Pathophysiological Aspects of the Development of Abdominal Aortic Aneurysm with a Special Focus on Mitochondrial Dysfunction and Genetic Associations
-
Volha I. Summerhill
 ,
Vasily N. Sukhorukov
,
Vasily N. Sukhorukov

Abstract
Abdominal aortic aneurysm (AAA) is a complex degenerative vascular disease, with considerable morbidity and mortality rates among the elderly population. The mortality of AAA is related to aneurysm expansion (the enlargement of the aortic diameter up to 30 mm and above) and the subsequent rupture. The pathogenesis of AAA involves several biological processes, including aortic mural inflammation, oxidative stress, vascular smooth muscle cell apoptosis, elastin depletion, and degradation of the extracellular matrix. Mitochondrial dysfunction was also found to be associated with AAA formation. The evidence accumulated to date supports a close relationship between environmental and genetic factors in AAA initiation and progression. However, a comprehensive pathophysiological understanding of AAA formation remains incomplete. The open surgical repair of AAA is the only therapeutic option currently available, while a specific pharmacotherapy is still awaited. Therefore, there is a great need to clarify pathophysiological cellular and molecular mechanisms underlying AAA formation that would help to develop effective pharmacological therapies. In this review, pathophysiological aspects of AAA development with a special focus on mitochondrial dysfunction and genetic associations were discussed.
Introduction
Abdominal aortic aneurysm (AAA) is associated with substantial morbidity and mortality worldwide, predominantly affecting older men aged 65 years and over [1, 2, 3]. Although women represent an increasing proportion (almost one-third) of the patients presenting to hospital with AAA rupture thus, they contribute to an increasing number of AAA-related deaths [4, 5]. Aortic aneurysm rupture causes profound internal bleeding and is usually lethal unless the repair is performed promptly. The mortality of ruptured AAA is over 80% [1].
It is considered that AAA is a permanent and irreversible dilatation in the infra-renal part of the aorta; the enlargement of the aortic diameter up to 30 mm and above. The natural course of AAA, the gradual expansion to ultimate rupture, significantly varies among individuals. Progressive AAA is typically asymptomatic until the rupture [6]. The standard of care for patients with large (≥55 mm in diameter), rapidly growing (>10 mm per year), and/or symptomatic AAAs is endovascular exclusion or open surgical repair, and these are the only therapeutic options currently available [7]. As for small AAAs (<55mm in diameter), surgical repair is not beneficial and therefore limited to regular imaging surveillance. A large randomized controlled clinical trial showed that survival rates were not increased after immediate elective repair of AAAs less than 5.5 cm in diameter [8]. Ultrasound-based screening aiming at those at risk (commonly men of 65-years and over) proven to be effective in reducing the risk of AAA-related mortality [9]. However, ultrasound screening does not provide possibilities to identify patients of high- or low risk for the disease progression. Therefore, there is an unmet clinical requirement to develop pharmacological or cell-based therapies that would restrict or prevent AAA formation, its progressive expansion, and rupture. The deficit in effective risk stratification strategies and of specific drug therapy drives significant interest in the pathogenesis of this condition. Even though the pathophysiological pathways underlying AAA have been broadly studied, a detailed pathophysiological understanding of AAA remains incomplete. In this review, we will discuss pathophysiological aspects of AAA development with a special focus on mitochondrial dysfunction and genetic associations.
Biological Processes Involved in AAA Formation
The pathophysiology of AAA is complex and associated with the degradation of the elastic media of the aortic wall, which leads to aortic dilatation and consecutive rupture. Numerous studies have been conducted on the molecular mechanisms implicated in AAA pathogenesis [10, 11, 12]. The following biological processes play a significant role in the development of this disorder: (i) chronic inflammation, (ii) degeneration and apoptosis of vascular smooth muscle cells (VSMCs), (iii) extracellular matrix (ECM) proteolysis, (iv) oxidative stress, (v) endothelial cell alteration, and (vi) formation of intraluminal thrombus. All these processes were described in detail by Golledge et. al. [13]. A large amount of evidence indicated the presence of innate and adaptive immune systems in AAA pathogenesis [12, 14, 15, 16].
