativistic effect controlling the properties of the heavier elements.
This self explanatory tutorial review aims to answer all the probable questions related to the relativistic effect on the properties of elements across the periodic table. It will play the role of a chemistry teacher to the students to satisfy the needs of the students. In fact, the corresponding author has presented and used this review material in class rooms and refresher courses many times to benefit the students and college teachers and they have learnt and understood the subject clearly. The authors do believe that this tutorial review will be immensely helpful in chemistry education on the present important topic at the university level.
3 Design of the study in a teaching learning environment and aim of the study
The undergraduate first year students with chemistry as the major (honours) subject were our study. The student strength was about 50–60 in each batch. To understand the baseline of student knowledge in the present topic, pre-tests were done. Post-tests were done to measure the growth of knowledge in the present topic among the students after presentation and teaching of the material. Pre-tests established that most of the students (more than 98 %) were not aware of the subject, ‘relativistic effect controlling the properties of elements and their compounds’. It was quite expected because the matter is ignored in the available inorganic chemistry text books and in the existing syllabus. From the observation of pre-tests, it was decided to start the teaching from the basic idea of relativistic effect. The following steps were adopted for introducing the fundamental concepts of relativistic effect on the properties of orbital electrons and the properties of the elements in a teaching learning environment.
Step 1:
Introduction.
Aim: To understand the relativistic effect on the mass of the moving electron in the atom.
Step 2:
Relation of the relativistic mass correction of the orbital electron with the atomic number (Z) of the element.
Aim: To establish the fact that the relativistic mass correction of the orbital electron is only important for the heavier elements, specially for the 6th and 7th period elements of periodic table.
Step 3:
Relation of the relativistic mass correction of the orbital electron with the principal quantum number (n) and azimuthal (orbital angular momentum) quantum number (l) of the electron.
Aim: To establish the fact that the relativistic mass correction of the orbital electron is more important for the smaller values of l (for a particular n and Z value) in the order: s (l = 0) > p (l = 1) > d (l = 2) > f (l = 3).
Step 3: Relativistic contraction of orbital (direct relativistic effect).
Aim: To establish the fact that the increased relativistic mass causes the contraction of orbital in the order: s (l = 0) > p (l = 1) > d (l = 2) > f (l = 3).
Step 4:
Relativistic expansion of orbital (indirect relativistic effect).
Aim: To establish the fact that for a particular n value and Z value, the relativistic contraction of s (l = 0) and p (l = 1) orbitals actually causes indirectly the expansion of d (l = 2) and f (l = 3) orbitals.
Step 5:
Relativistic effect (both direct and indirect) on the properties of the heavier congeners across the periodic table.
Aim: To illustrate by giving examples of the different special properties of the heavier congeners (i.e. the elements in the 6th and 7th periods) in the s-, p- and d-block elements due to the relativistic effect.
Step 6:
Relativistic expansion of 4f-orbital (lanthanides) versus 5f-orbital (actinides).
Aim: To rationalise the difference in the properties of lanthanides and actinides in terms of the relativistic effect.
Step 7:
Spin–orbit (SO) coupling.
Aim: To establish the fact that spin–orbit (SO) coupling (third relativistic effect) can destroy the degeneracy of the orbitals (e.g. p, d, f) with the nonzero values of l for the heavier elements. This effect imparts some special properties to the heavier congeners.
All these aspects have been discussed in the subsequent sections. These were presented in the class rooms in terms of class lecture by using both blackboard and slide presentation. The matter was taught in a question-answer relationship (QAR) strategy. It took four class hours (4 h) for presentation and one class hour for interaction. The students also participated in group discussions. The material was also used in the online refresher course training programmes of college teachers.
Step 8:
Post-tests to measure the extent of learning by the students.
This was done by conducting both the written and oral closed-book examinations after presentation, interaction and teaching of the subject. The results revealed that more than 80 % of the students learnt the subjects very well. For the remaining 20 % students, two additional tutorial classes were arranged to clear their doubts in the subject. Feedback from the students indicated that almost all the students realised the importance of the topic to be taught for understanding the chemistry of 6th and 7th period elements at the undergraduate level. About 70 % of the students expressed the view that they would teach the matter in the class rooms in the same style if they are given the task to teach chemistry at the undergraduate level.
4 Relativistic effects on the properties of orbital electrons
In terms of the relativistic theory, the mass (m) – velocity (v) correction for a moving electron in an atom indicates that mass (m) of the electron increases with respect to its rest mass (m 0) with the increase of its velocity (v) as follows (Das et al., 2020, 2023):
For the H-like atom (one electron system), it gives:
where α (Sommerfeld fine structure constant) is defined as follows:
Relativistic contraction of orbital:
For hydrogen (1s 1), the speed of 1s electron is 2.2 × 106 m s−1 = 1 a. u. of velocity = c/137 ≈ 0.0073c where c = velocity of light ≈ 3 × 108 m s−1 and the relativistic mass of the electron becomes only 1.00003 times the rest mass (m/m 0 = γ = 1.00003). For gold (Z = 79), speed of its 1s-electron is 79c/137 = 0.576c and its relativistic mass (m) becomes about 1.23m 0 (i.e. γ = 1.23) and the orbital radius shrinks by a factor 1.23 (i.e. 23 %) (Das et al., 2020). It gives almost the same result for mercury (Z = 80), the next element of gold in PT.
The electron velocity (v) decreases with the increase of n (principal quantum number) and the relativistic effect becomes more important for the smaller values of n and higher values of Z (i.e. heavier elements) as the electron velocity is directly proportional to Z and inversely proportional to n.
From the above relationship, it is evident that for the element with Z > 137, velocity of the 1s electron (=Zcα = Zc/137) becomes higher than that of light but in terms of the basic postulates of the theory of relativity, no object can have the velocity greater than that of light. Hence, no element with Z > 137 can exist (i.e. limiting value of Z = 137). In this connection, it may be mentioned that the heaviest experimentally known element is oganesson (Og, Z = 118, Gr. 18 of the inert gas elements, in the 7th period). An extended periodic table for Z = 1–172 has been proposed based on the theoretical calculation (Pyykkö, 2011). But, the elements with Z = 119–172 are so far unknown because of their nuclear instability.