Vascular inflammation is the major pathophysiological feature of the aortic aneurysm developmental process [13]. Multiple exogenous immune cells, such as lymphocytes, macrophages, mast cells, neutrophils, and natural killer (NK) cells, gradually infiltrate into the outer part of the aorta and often into surrounding tissue, thus, inducing the inflammatory response. The functional attributes of different populations of inflammatory cells in AAA tissue are unclear, but some evidence emerged indicating the role of T-cells and NK cells in the pathogenesis of AAA. It was demonstrated that T-cells can secrete proinflammatory cytokines [17] and NK cells have exhibited increased cytotoxicity that is a contributing factor in the generation or potentiation of inflammation in patients with AAA [18]. The infiltration of inflammatory cells promotes SMC production and activation leading to a secretion of several matrix metalloproteinases (MMPs), which are considered to be crucial enzymes causally linked to AAA formation and progression [19]. In particular, some studies support MMP2 and MMP9 as potentially important factors [20, 21]. These MMPs destroy and destabilize the mechanical property of the aortic wall by modulating interstitial elastin and collagen that results in the apoptotic loss of medial SMCs and ECM destruction, thus, promoting dilation and rupture of AAA. Interestingly, there is evidence suggesting that the chronic inflammation seen in AAAs is a result of a disturbed autoimmune response against autologous components of the aortic wall [22]. Moreover, new vessel formation (angiogenesis) was demonstrated within the adventitia of human AAA biopsies and implicated in promoting the influx of inflammatory cells [23]. Additionally, a number of inflammatory genes were found to be up-regulated in AAA murine models [24] and that supports the inflammation-mediated concept of AAA further.
Furthermore, many studies showed that promoting inflammation oxidative stress is a great contributor to AAA pathogenesis [25, 26, 27]. Human and animal studies suggested that oxidative stress is implicated in the vascular degeneration seen in AAA [28, 29]. The evidence coupling up oxidative stress with the increased matrix proteolysis, SMC apoptosis, altering mechanical forces, and further augmenting the cycle of inflammation was extensively reviewed [30]. The association of inflammatory cells with the elastic lamina disruptions and the presence of ROS is indicative that the natural course of AAA is a slow process, which eventually reaches a stress point resulting in the rupture of the aneurysm [31, 32]. Oxidative stress is excessive ROS production that prevails over antioxidant protection. In particular, overproduction of ROS occurs as a result of the imbalance between the activity of endogenous pro-oxidative enzymes (NADPH oxidase (NOX), xanthine oxidase (XO), and the mitochondrial respiratory chain) and the antioxidant enzymes (glutathione peroxidase (GP), haem oxygenase (HO), superoxide dismutase (SOD), thioredoxin (TRX), and catalase) [33, 34].
An important factor, which is also involved in AAA pathogenesis is alterations in the activity of the renin-angiotensin system signaling pathway [35]. The impacts of angiotensin II on vascular cells have been extensively studied [23, 36, 37] and include several cellular mechanisms, such as generation of ROS, stimulation of MMPs, and infiltration of inflammatory cells. Interestingly, angiotensin II can mediate mitochondrial dysfunction in vascular endothelial cells [38, 39]. As for the molecular mechanism by which angiotensin II can generate an increased production of mitochondrial ROS, the involvement of NADPH oxidase-derived cytosolic ROS was shown [40]. Moreover, angiotensin II was demonstrated to inhibit mitochondrial sirtuin 3 and SOD2 through S-glutathionylation and acetylation, respectively, thus increasing mitochondrial ROS production [41]. Noteworthy, ROS generated by dysfunctional mitochondria can initiate an inflammatory response via the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway activation, which serves as a pivotal mediator of innate and adaptive inflammatory responses [42, 43]. It was also demonstrated that angiotensin II can regulate the morphology of mitochondria. The stimulation of angiotensin II caused mitochondrial fission, which was associated with dynamin-related protein-1 Drp1 Ser616 phosphorylation in VSMC culture [44]. Mitochondrial fission and fusion are regulated by several different proteins of which GTPases, Drp1 and mitochondrial fusion 1/2 proteins are key mediators of fission and fusion [45]. The association of mitochondrial dysfunction with the formation of AAA will be discussed further in the next section.
In addition, it is commonly believed that AAA development is associated with atherosclerosis [31, 46]. The investigation of human AAA biopsies has consistently found concurrent intimal atherosclerosis and most AAAs were considered to be caused by atherosclerosis [47]. Along these lines, atherosclerosis is an essential independent risk factor for AAA, as patients with AAA often have atherosclerosis. However, it is still not fully understood whether this alliance is contributory or a result of frequently shared risk profiles. Investigated AAAs have common risk factors with atherosclerosis, such as smoking, hypertension, and male gender [47, 48]. Some authors suggested that these pathologies are more discrete than commonly thought, pointing out the controversial role of atherosclerosis in AAA [25]. There is a large amount of evidence indicating that known high-risk factors for atherosclerosis, such as diabetes mellitus, hypercholesterolemia, and obesity, are less prominent in AAA; whereas, smoking, male gender, and ethnicity are particularly important in AAA development but less pronounced for atherosclerosis [47, 49, 50, 51]. Hypertension was found to have only a weak association with AAA [50]. Remarkably, out of the risk factors known to be linked to AAA presence, only smoking and diabetes mellitus were revealed to predict AAA progression [50]. Importantly, the understanding of the atherosclerosis significance in AAA can be of therapeutic relevance. Human studies presented conflicting evidence on whether statins or angiotensin-converting enzyme inhibitors, which are effective in atherosclerosis, can also prevent AAA progression [52, 53, 54].
The biological processes involved in AAA formation are summarized in Figure 1.
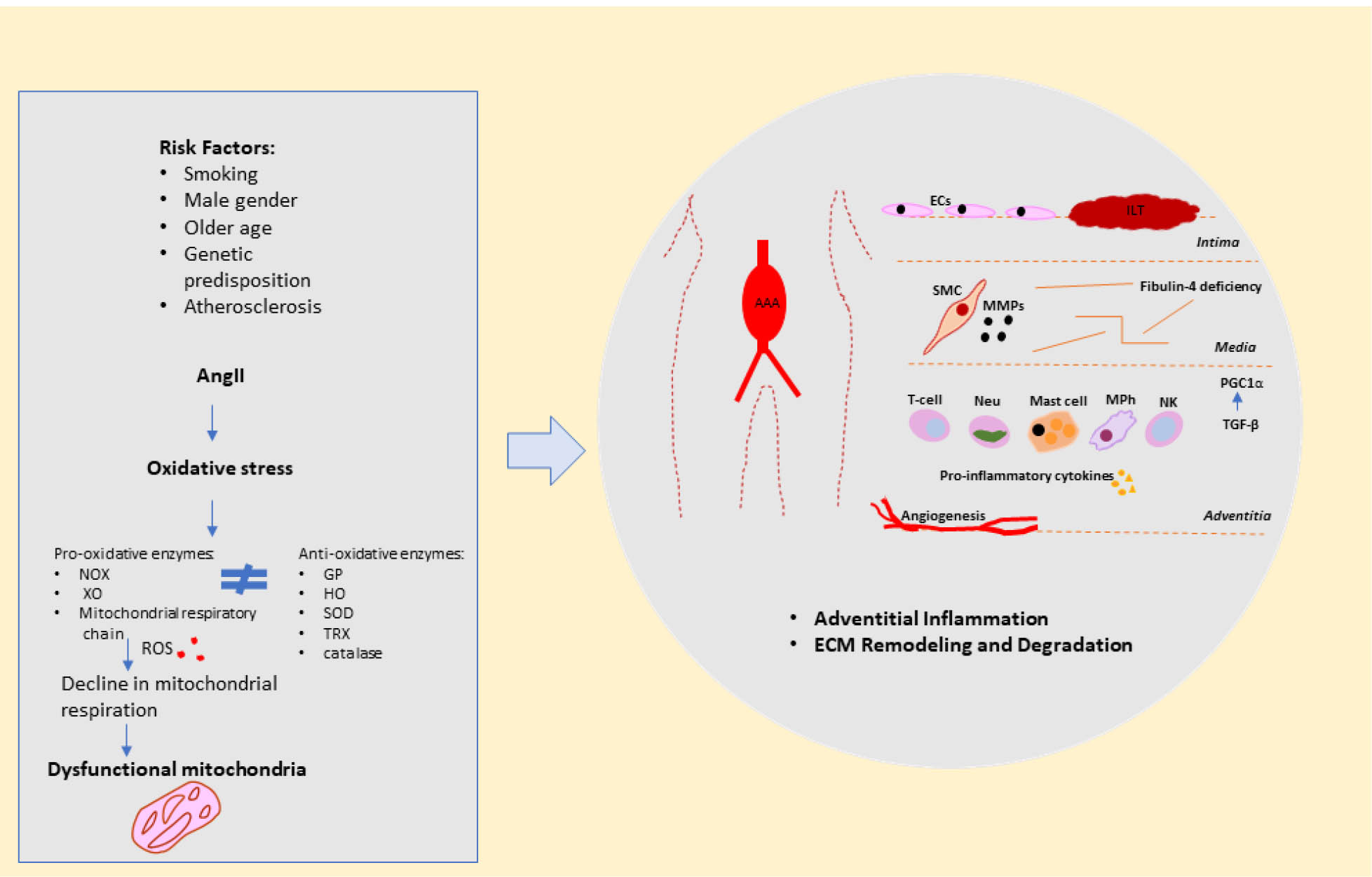
Schematic representation of biological processes involved in AAA formation.