Relativistic contraction of orbitals (r rel = r/γ) is more important in the heavier elements for the orbitals with the lower n and l values where the velocity of the electron is higher and the relativistic contraction of the orbitals follows the order: s (l = 0) > p (l = 1) > d (l = 2) > f (l = 3). In fact, relativistic contraction of the s- and p- orbitals is more important and this orbital contraction is called the direct relativistic effect (Burke & Grant, 1967; Das et al., 2023; Pyper, 2020). Figure 1 compares the RDF (radial distribution function) curves of the relativistic and corresponding non-relativistic 3s-orbitals to illustrate the relativistic contraction of orbitals for a hydrogen like atom with Z = 80. These contracted s- and p-orbitals effectively shield the d- and f- orbital electrons and consequently, they experience the less electrostatic attraction by the positive nuclear charge and they actually undergo expansion and this relativistic expansion of the d- and f- orbitals is called the indirect relativistic effect that appears as the consequence of direct relativistic effect. Thus the order of relativistic orbital contraction (direct relativistic effect) is: 6s > 5s > 4s and ns > np (n = 4, 5, 6) and the order of relativistic expansion of the orbitals (indirect relativistic effect) is: 5d > 4d > 3d; 5f > 4f.
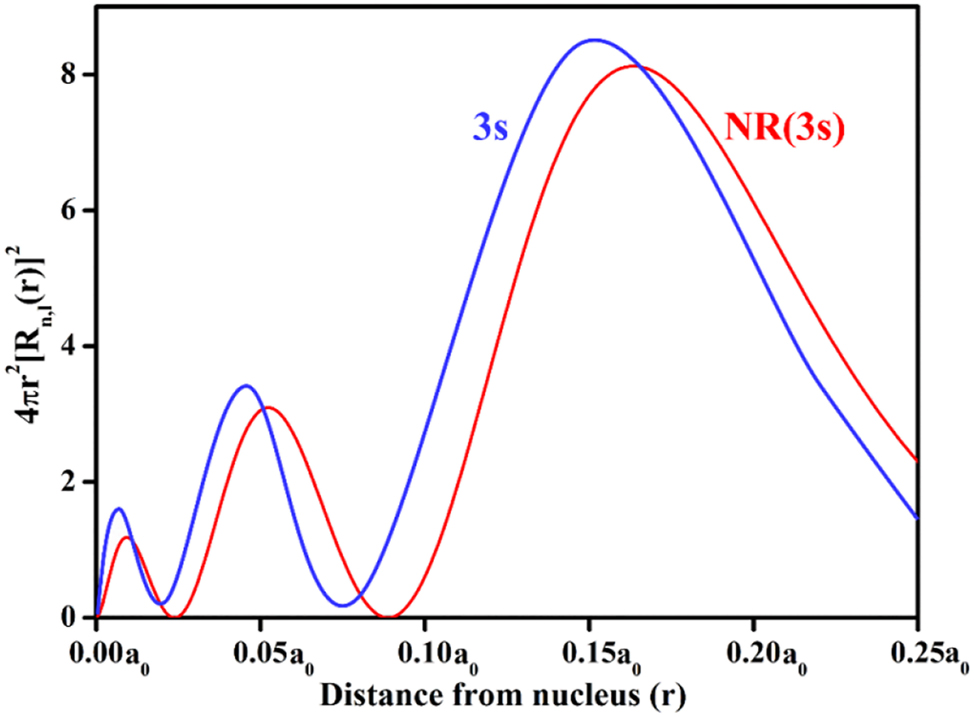
Relativistic orbital shrinkage by comparing the non-relativistic (NR) and relativistic RDF (radial distribution function) curves (blue curve for relativistic and red curve for NR) of 3s orbital of a hydrogen like atom with Z = 80 (Burke & Grant, 1967; Das et al., 2023).
Spin–orbit (SO) coupling splits the orbitals (with the nonzero l value) into two sets for a particular l-value with the possible j values: j = l ± s and each set can accommodate maximum 2j + 1 electrons. The orbitals with the lower j-value are more stable for a particular l- and n-value. The energy order is: np
3/2 > np
1/2; nd
5/2 > nd
3/2 and nf
7/2 > nf
5/2. Bismuth (Bi, 6th period p-block element, Gr. 15 in PT) having the valence shell electronic configuration (6s)2(6p
1/2)2(6p
3/2)1 shows the first ionisation energy relatively small as the outermost (6p
3/2)1 electron is destabilised to some extent. This is why, Bi(I) (+1 oxidation state) gains the stability. The s
1/2 and p
1/2 orbitals possessing the same angular dependence are spherical in shape while the p
3/2 orbital is doughnut-shaped as in the
All these relativistic effects depend on Z 2 (Z = atomic number measuring the full nuclear charge) for the valence shell electrons in many electron atoms (Pyykkö, 2012). It explains that the relativistic effects on the valence electrons are more pronounced for the heavier congeners in a particular group of PT. In fact, these effects become significant when Z (atomic number) reaches 60–70 (ca. half of the limiting value of Z = 137).
5 Importance to discuss the relativistic effects on the chemistry of heavier elements
In teaching the properties of the heavier elements, specially the 6th and 7th period elements, including the lanthanides and actinides, discussion of relativistic effect appears to be important. In fact, without introducing the relativistic effect controlling the properties of the heavier elements, it becomes difficult to rationalize their properties. To answer the important questions listed in Section 2, it requires the discussion of relativistic effect to control the properties of the heavier elements in PT. But unfortunately, in most of the inorganic chemistry text books, this aspect has been grossly overlooked. Here these aspects have been illustrated in the next section.

Qualitative illustration of the relativistic effect on the energy of optical transition, (n − 1)d → ns for the Gr. 11 elements (Cu, Ag and Au). Relativistic effect not important for the lightest congener Cu but most important for the heaviest congener Au showing the highest degree of relativistic destabilisation of 5d orbital and relativistic stabilisation of 6s orbital.
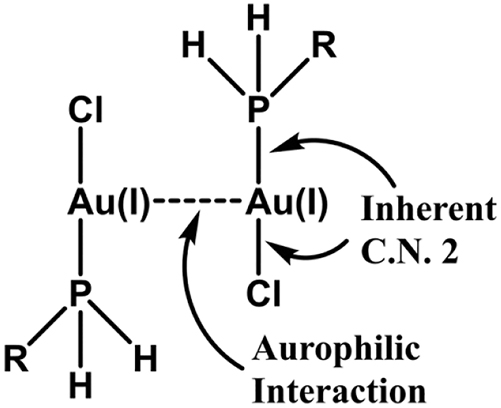
Illustration of aurophilic interaction leading to dimerisation in an Au(I) complex.
6 Illustration of relativistic effects on the properties of heavier elements
Inert pair effect and stability order of higher oxidation states in the p-block congeners: Inert pair effect is well noted for the Gr. 13–15 elements. Gr. 13 (stability order of the +1 state with respect to that of the +3 state): Tl (6s 26p 1) > In (5s 25p 1) > Ga (4s 24p 1); Gr. 14 (stability order of the +2 state with respect to that of the +4 state): Pb (6s 26p 2) > Sn (5s 25p 2) > Ge (4s 24p 2); Gr. 15 (stability order of the +3 state with respect to that of the +5 state): Bi (6s 26p 3) > Sb (5s 25p 3). This can be rationalised in terms of the relativistic order of stabilisation of the ns 2 valence electron pair (Das et al., 2020). The relativistic stabilisation order is: 6s 2 > 5s 2 > 4s 2. The relativistic effect is not important for the lighter congeners (smaller Z) and their ns 2 electron pair not being so tightly bound can participate in bonding and their higher oxidation states become more stable. It is the reason behind the inert pair effect noted for the heavier p-block congeners. It explains the order of oxidising power: Bi(V) > Sb(V); Pb(IV) > Sn(IV) and Tl(III) > In(III).
High voltage of commercial lead-acid battery: Lead-acid battery is a secondary cell where the discharging cell reaction is:
It has been theoretically shown that the contribution of relativistic effect is about 80 % to the observed E 0(cell) (Ahuja et al., 2011; Pyykkö, 2012). The thermodynamic driving force behind the above cell reaction arises from the relativistic stabilization of the 6s-orbital to favour the reduction of Pb(IV) to Pb(II) (inert pair effect). In fact, our domestic inverters using the lead-acid battery work because of the relativistic effect in lead and our cars also using the lead-acid battery start because of the same effect.
Reactivity of cesium (Cs, Z = 55, [Xe] 6s 1 ) vs. francium (Fr, Z = 87, [Rn]7s 1 ) – bucking the periodic trend: Relativistic stabilisation of the 7s 1 orbital of Fr is more important compared to that of the 6s 1 orbital of its immediate lighter congener Cs (the 6th period alkali metal). In fact, the ionisation energy and electron affinity of Cs are less than those of Fr because of the more pronounced relativistic effect in the heavier congener Fr. It makes Cs more reactive than its heavier congener Fr and it bucks the normal periodic trend of properties in Gr. 1 of alkali metals. Thus, because of the relativistic effect, Cs not Fr appears as the most reactive metal in the periodic table (Royal Society of Chemistry, 2019).
Apparent anomalies in electronic configurations in the pairs: Mo (4d 5 5s 1 , 4d-member) vs. W (5d 4 6s 2 , 5d-member); Pd (4d 10 5s 0 , 4d-member) vs. Pt (5d 9 6s 1 , 5d-member) (Das et al., 2020, 2023; Tatewaki et al., 2017): For the 4d-members (second transition series), the 5s electron moves to the 4d orbital to give the half-filled 4d-level (Mo) and full filled 4d-level (Pd) to earn the stabilisation in terms of the exchange energy but this does not happen for their corresponding heavier 5d-congeners (third transition series members) for which the 6s-orbital is relativistically so stabilised/contracted that the transfer of electrons from the 6s-orbital to the relativistically destabilised 5d-orbital becomes energetically unfavourable.
Colours of Cu (golden reddish lustre), Ag (white) and Au (golden yellow): The electron transitions responsible for their observed colours are (Figure 2): 3d → 4s (Cu), 4d → 5s (Ag) and 5d → 6s (Au) (Das et al., 2017, 2020, 2023; Greenwood & Earnshaw, 2005). More correctly, the optical transitions occur from the Fermi level present in the (n − 1)d band to the ns band. Because of the most pronounced relativistic effect in gold among these three congeners, the 6s orbital is contracted (i.e. stabilised) maximum and the 5d orbital is expanded (i.e. destabilised) maximum and the corresponding electronic transition occurs at a lower energy than that of silver for which the relativistic effect is less important. In fact, gold absorbs blue light in the visible range (5d → 6s transition: energy difference about 2.4 eV that corresponds to blue light of 516 nm wavelength; cf. E = hν = hc/λ, 1 eV = 1.6 × 10−19 J, E (eV) ≈ 1240/λ (nm)) and looks yellow which is the complementary colour to blue while silver absorbs in the UV-range (4d → 5s transition: energy difference 3.7 eV that corresponds to the UV light of 335 nm wavelength) and looks white (i.e. all the incident visible lights are reflected back). Theoretical calculation indicates that the non-relativistic gold (NR-Au) also absorbs in the UV region and it is expected to be white. For silver, the relativistic effect also reduces the energy gap for the optical transition, but still the energy difference corresponds to the UV light. The energy difference for the 5d → 6s electronic transition in gold is relativistically highly reduced and it becomes very close to that of 3d → 4s transition (ca. 2.3 eV, i.e. absorbing blue-green light of 539 nm wavelength) in copper for which the relativistic effect is not important. It gives the characteristic golden reddish lustre of copper (Das et al., 2023; Greenwood & Earnshaw, 2005).
Colours in the compounds of Bi(V) vs. Sb(V) in Gr. 15 and Pb(IV) vs. Sn(IV) in Gr. 14: The difference in colour: sodium bismuthate (NaBiO3) (yellowish brown colour) versus sodium antimonate (NaSbO3) (white); lead dioxide (PbO2) (dark brown maroon colour) versus tin dioxide (SnO2) (white) can be rationalised by considering the LMCT (ligand to metal charge transfer) transition and the relativistic stabilization of the lowest unoccupied molecular orbital (LUMO) enriched with the character of the heavy metal 6s-orbital (Das et al., 2023). The optical electron transition occurs from the HOMO enriched with the 2p-orbital of oxygen to the LUMO (i.e. 2p(O) → 5s/6s of the central metal) which is relativistically stabilised more for the heavier congener. In the same way, the difference in colour: BiPh5 (violet) versus SbPh5 (colourless) and [PbCl6]2− (yellow) versus [SnCl6]2− (colourless) can be interpreted (El-Issa et al., 1991). Thus colour of Bi(V) and Pb(IV) compounds is due to the relativistically red shifted LMCT transition towards the visible range. On the other hand, for the corresponding compounds of the respective lighter congeners (Sb and Sn), the relativistic stabilisation of the corresponding LUMO enriched with the 5s-orbital is not so important and consequently, the HOMO-LUMO energy gap is relatively larger (>3.2 eV that corresponds to the violet light with λ ≈ 390 nm). In reality, their LMCT transitions occur not in the visible range but in the UV range and consequently, they look colourless or white.