Abbreviations: AAA, abdominal aortic aneurysm; AngII, angiotensin II; EC, endothelial cells; ECM, extracellular matrix; GP, glutathione peroxidase; HO, haemoxygenase; ILT, intraluminal thrombus; MMPs, matrix metalloproteinases; MPh, macrophage; Neu, neutrophil; NK, natural killer cell; NOX, NADPH oxidase; PGC1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; RAS, reninangiotensin system; ROS, reactive oxygen species; SMC, smooth muscle cell; SOD, superoxide dismutase; TGF-β, transforming growth factor beta; TRX, thioredoxin; XO, xanthine oxidase.
Mitochondrial Dysfunction as an Important Contributor to the Formation of AAA
In general, the impairment of mitochondrial function is significant in the progression of ageing and age-related disorders [55]. Reduced mitochondrial respiration, as a result of mitochondrial dysfunctions, can manifest itself both in normal ageing of blood vessels and in age-related cardiovascular diseases including, atherosclerosis and the formation of the aneurysm [56]. There is a growing body of evidence indicating that disturbed mitochondrial functions are associated with AAA in both animal models and humans. These alterations include not only increased mitochondrial ROS production [57] but also dysregulation of mitochondrial biogenesis [47, 58] and mitochondria-dependent cellular apoptosis of aortic SMCs [59]. The oxidative damage of mitochondrial DNA (mtDNA) induces respiratory chain dysfunction via disruption of oxidative phosphorylation leading to reduced adenosine triphosphate (ATP) production, the molecular unit carrying intracellular energy, and further increased generation of ROS. Considerable data is indicating a decline in oxidative phosphorylation capacity of human mitochondria with age. Thus, mitochondrial dysfunction leading to the impaired mitochondrial respiration was found to be associated with age-related pathological changes, including both atherosclerosis and AAA formation. For instance, significantly reduced copy number of mtDNA, mitochondrial respiration, and the expression of specific electron transport chain complexes was demonstrated in VSMCs obtained from human atherosclerotic plaques [60, 61]. The differential expression of several genes related to mitochondrial function and oxidative phosphorylation was shown to be involved in AAA pathogenesis [62].
Evidence has emerged that the maintenance of mitochondrial homeostasis advocates mechanisms involving mitochondrial dynamics or mitochondrial quality control that also participate in fundamental processes including ageing and AAA. The mitochondrial dynamics comprises of the following events: mitochondrial movement along the cytoskeleton, the regulation of mitochondrial architecture, and connectivity mediated by continuous fusion/fission [63]. Fission is involved in the selection of dysfunctional mitochondria; therefore, it is important for maintaining mitochondrial quality and integrity. Reduced mitochondrial fusion leads to loss of mtDNA and, as a consequence, to respiration deficient mitochondria [64]. In turn, respiration deficient mitochondria are selectively removed by mitophagy, an autophagy-lysosome system that degrades dysfunctional mitochondria by lysosome fusion [65]. Thus, consistent fusion and fission play a vital role in mitochondrial homeostasis and energy adaptation. It was demonstrated that inefficient fission and fusion contribute to ageing [66]. Besides, mitochondrial fission is involved in the development of AAA, as suggested by recent data [67]. Moreover, mitochondrial mitophagy was observed to decline in ageing [68], as well as to be linked to the development of human AAA [69]. Inefficient removal of damaged mitochondria can lead to the excessive activation of inflammatory signaling pathways and subsequently to chronic systemic inflammation [70, 71]. In this way, in response to damaging stimuli, such as infectious pathogens and cellular debris, severe mitochondrial dysfunction mediates NLRP3 inflammasome hyperstimulation and mitochondrial ROS production causing caspase-1 activation. The presence of various identifiable causative pathogens, including Staphylococcus, Streptococcus, Salmonella, Escherichia coli, Chlamydia subspecies, Cytomegalovirus and other microorganisms was reported in AAA patients [72, 73, 74, 75]. Activated caspase-1 generates mature pro-inflammatory interleukin (IL)-1β and IL-18 promoting inflammatory cell death and chronic inflammation associated with ageing and age-related diseases [76]. Notably, the inflammasome activation leads to caspase-1-dependent mitochondrial damage and inhibits mitophagy [77]. Therefore, the appropriate mitochondrial functioning plays a crucial role in the innate immune system signal transduction with mitophagy as a key regulatory mechanism that maintains tissue homeostasis by limiting excessive inflammation. Innate immunity is involved in AAA progression [12]. Moreover, according to the oxy-“inflammaging” theory, oxidative stress impairs proinflammatory processes contributing to the further progression of various age-related diseases [78]. Excessive production of mitochondrial ROS, as a result of mitochondrial dysfunction, is a critical factor in the manifestation of “inflammaging”, the chronic inflammation profile accompanying ageing [79]. The accumulation of damaged macromolecules during ageing by both, the increased production of ROS and/or by inadequate elimination due to mitophagy/autophagy decline are at least partially responsible for this chronic inflammation profile [79] [80]. In support of “inflammaging” being implicated in AAA, a large population-based study has found a strong positive relationship between AAA clinical incidence and circulating biomarkers of inflammation (C-reactive protein, white cell count, and fibrinogen) [81].