Astonishingly high electron affinity and electronegativity of gold, formation of auride (Au − ) and platinide (Pt 2− ) salts, relatively higher electron affinity of the noble metals: The electronic configuration of gold is: 1s 2 2s 2 2p 6 3s 2 3p 6 4s 2 3d 10 4p 6 5s 2 4d 10 5p 6 6s 1 4f 14 5d 10, i.e. [Xe]4f 145d 106s 16p 0. The half filled 6s-orbital of gold is relativistically highly contracted (i.e. stabilised) and accommodation of an electron in its 6s 1 orbital to give the 6s 2 configuration is energetically highly favourable. This relativistic stabilisation of the 6s 2 pair is the driving force behind the observed high electron affinity and electronegativity of Au. Energetically favourable formation of auride, Au + e → Au− (5d 106s 2, stable electronic configuration) explains the stability of auride salts (e.g. CsAu, RbAu, etc) with the large cations (e.g. Cs+, Rb+, etc.) of comparable size (cf. large cation and large anion combination gives the better crystal packing to give the higher lattice energy to stabilise the salt) (Das et al., 2020; Jansen, 2008). Here, stability of cesium platinide (Cs2Pt) can be rationalised in terms of the electronic configuration of Pt2− which is isoelectronic with Au− (Jansen, 2005). In fact, very often, the noble metals (i.e. high ionisation energy) show the high electron affinity and electronegativity because of the relativistic effect.
Liquidity of mercury at room temperature – low melting and boiling point of mercury: Mercury (Hg: [Xe]4f 145d 106s 26p 0) records the lowest m. p. (−39°C) among the neighbouring metals (321°C for Cd, 1064°C for Au, 304°C for Tl) in PT and mercury exists in a liquid state at the common working condition. In fact, it is the only metal that exists in liquid state. The symbol Hg is derived from the Greek word hydrargyrum meaning ‘watery silver’. Due to the very high relativistic contraction, the filled 6s 2-orbital is relativistically highly stabilised (inert pair) and the filled 6s 2-orbital does not participate in metallic bonding (Calvo et al., 2013; Das et al., 2020, 2023; Norrby, 1991). Because of the absence of electron sharing covalent interaction among the neighbouring Hg-atoms, only the weak London dispersion forces prevail among the Hg-atoms. It makes the cohesive force very weak and mercury shows the so low melting point. Theoretically, it has been predicted that the non-relativistic mercury (NR-Hg) is solid and its melting point is much higher than that of real mercury. In terms of band theory of metallic bonding, the filled contracted 6s-band (relativistically stabilised) cannot overlap with the high energy vacant 6p-band of mercury to produce the partially filled 6s-6p metallic band (cf. the band structure of Gr. 2 alkaline earth metals having the partially filled ns-np metallic band) and the filled contracted 6s-band of mercury cannot contribute to metallic bonding and electrical conductivity. Among the Hg atoms in its liquid state, only the weak van der Waals force works and this is why, it also shows the relatively low boiling point (356.7 °C) and it can be easily purified by distillation.
Low melting point of gallium (Ga, Z = 31) not because of the relativistic effect: In PT, gallium (Ga, Z = 31) shows unexpectedly also a very low melting point (29.8 °C) compared to those of its lighter and heavier congeners (660 °C for Al, 157 °C for In) but it should not be confused with the case of mercury. Here the chemistry is different because the relativistic effect is not definitely important for the lighter element like gallium (Z = 31). The low melting point of gallium is due to its unusual crystal structure consisting of discrete diatomic molecules (Ga2) rather than a metallic structure of high coordination number as in closed packing model of metallic structure. In its crystal structure, each Ga atom has one closest neighbour at a distance of 244 pm but other neighbours are far away (>270 pm). Ga experiencing the first d-contraction (secondary periodicity) probably keeps the inner core [Ar]3d 104s 2 as the nonbonding one and the single p-electron outside this core participates in bonding to produce the discrete molecular structure (Ga2). In the molecular crystal structure, the cohesive force is weak and consequently, the melting point is also low (Das et al., 2020). Here it may be mentioned that its boiling point (2205 °C) follows the normal periodic trend (cf. b.p. of Al 2520 °C) and it justifies the fact that the low melting point of gallium is due to its unusual molecular lattice structure (cf. for mercury, both the melting and boiling points are low). It is quite interesting to note that at common room temperatures, the element looks as a shiny solid metallic block, but when placed in our palm, it will liquefy because its melting point is a few degrees below our body temperature. Very recently, the different properties of liquid gallium have been discussed (Crow, 2023).
Difference in the physical states and electrical conductivity of Au (6s 1 6p 0 ) and its nearest neighbour Hg (6s 2 6p 0 ) in PT: In the light of band theory of metallic bonding, the filled 6s-band of 80Hg(6s 2) cannot stabilise the metallic bonding interaction but in 79Au (6s 1), the half-filled 6s-band even without overlapping with the vacant 6p-band can itself effectively contribute to the metallic bonding. This stronger metallic bonding is responsible for the higher melting point of gold (Das et al., 2023; Löffelsender et al., 2022). The half-filled 6s-band of gold can act as a conduction band for electrical conductivity while the filled 6s-band of mercury cannot act as a conduction band and it shows a poorer conductivity (10.5 kS m−1 for Hg; 425 kS m−1 for Au).
Chemistry of selectivity in amalgamation – easy amalgamation with Au, Ag and Na but not with most of the common transition metals: It is well known that liquid mercury can easily dissolve many one electron donor metals like Na, Ag, Au, etc to form amalgams. But, strikingly, mercury does not amalgamate most of the common transition metals generally acting as the two electron donors (Das et al., 2023; Norrby, 1991). This selectivity in amalgamation can be explained at least qualitatively, by considering the electron sharing covalent type interaction between mercury and the dissolved metal (M) in the pair Hg-M. The diatomic molecular species like Hg2 2+ and Au2 forming a 2c–2e bond (6σ 2) are stable because of the relativistic stability of the sigma bonding MO. In Hg-M alloy, if M is a one electron donor as in the molecular species HgAu(g), HgAg(g) and HgNa(g), it produces a 2c–3e bond of bond order 0.5 to impart the stability to the bimolecular species. In HgAu(g), by using the 6s 2 (Hg) and 6s 1 (Au) valence electrons, it gives the bond order 0.5 from 6σ 26σ*1. But in the bimolecular species Hg-M (M a representative transition metal acting as a two electron donor), the bond order becomes 0 as in Hg2 (6σ 26σ*2). In purification of gold, it is first amalgamated and then gold can be separated by simple heating the amalgam (cf. bond order and stability order in the bimolecular species: 1 in Au2 (6σ 26σ*0) > 0.5 in HgAu (6σ 26σ*1) > 0 in Hg2 (6σ 26σ*2)). The relationship between Au 2 and Hg may be compared with the relationship between H 2 and He in PT. Thus possibility of amalgamation can be qualitatively rationalised by considering the stability of the HgM molecular species through the electron sharing covalent interaction. It may be mentioned that besides the covalent interaction, many other weak forces including the London dispersion force contribute in amalgamation (Das et al., 2020, 2023).
Relative stability of the dimeric species Au 2 , Ag 2 and Cu 2 : The relatively higher bond dissociation energy (BDE) of the Au–Au bond with respect to that of the Ag–Ag or Cu–Cu bond can be explained in terms of stabilisation of the sigma bonding MO constituted by the relativistically stabilised 6s 1 orbital of Au (Das et al., 2023; Norrby, 1991). Relativistic stabilisation order of the dimeric species M2 (nσ 2 nσ*0) (M = Au, Ag and Cu) in gas phase follows the relativistic stabilisation order of the sigma bonding MO constituted by the ns 1 orbital: 6σ 2 (Au2) > 5σ 2(Ag2) > 4σ 2 (Cu2).
Chemical inertness of gold (6s 1 ) and mercury (6s 2 ); relationship between gold and mercury in PT: Due to the relativistic contraction, these 6s-valence electrons are highly stabilised and it explains, in general, the chemical inertness of gold and mercury. It may be noted that their non-relativistic analogues (i.e. NR-Au and NR-Hg) are more reactive. In fact, in terms of chemical inertness, Hg with the electronic configuration 5d 106s 2 can be described as a ‘noble liquid’ in comparison with the noble gases (cf. helium of 1s 2 configuration). On the other hand, Au can attain the electronic configuration (6s 2) of Hg (noble liquid metal) by accepting an electron in the relativistically stabilised 6s 1 orbital as in the auride salts (Das et al., 2020, 2023; Norrby, 1991). The relationship between Au and Hg in PT may be compared with the relationship between hydrogen (H: 1s 1 vs. Au: 6s 1) and helium (He: 1s 2 vs. Hg: 6s 2) in terms of both the reactivity and electronic configuration or halogens (Gr 17) (cf. auride vs. halide) and inert gases (Gr. 18) in terms of reactivity.
Relative stabilities of the dimeric species Hg 2 2+ (mercurous ion) vs. Cd 2 2+ (cadmous ion) and Zn 2 2+ (zincous ion): Because of the relativistic contraction, the bonding MO (6σ 2) constituted by the 6s 1 orbitals of Hg+ ions in the dimeric species Hg2 2+ (mercurous ion) is strongly stabilised (i.e. 6σ26σ*0, bond order 1). Such relativistic stabilisation is not possible in the cases of Cd2 2+ (cadmous, 5σ 2) ion and Zn2 2+ (zincous, 4σ 2) ion, i.e. the dimeric species of lighter congeners of Gr. 12 in PT (Das, 2020; Das et al., 2020, 2023).
Preferred linear coordination (C. N. 2) geometry of Au(I) and Ag(I) of Gr. 11 and Hg(II) of Gr. 12 of (n − 1)d 10 ns 0 np 0 electronic configuration: The illustrative examples are: [Ag(NH3)2]2+ (cf. Tollens’ reagent), [Ag(CN)2]−, [Au(CN)2]− (cf. hydrometallurgical extraction of silver and gold through the cyanide leaching process), etc. Their strong preference for the linear coordination geometry cannot be interpreted by the classical sp-hybridisation because of two reasons: sp 3-hybridisation instead of sp-hybridisation should be more favoured because of two additional metal – ligand interactions in sp 3-hybridisation and in the simple sp-hybridisation scheme for the (n − 1)d 10 ns 0 np 0 electronic configuration, the filled d-orbitals will electrostatically disfavour the linear coordination geometry. For example, the filled
Closed-shell interaction in aurophilicity and metallophilicity: In many Au(I) (5d 106s 0) complexes, Au(I) even after maintaining its desired inherent coordination number 2, shows an additional tendency to make the closed-shell Au(I)(d 10)–Au(I)(d 10) supramolecular type interaction (ca. 30 kJ mol−1, comparable to that of H-bonding). For example, in the dimer, (PR3)(Cl)Au(I)–Au(I)(PR3)(Cl), the additional Au(I)–Au(I) bond beyond the inherent 2 coordination number of Au(I) is a representative example of aurophilicity. This tendency to form the additional Au(I)–Au(I) bond is called aurophilicity (metallophilicity in general) (Das et al., 2021; Huheey et al., 2007; Schmidbaur & Schier, 2008). Figure 3 illustrates the dimerisation of Au(I) complex through the aurophilic interaction. Other examples of such metallophilic interactions are: Au(I)–Hg(II), Au(I)–Ag(I), Au(I)–Tl(I), Au(I)–Pb(II). It may be mentioned that in such metallophilic interactions, the other cations like Ag(I), Tl(I), Pb(II), Au(I), Hg(II) etc. also retain their required inherent coordination number and after satisfying their inherent coordination number, they introduce the metallophilicity. It again implies that the weaker metallophilic interaction is not attained at the cost of preferred inherent coordination number and metallophilicity imparts only an additional weak intermolecular bonding interaction (supramolecular type interaction) with the Au(I) centre. The several aurophilic Au(I)–Au(I) interactions stabilse the hypervalent [C(AuPR3)6]2+ (hedgehog cation, octahedral coordination of carbon) and [N(AuPR3)5]2+ (trigonal bipyramidal structure, 5 C. N. of nitrogen) compounds bearing the weak Au–C and Au–N bonding respectively (Das et al., 2023).
The additional bonding power of Au(I) (5d 106s 0) in the aurophilic interactions can be rationalised by considering the relativistic expansion/destabilisation of 5d-orbital and relativistic contraction/stabilisation of 6s-orbital of gold. It favours the transfer of some electrons to the relativistically stabilised/contracted 6s-orbital from the relativistically destabilised/expanded 5d-orbital and the excitation energy for the process: (n − 1)d 10 ns 0 → (n − 1)d 9 ns 1, is small for gold (ca. 180 kJ mol−1) (cf. Figure 2) (Das et al., 2020, 2023). Thus the energetically favoured promoted electrons into the 6s-orbital of Au(I) can participate in the additional bonding interaction (i.e. aurophilic interaction) with the suitable neighbouring metal centre for which the relativistic effect is also important (Das et al., 2020, 2023).