Several independent research groups have examined the relationship between mitophagy and mitochondrial mutations. The causative role of m.12338T>C mutation in decreased mitophagy was exhibited by decreased levels of autophagy protein light chain 3 and deposition of autophagic substrate p62 in the mutant cybrid cells as compared with control cell lines. These data established the direct link between mitochondrial dysfunction caused by complex I mutation and mitophagy, which determines the critical function of mitochondrial dynamics [82]. Numerous studies observed ageing-related accumulation of mtDNA mutations [83, 84, 85, 86]. The accumulation of mtDNA mutations causing a decline in mitochondrial integrity and function is implicated in the cellular dysfunction associated with ageing and age-related pathology. The development of cytoplasmic cybrid cell line models made it possible to study the functional significance of mtDNA mutations. Using cybrids carrying point mutations in mtDNA, the significant accumulation of heteroplasmic mtDNA mutations in ageing mitochondria was demonstrated [87]. The results obtained in this study showed that ageing-related mtDNA mutations caused mitochondrial dysfunction by altering the oxidative phosphorylation machinery. These findings were consistent with previously reviewed data demonstrating that altering the expression of oxidative phosphorylation complexes, mutations in mtDNA can lead to mitochondrial dysfunction and enhanced production of ROS [88]. Furthermore, if high levels of mtDNA mutations can lead to mitochondrial dysfunction, the impact that mtDNA mutations can have on a cell would be to impair its function ultimately leading to death. For instance, the respiratory chain deficiency triggered by the reduced mtDNA expression was shown to be associated with increased apoptosis in TFAM (nuclear-encoded mitochondrial transcription factor) knockout animal models [89]. The cellular damage and apoptosis-related to incorrect functioning of the respiratory chain and energy production caused by mutations in the mitochondrial genome are tightly implicated in AAA development [90]. It should be noted that, in most cases, mtDNA mutations are not specifically linked to certain pathological conditions. Mitochondrial mutations associated with cardiovascular diseases, such as atherosclerosis [91], type 2 diabetes [92], and acute myocardial infarction [93] were identified. Therefore, mitochondrial dysfunction may manifest itself in different pathologies, AAA including.
Mitochondrial DNA typically has higher mutation rates compared to nuclear DNA, as a result of its proximity to the site of ROS production, the deficiency of protective histone proteins, and the lack of a comprehensive DNA repair system [94]. Mutations of mtDNA are inherited by the maternal line and then reproduced by dividing mitochondria encompassing mutant mtDNA. This determines the high mutability rates of mtDNA, as well as the lifelong accumulation of somatic mutations of mtDNA [84, 86]. In each cell there is a high copy number of mtDNA which can exist in heteroplasmy containing both wild type and mutant mtDNA, or in homoplasmy containing identical mtDNA. In general, pathogenic mtDNA mutations are heteroplasmic [95]. MtDNA mutations could be caused by either replication errors or increased ROS production and, if a mutated mtDNA molecule undergoes subsequent replication and clonal expansion within a cell, this cell may become respiratory chain deficient [86, 96]. It is worth mentioning that the relationship between mtDNA mutations and oxidative stress in ageing and age-related diseases were extensively studied over several decades. The widely accepted mitochondrial free radical theory of ageing, which remained to be the predominant hypotheses in the field for many years, assumed the presence of a positive feedback loop (a “vicious cycle”). MtDNA accumulates genetic damage caused by the production of ROS during cellular respiration, in turn, dysfunctional mitochondria leak free radicals into the cytosol creating more mtDNA mutations [97]. However, some contradictory evidence on this theory emerged in the past decade. A study suggested some factors that are mainly responsible for the majority of point mutations accumulating in mtDNA, including replication errors by DNA polymerase γ and/or spontaneous base hydrolysis [98]. Other experimental studies harbouring conflicting evidence on oxidative damage of mtDNA as a major driver of ageing were discussed in several reviews [99, 100]. Taken together, the impaired mitochondrial function due to oxidative damage of mtDNA, dysregulated mitochondrial biogenesis and dynamics can lead to disruption in the cell homeostasis, development of inflammation, excess of ROS, and loss of VSMCs, the hallmark pathophysiological processes of AAAs. Presented in this section, the information on mitochondrial dysfunction implicated in the AAA pathogenesis may appear to be rather speculative but it is hoped that it will encourage further investigation of this issue.
Mitochondrial Dysfunction-Associated Potential Molecular Mechanisms Underlying Aortic Aneurysm Formation
Several lines of evidence suggest an important role for Fibulin-4 deficiency associated with the formation of the aortic aneurysm [101, 102]. Fibulin-4 is a glycoprotein located in microfibril bundles and known to play a significant role in the formation of elastic fibres and maintenance of arterial wall integrity [103]. In an attempt to provide a comprehensive understanding of the molecular mechanisms underlying aortic aneurysm formation, a recent study by van der Pluijm et. al. has demonstrated a potential pathway linking the decreased expression levels of Fibulin-4 with ROS mitochondrial dysfunction, disturbed transforming growth factor-beta (TGFβ) signaling, and the dysregulated activity of peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC1α) [104]. This study used a combination of proteomics, genomics, and verifying functional experiments on thoracic aortas of aneurysmal mice with a four-fold reduction in the expression of Fibulin-4. The defective mitochondrial function, i.e., decreased mitochondrial respiration, as well as altered energy metabolism in aortic SMCs, were observed. It was found that Fibulin-4-deficient mice had increased TGFβ signaling. This was also confirmed by another study, in which the reduced levels of Fibulin-4 resulted in the increased TGFβ activation [105]. However, the mechanism by which Fibulin-4 deficiency induces excessive TGFβ signaling remains unknown. Moreover, the increased TGFβ signaling decreased PGC1α transcriptional levels in Fibulin-4-deficient VSMCs and that is indicative that the increased TGFβ signaling is responsible for the decreased PGC1α expression levels in Fibulin-4-deficient aortas and VSMCs, hence, making an impact on mitochondrial respiration [104]. PGC1α is a transcriptional co-activator, which is expressed in tissues with high oxidative potential regulating mitochondrial function and energy metabolism in cells [106]. Of note, TGFβ signaling negatively regulating PGC1α expression levels was previously reported [107, 108]. It was also established that reduced PGC1α levels are involved in the reduced cellular metabolism and mitochondrial function [109]. PGC1α is engaged in fatty acid mitochondrial oxidation for energy in the cell via direct co-activation of the peroxisome proliferator-activated receptors (PPARs [α, β, and γ]), the major regulators of the cellular metabolic process [106]. PPARα and PPARγ play a central role in regulating the expression of proteins involved in extra- and intra-mitochondrial transport of fatty acids and oxidation. While PPARβ regulates the antioxidant synthesis, so that their reduced expression and activity may increase further ROS production. In this respect, the increased levels of ROS were observed in aneurysmal aortas of Fibulin-4-deficient mice [104]. Decreased expression of PGC1α protein was seen in patients with AAA indicating that PGC1α expression may play a regulatory role in both thoracic and abdominal aneurysms of the aorta [110]. In addition, improving mitochondrial function via PGC1α activation exhibited a favorable effect on ageing and longevity [111, 112].
Furthermore, in the study of van der Pluijm et. al., consistent with the reduced mitochondrial respiration, VSMCs derived from Fibulin-4-deficient mice exhibited reduced uncoupled oxygen utilization and increased acidification rates that may be indicative of the increased glycolysis. While PGC1α activation increased both basal and maximum oxygen utilization in VSMCs of Fibulin-4-deficient mice and improved cell proliferation proposing that the reduced proliferation phenotype can possibly be provoked by the increased TGFβ levels through a reduction in PGC1α levels. Similarly, another study reported that the lowering of TGFβ levels in Fibulin-4-deficient VSMCs amended their reduced proliferation rate [113]. In addition, the disruption of TGFβ signaling can attenuate proliferation and apoptosis of VSMCs and AAA formation [114]. Thus, the evidence presented above showed that Fibulin-4 deficiency can lead to the increased TGFβ1 signaling, which can cause not only alterations in protein composition affecting mainly ECM and cytoskeleton proteins but also mitochondrial proteins (although mitochondrial structure and appearance remained unaffected, altered mitochondrial function and a reduction in mitochondrial size were observed) [104]. That may help to explain reduced mitochondrial respiration found in cells derived from different aneurysm phenotypes that also display increased TGFβ signaling, including Loeys-Dietz Syndrome, Arterial Tortuosity Syndrome, and Marfan Syndrome [102]. However, the exact mechanisms by which a deficiency in a protein involved in ECM structural integrity can lead to altered mitochondrial function and metabolism need to be elucidated. Finding mutual causal pathways with other TGFβ-linked vasculopathies may offer new options for preventative and therapeutic strategies for AAA.