Stability order of the higher oxidation states for the elements in a particular group of d-block: The relativistic expansion of d-orbitals (indirect relativistic effect) is more pronounced for the heavier congeners in the d-block elements. Consequently, the 5d-valence electrons experiencing the relativistic destabilisation maximum can be most easily ionised to attain the higher oxidation states for the elements of 5d-series (i.e. 3rd transition series). Besides this, as the energy difference between the 5d- and 6s-orbitals becomes smaller because of the relativistic effect (cf. Figure 2), the 5d-electrons can participate energetically more easily for the bonding purposes along with the 6s-valence electrons compared to the 4d-electrons of the 4d-series (i.e. 2nd transition series) because the relativistic effects are not so important for the 4d-series elements and in fact, energy difference between the 4d- and 5s-orbitals is relatively higher (cf. Figure 2) (Das et al., 2023; Riedel & Kaupp, 2009). It is needless to say that the relativistic effect is not practically important for the lightest 3d-congeners. Thus the relativistic effect explains the higher stabilities of the higher oxidation states of the heavier congeners. The stability orders for the congeners are (Das et al., 2020): Cr(VI) < Mo(VI) < W(VI) (Gr. 6); Mn(VII) < Tc(VII) < Re(VII) (Gr. 7); Ni(IV) < Pd(IV) < Pt(IV) (Gr. 10); Cu(III) < Ag (III) < Au(III) (Gr. 11); etc. The relativistic effect can explain the relative ease of oxidative addition (+1 state to +3 state) in the organometallic complexes in the order: Ir(I) > Rh(I) > Co(I). The ease of oxidative addition of Vaska complex [IrI(CO)Cl(PPh3)2] (square planar 16e complex) is well documented. In fact, the higher stability of higher oxidation state for a heavier congener makes it less oxidising and it explains the oxidising power order of the tetrahedral oxyanions: CrO4 2− > MoO4 2− > WO4 2−; MnO4 − > TcO4 − > ReO4 −.
Difference in colours of oxyanions: CrO4 2− (yellow) but MoO4 2− (colourless) and WO4 2− (colourless); MnO4 − (pink) but TcO4 − (colourless): Colours of these species arise from the LMCT transition (i.e. HOMO enriched with the 2p orbitals of oxide to LUMO enriched with the d-orbitals of metal). Simply, the LMCT transition can be represented as: 2p (O)→3d (Cr, Mn)/4d (Mo, Tc)/5d(W, Re). In terms of relativistic destabilisation of the (n − 1)d orbital, the energy order is: 5d > 4d > 3d. This is why, the said LMCT transition is red shifted towards the visible range for the lighter congener to give the colour while for the heavier congeners, the transitions occur in the UV-region and they are white/colourless (Das & Das, 2016b in Vol. 5).
Stability of Hg(IV) in HgF 4 : For the Gr. 12 elements (i.e. Zn, Cd and Hg) having the electronic configuration (n − 1)d 10 ns 2, the common stable oxidation state is +2. The valence d-electrons (i.e. 5d-electrons) of the heaviest congener (i.e. Hg) are most relativistically destabilised and consequently, mercury is expected to be the most probable element among the Gr. 12 elements to attain the +4 oxidation state in the light of relativistic destabilisation of the 5d-orbitals (Das et al., 2020, 2023). The d 8-configuration of HgIV can also enjoy a high crystal field stabilisation energy (CFSE) in a square planar complex like [HgF4] because of the large crystal field splitting power of highly positive oxidation state as in Hg(IV) (cf. effect of oxidation state on the magnitude of crystal field splitting). In fact, the characterisation of square planar [HgF4] complex in 2007 (Wang et al., 2007) supports the validity of the relativistic effect. Obviously, to stabilise Hg(IV), it requires the non-oxidisable hard ligands like fluoride. Existence of [HgF4] (d 8 for HgIV) claims the fact that mercury can also be considered as a member of 5d-transition series.
Highest possible oxidation state among the metals: For the 5d-series metal iridium (Ir), the relativistically highly destabilised 5d electrons are relatively ionised more easily to produce the species IrO4 (5d 16s 0) and IrO4 + (5d 06s 0) to give the stability of +VIII and +IX oxidation states of Ir respectively (Pyykkö & Xu, 2015). This +IX oxidation state is probably the highest oxidation state identified for the metals. Obviously, for its lighter congeners (Co and Rh) for which the relativistic destabilisation of the valence d-orbital is not so pronounced, attainment of such high oxidation states is not possible (Das et al., 2020, 2023).
Difference between the lanthanides and actinides: The relativistic expansion of the 5f-orbitals is more pronounced for the actinides compared to that of the 4f-orbitals of the lanthanides. This is why, the 5f-orbitals of the actinides are exposed more to the ligands. It may be noted that in terms of the non-relativistic model, the 4f orbitals and 5f orbitals are quite similar (Pyykkö, 1978). Because of the higher relativistic expansion of the 5f-orbitals towards the incoming ligands, the crystal field effect (i.e. ligand field effect) becomes more important for the actinides and in fact, the actinides behave like the transition metals to some extent in terms of their variable oxidation states and electronic spectra. For the lanthanides, the crystal field effect is not important because the relativistically less expanded 4f-orbitats are more deeply seated. The difference in relativistic effect on the 4f and 5f orbitals can explain the difference in spectral, magnetic and other properties of the lanthanides and actinides (Das & Das, 2016b in Vol. 5; Das & Das, 2019, Das et al., 2023). Relaxation of the Laporte orbital selection rule for the 5f → 5f transitions of the actinides is attained better due to the vibronic coupling from the metal-ligand bond vibration and their spectral bands are more intense and broader compared to those of the lanthanides that mainly show the sharp line-like spectra (Das & Das, 2016b in Vol. 5; Das et al., 2023). The allowed ligand to metal charge transfer (LMCT) transitions of the actinides may give the intense absorption bands in some cases to give the characteristic colour while for the lanthanides, the metal–ligand interaction is less pronounced and in fact, such CT bands are absent. In the actinide complexes, the orbital contribution to the observed magnetic moment can be quenched to some extent (but definitely less than that in the transition metal complexes) by the ligand field effect but such quenching effect is not observed for the lanthanides for which the ligand field effect is not important. Lanthanide contraction measured in terms of the shrinkage in the Ln—X bond length in LnX3 (Ln = La to Lu) partly (ca. 10–20 %) arises from the relativistic contraction of the 6s-valence orbitals of lanthanides (Seth et al., 1995). Definitely, relativistic effect in the actinide contraction is more important (Das et al., 2023). Here it is important to note that the actinides (An3+) are more polarisable (i. e. softer in HSAB concept; Hard and soft, acids and bases) than the lanthanides (Ln3+) because the relativistically more expanded 5f-orbital electrons can be more easily perturbed by the external field. Thus the difference between the lanthanides and actinides arises from the difference in relativistic effect experienced by the 4f-orbitals of lanthanides and 5f-orbitals of actinides.
7 Conclusions
The examples illustrate that without considering the relativistic effect, understanding the chemistry of heavier elements across the periodic table remains incomplete. In fact, if the relativistic effect on the properties of the heavier elements were absent, then properties of the heavier elements (e. g. lead, gold, platinum, etc.) would be different and our real world and civilisation would not be developed in the present form. Unfortunately, this important aspect is not properly addressed in most of the available inorganic chemistry text books and most of the phenomena discussed here are explained by considering the effects of lanthanide contraction. Only in few books, the relativistic effect on the properties of the heavier elements is just mentioned in brief without any detail discussion. More space should be provided to discuss the relativistic effect in the text books. This topic should be given proper importance in the UG and PG chemistry courses. This mini tutorial review reflects the corresponding author’s experience in class rooms and refresher courses (training programmes of college and university teachers). This material has been used successfully by the corresponding author in teaching the present topic in class rooms and refresher courses. This self explanatory review article aims to answer all the probable questions related to the present topic and it is designed in a way to act itself as a teacher for this topic.
This article is dedicated to Prof. Pekka Pyykkö, Chemistry Department, University of Helsinki for making a monumental amount of contribution in understanding the relativistic effect in chemistry.
Acknowledgments
The authors express thankfulness to the authority of Visva Bharati University for providing the required facilities. The authors appreciate the encouraging attitude of Dr. M. Seikh and Prof. P. Sarkar, Department of Chemistry, Visva Bharati.
-
Research ethics: Not applicable.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission. All the coauthors have contributed equally.
-
Competing interests: The authors state no conflict of interest.
-
Research funding: Not applicable.
-
Data availability: Not applicable.
References
Ahuja, R., Blomqvist, A., Larsson, P., Pyykkö, P., & Zaleski-Ejgierd, P. (2011). Relativity and the lead-acid battery. Physical Review Letters, 106, 018301. https://doi.org/10.1103/PhysRevLett.106.018301 Search in Google Scholar PubMed
Burke, V. M., & Grant, I. P. (1967). Effect of relativity on atomic wave functions. Proceedings of the Physical Society, 90, 297–314. https://doi.10.1088/0370-1328/90/2/301 10.1088/0370-1328/90/2/301Search in Google Scholar
Calvo, F., Pahl, E., Wormit, M., & Schwerdtfeger, P. (2013). Evidence for low-temperature melting of mercury owing to relativity. Angewandte Chemie International Edition in English, 52, 7583–7585. https://doi.org/10.1002/anie.201302742 Search in Google Scholar PubMed
Crow, J. M., (2023). The liquid metals giving catalysis a new phase. Chemistry World. https://www.chemistryworld.com›4017659.Search in Google Scholar
Das, A. K. (2020). Thermodynamic and kinetic aspects of the stability of Sir P. C. Ray’s mercurous nitrite compound. Resonance, 25, 787–799. https://doi.org/10.1007/s12045-020-0996-9 Search in Google Scholar
Das, A. K., & Das, M. (2016a). Fundamental concepts of inorganic chemistry (2nd reprint, 1st ed., Vol. 4, pp. 231–234). New Delhi: CBS Publishers & Distributors, ISBN 978-81-239-2351-2.Search in Google Scholar
Das, A. K. & Das, M., (2016b), Fundamental concepts of inorganic chemistry (2nd reprint, 1st ed., Vol. 5, pp. 1034–1042, 1046). New Delhi: CBS Publishers & Distributors, ISBN: 978-81-239-2352-9.Search in Google Scholar
Das, A. K., & Das, M. (2019). Fundamental concepts of inorganic chemistry (3rd reprint, 1st ed., Vol. 6, pp. 1148–1154). New Delhi: CBS Publishers & Distributors, ISBN 978-81-239-2353-6.Search in Google Scholar
Das, A. K., Das, M., & Das, A. (2017). An introduction to nanomaterials and nanoscience (1st ed., p. 95). New Delhi: CBS Publishers & Distributors, ISBN 978-93-85915-67-3.Search in Google Scholar
Das, A. K., Das, M. & Das, A., (2020), Fundamental concepts of inorganic chemistry (3rd ed., Vol. 1, pp. 101–102, 592–599). New Delhi: CBS Publishers & Distributors. ISBN: 978-93-89565-97-3.Search in Google Scholar
Das, A. K., Das, M., & Das, A. (2021). Fundamental concepts of inorganic chemistry (3rd ed., Vol. 2, pp. 164–165). New Delhi: CBS Publishers & Distributors, ISBN 978-93-89688-02-3.Search in Google Scholar
Das, A., Das, U., & Das, A. K. (2023). Relativistic effects on the chemical bonding properties of the heavier elements and their compounds. Coordination Chemistry Reviews, 479, 215000. https://doi.org/10.1016/j.ccr.2022.215000 Search in Google Scholar
Dirac, P. A. M. (1929). Quantum-mechanics of many-electron systems. Proceedings of the Royal Society of London A, 123, 714–733.10.1098/rspa.1929.0094Search in Google Scholar
El-Issa, B. D., Pyykkö, P., & Zanati, H. M. (1991). MS X.alpha. Studies on the colors of pentaphenylbismuth, hexachloroplumbate(2-) and tetrathiotungstate(2-): Are relativistic effects on the LUMO important? Inorganic Chemistry, 30, 2781–2787. https://doi.org/10.1021/ic00013a015 Search in Google Scholar
Greenwood, N. N. & Earnshaw, A., (2005), Chemistry of the elements (2nd ed., pp. 599, 1177). New Delhi: Elsevier, ISBN: 81-8147-806-1 (Indian Reprint).Search in Google Scholar
Huheey, J. E., Keiter, E. A., Keiter, R. L., & Medhi, O. K. (2007). Inorganic chemistry: principles of structure and reactivity (4th ed., pp. 287–290, 497). New Delhi: Pearson Education, ISBN 81-7758-130-9.Search in Google Scholar
Jansen, M. (2005). Effects of relativistic motion of electrons on the chemistry of gold and platinum. Solid State Sciences, 7, 1464–1474. https://doi.org/10.1016/j.solidstatesciences.2005.06.015 Search in Google Scholar
Jansen, M. (2008). The chemistry of gold as an anion. Chemical Society Reviews, 37, 1826–1835. https://doi.org/10.1039/B708844M Search in Google Scholar PubMed
Löffelsender, S., Schwerdtfeger, P., Grimme, S., & Mewes, J. M. (2022). It’s complicated: On relativistic effects and periodic trends in the melting and boiling points of the group 11 coinage metals. Journal of the American Chemical Society, 144, 485–494. https://doi.org/10.1021/jacs.1c10881 Search in Google Scholar PubMed