Relying on studies demonstrating that TGFβ signaling negatively regulates PGC1α levels in several human aneurysm syndromes [107, 108], some authors suggest that this may be due to mitochondrial structural abnormalities [115]. However, a decrease in the copy number of mtDNA, mitochondrial respiration, and expression of PGC1α and several other regulatory genes of mtDNA synthesis was also observed in arterial ageing associated with ROS and loss and/or senescence of VSMCs [56]. In agreement with this study, an intriguing point of view was expressed that the changes seen in Fibulin-4 deficient mice may have occurred as a result of VSMC loss and/or senescence in the arterial wall accompanied by the distorted mitochondrial function and increased generation of ROS [115].
Genetic Associations for AAA
Population-based studies have established that a combination of environmental and genetic factors which included smoking, older age, male sex, white ethnicity, and a family history of AAA determine the risk of developing AAA [47, 116]. Moreover, the robust evidence suggests that genetic variations contribute to the formation of AAA.
DNA Linkage Studies of AAA
It was estimated that heritability of around 70% is an important contributor to the overall AAA susceptibility [117] and that is indicative that genetic factors play a major role in the initiation and progression of AAA. The high prevalence of AAA was found among brothers supporting the contribution of genetic factor to AAA pathogenesis [118]. This study also established that the familial AAA cases were more likely to have AAA rupture compared with the sporadic cases. Some studies on families, in which at least two relatives had AAA, revealed that the risk for developing an AAA is eight-fold higher for a first-degree relative than among the general population [119]. Family-based DNA linkage studies have provided unbiased means of identifying genetic risk factors for AAA [120, 121]. Using the affected-relative-pair approach, these studies discovered two genetic loci located on chromosomes 4q31 and 19q13 containing several plausible and physiologically relevant candidate genes, such as low-density lipoprotein receptor-related protein 3 (LRP3), peptidase D (PEPD), hepsin (HPN), interleukin 15 (IL-15), GRB2-associated binding protein 1 (GAB1), an endothelin receptor type A (EDNRA), thus providing the evidence for genetic heterogeneity and the presence of susceptibility loci for AAA. These genomic regions were assigned as AAA susceptibility loci in Online Mendelian Inheritance in Man (OMIM).
GWASs of AAA
According to current consensus, in some cases, the clinical phenotype, to a large extent, can be determined by the presence of a single nucleotide substitute in the nuclear genome. Nevertheless, it is usually difficult to establish a causal relationship between a particular variant of the nuclear genome and its corresponding phenotypic manifestation, therefore, identifying genetic predisposition to chronic pathologies, AAA including, can be challenging. Using single SNPs in AAA-relevant genes, almost 100 genetic associations have been reported [122]. In the past decade, the genome-wide associations study (GWAS) approach emerged as a targeted search for associations between single nucleotide polymorphisms (SNPs) and phenotypic traits of major human diseases, including AAA. A recent meta-analysis of GWASs has identified four novel SNPs specifically associated with AAA, such as 1q32.3 (SMYD2), 13q12.11 (LINC00540), 20q13.12 (near PCIF1/MMP9/ZNF335), and 21q22.2 (ERG) [123]. These associations were in agreement with known AAA biological pathways, implicating lipoprotein metabolism, inflammation, and ECM degradation but other potential pathomechanisms relating to transcription factor ERG and fibroblast growth factor 9 (FGF9) were also identified. The latter has a wide range of mitogenic and cell survival activities in VSMCs [124]. Besides, the latest GWAS study identified 14 novel loci of AAA risk variants, bringing the total number of known significant AAA loci to 24 [125]. Moreover, relying on the fact that some genetic and environmental risk factors are related to both atherosclerosis and AAA, the gene regulatory function of rs2836411, the non-coding SNP located in an intron of the ERG gene, attracted special attention in a recent study [126]. It was found that ERG expression is likely to be affected by rs2836411 by altering both enhancer activity and local chromatin interactions. In atherosclerosis, the ERG gene is involved in vascular development, angiogenesis, and inflammation, hence, modulating ERG expression levels mechanistically, rs2836411 could also contribute to AAA pathogenesis. ERG is a transcription factor expressed in both hematopoietic and endothelial cells and mediates several key processes of vascular development, including endothelial specification, angiogenesis, and vessel stability [127]. An additional role of ERG in AAA formation is the observation that angiogenesis is increased in the aneurysm wall [128]. Previous GWASs also reported a substantial number of other significant AAA-associated SNPs and genes from genomic data. The summary of several SNPs and their respective genes studied for conferring susceptibility to AAA was presented in Table 1.