Norrby, L. J. (1991). Why is mercury liquid? Or, why do relativistic effects not get into chemistry textbooks? Journal of Chemical Education, 68, 110–113. https://doi.org/10.1021/ed068p110 Search in Google Scholar
Orgel, L. E. (1958). Stereochemistry of metals of the B sub-groups. Part I. Ions with filled d-electron shells. Journal of the Chemical Society, 4186–4190. https://doi.org/10.1039/JR9580004186 Search in Google Scholar
Pitzer, K. S. (1979). Relativistic effects on chemical properties. Accounts of Chemical Research, 12, 271–276. https://doi.org/10.1021/ar50140a001 Search in Google Scholar
Pyper, N. C. (2020). Relativity and the periodic table. Phil. Trans. R. Soc. A, 378, 20190305. https://doi.org/10.1098/rsta.2019.0305 Search in Google Scholar PubMed
Pyykkö, P. (1978). Relativistic quantum chemistry. Advances in Quantum Chemistry, 11, 353–409. https://doi.org/10.1016/S0065-3276(08)60241-5 Search in Google Scholar
Pyykkö, P. (1988). Relativistic effects in structural chemistry. Chemistry Review, 88, 563–594. https://doi.org/10.1021/cr00085a006 Search in Google Scholar
Pyykkö, P. (2011). A suggested periodic table up to Z ≤ 172, based on Dirac–fock calculations on atoms and ions. Physical Chemistry Chemical Physics, 13, 161–168. https://doi.org/10.1039/C0CP01575J Search in Google Scholar
Pyykkö, P. (2012). Relativistic effects in chemistry: More common than you thought. Annual Review of Physical Chemistry, 63, 45–64.10.1146/annurev-physchem-032511-143755Search in Google Scholar PubMed
Pyykkö, P., & Desclaux, J. P. (1979). Relativity and the periodic system of elements. Accounts of Chemical Research, 12, 276–281. https://doi.org/10.1021/ar50140a002 Search in Google Scholar
Pyykkö, P., & Xu, W. H. (2015). The formal oxidation states of iridium now run from −III to +IX. Angewandte Chemie International Edition, 54, 1080–1081. https://doi.org/10.1002/anie.201410615 Search in Google Scholar PubMed
Riedel, S., & Kaupp, M. (2009). The highest oxidation states of the transition metal elements. Coordination Chemistry Reviews, 253, 606–624. https://doi.org/10.1016/j.ccr.2008.07.014.Search in Google Scholar
Royal Society of Chemistry. (2019). IYPT 2019 Elements 087: Francium: Not the most reactive group 1 element. In Explorations of everyday chemical compounds. November 6, 2019. Compound Interest: Chemistry Infographics. https://www.compoundchem.com › 2019/11/06 › iypt.Search in Google Scholar
Schmidbaur, H., & Schier, A. (2008). A briefing on aurophilicity. Chemical Society Reviews, 37, 1931–1951. https://doi.org/10.1039/B708845K Search in Google Scholar PubMed
Seth, M., Dolg, M., Fulde, P., & Schwerdtfeger, P. (1995). Lanthanide and actinide contractions: Relativistic and shell structure effects. Journal of the American Chemical Society, 117, 6597–6598. https://doi.org/10.1021/ja00129a026 Search in Google Scholar
Tatewaki, H., Yamamoto, S., & Hatano, Y. (2017). Relativistic effects in the electronic structure of atoms. ACS Omega, 2, 6072–6080. https://doi.org/10.1021/acsomega.7b00802 Search in Google Scholar PubMed PubMed Central
Thayer, J. S. (2005). Relativistic effects and the chemistry of the heaviest main-group elements. Journal of Chemical Education, 82, 1721–1729. https://doi.org/10.1021/ed082p1721 Search in Google Scholar
Wang, X., Andrews, L., Riedel, S., & Kaupp, M. (2007). Mercury is a transition metal: The first experimental evidence for HgF4. Angewandte Chemie, International Edition in English, 46, 8371–8375. https://doi.org/10.1002/anie.200703710 Search in Google Scholar PubMed
© 2023 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Frontmatter
- Review Article
- Relativistic effects on the chemistry of heavier elements: why not given proper importance in chemistry education at the undergraduate and postgraduate level?
- Research Articles
- Designing a learning environment based on the spiral of skills to overcome the didactic obstacles associated with teaching the Daniell cell
- Analysis of the relationship between students’ argumentation and chemical representational ability: a case study of hybrid learning oriented in the environmental chemistry course
- Good Practice Report
- What makes representations good representations for science education? A teacher-oriented summary of significant findings and a practical guideline for the transfer into teaching
- Research Article
- Student exploration of the Henderson-Hasselbach equation and pH readings to determine the pK a value of 4,4′-trimethylenedipiperidine (TMDP)
- Good Practice Report
- Chemistry saving lives: using First World War Hypo helmets to avoid chlorine poisoning
- Research Article
- An exploration of the proton NMR problem-solving approaches of undergraduate students
- Good Practice Reports
- Chemical Quest: general knowledge and popular culture quizzes about the elements in a board game for the class
- Lessons learned from a case study on teaching the socioscientific issue of ethanol, used as an ingredient of sanitizers, to promote students’ learning of and about chemistry during the COVID-19 pandemic
Articles in the same Issue
- Frontmatter
- Review Article
- Relativistic effects on the chemistry of heavier elements: why not given proper importance in chemistry education at the undergraduate and postgraduate level?
- Research Articles
- Designing a learning environment based on the spiral of skills to overcome the didactic obstacles associated with teaching the Daniell cell
- Analysis of the relationship between students’ argumentation and chemical representational ability: a case study of hybrid learning oriented in the environmental chemistry course
- Good Practice Report
- What makes representations good representations for science education? A teacher-oriented summary of significant findings and a practical guideline for the transfer into teaching
- Research Article
- Student exploration of the Henderson-Hasselbach equation and pH readings to determine the pK a value of 4,4′-trimethylenedipiperidine (TMDP)
- Good Practice Report
- Chemistry saving lives: using First World War Hypo helmets to avoid chlorine poisoning
- Research Article
- An exploration of the proton NMR problem-solving approaches of undergraduate students
- Good Practice Reports
- Chemical Quest: general knowledge and popular culture quizzes about the elements in a board game for the class
- Lessons learned from a case study on teaching the socioscientific issue of ethanol, used as an ingredient of sanitizers, to promote students’ learning of and about chemistry during the COVID-19 pandemic