The summary of several SNPs and their respective genes studied for conferring susceptibility to AAA.
| SNP ID | Gene/Locus | Biological pathway involved | References |
|---|---|---|---|
| rs1626340 | TGFBR1 and TGFBR2 | Vascular remodeling | [129] |
| rs1036095 | |||
| rs4522809 | |||
| rs7025486 | DAB2IP | Cell growth and survival | [130] |
| Lipid metabolism | |||
| rs7635818 | CNTN3 | Cell adhesion | [131] |
| rs3025058 rs2252070 | MMP3 and MMP13 | Extracellular matrix | [132] |
| rs1800896 | IL-10 | Inflammation | [133] |
| rs10757278-G | CDKN2BAS1 (ANRIL) | Inflammation | [134] |
| rs1466535 | LRP1 | Lipid metabolism | [135, 136] |
| rs1795061 | SMYD2 | Vascular development and EC/SMC differentiation | [123] |
| rs599839 | SORT1 | Altering circulating lipoprotein profiles | [137] |
| rs6511720 | LDLR | LDL-cholesterol homeostasis | [138] |
| rs5186 | AGTR1a | Renin-angiotensin system | [139] |
| rs4646994 | ACE | Renin-angiotensin system | [140] |
Note: all mutations had a significant association with an increased risk of AAA and can be found in a single nucleotide polymorphism database (www.ncbi.nlm.nih.gov/projects/SNP). ACE, angiotensin-converting enzyme; AGTR1a, angiotensin-renin receptor 1 alpha; CDKN2BAS1, antisense RNA 1 gene; CNTN3, contactin-3; DAB2IP, GTPase activating protein; EC, endothelial cell; IL-10, interleukin 10; LDLR, low-density lipoprotein receptor; LRP1, low-density lipoprotein receptor-related protein 1 gene; MMP, matrix metalloproteinase; SMYD2, histone methyltransferase; SORT1, sortilin-1.
The information that was made available by GWAS of AAA outlines the role of common genetic variation in disease susceptibility. This can be helpful for the identification of the most promising molecular targets and candidate genes and their potential biological significance for further comprehensive research of AAA pathogenesis and therapy. Nevertheless, there are some limitations of GWAS that should be taken into account in order to place its results in the appropriate clinical context. First, one has to make sure that variants identified by GWAS are causal and it can take considerable effort to find functional SNPs. In fact, out of all GWAS-identified SNPs associated with AAA risk, only a limited number is actually causal. Second, GWAS fail to detect to what extent interactions between several variants or gene regions can affect the disease phenotype. Finally, GWAS has limited clinical predictive value and applicability. The latter is because of both the insignificant effect sizes and heritability of the majority of associations, which could be offset simply by environmental factors.
Concluding Remarks
With an ageing population, the prevalence of AAA may become more common in the future. In this review, we have attempted to define some of the important aspects that have emerged from the growing body of research on AAA pathogenesis. The development of AAA is a result of several factors, including a reduction in VSMCs, activation of MMPs, ECM disruption, and inflammatory cell infiltration. Moreover, several lines of evidence point toward mitochondrial dysfunction implicated in AAA development, hence, its role in AAA pathogenesis should not be underestimated. Potential mitochondrial dysfunction-associated molecular mechanisms underlying aortic aneurysm formation were described in this review. Besides, there has been a dramatic expansion in the research on genetic associations of AAA. Despite the considerable advances in the field, many uncertainties in mechanisms of AAA formation remain to be elucidated. The lack of understanding hinders the development of a comprehensive approach for better management of patients with AAAs in clinical settings.
Author contributions:
V.I.S. drafted the main text; V.N.S., L.V.N., A.H.E., and I.A.S. reviewed the literature and contributed to the concept of the manuscript; V.I.S. prepared the figures and table; A.N.O. and I.A.S. critically revised the final version of the manuscript; all authors read and agreed with the final version of the manuscript.
Funding:
This work was supported by the Russian Science Foundation (Grant # 20-45-08002).
Conflict of Interests:
Authors state no conflict of interest
Data Availability Statement:
The datasets analysed during the current study are available from the corresponding author on reasonable request.
References
1 Wang H, Naghavi M, Allen C, Barber RM, Bhutta ZA, Carter A, et al.; GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016 Oct;388(10053):1459–544.10.1016/S0140-6736(16)31012-1Search in Google Scholar
2 GBD 2015 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016 08;388(10053):1603–58.10.1016/S0140-6736(16)31460-XSearch in Google Scholar
3 Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011 Feb;8(2):92–102.10.1038/nrcardio.2010.180Search in Google Scholar PubMed
4 Powell JT, Sweeting MJ, Thompson MM, Ashleigh R, Bell R, Gomes M, et al.; IMPROVE Trial Investigators. Endovascular or open repair strategy for ruptured abdominal aortic aneurysm: 30 day outcomes from IMPROVE randomised trial [Internet]. BMJ. 2014 Jan;348 jan13 2:f7661. [cited 2020 Oct 19] Available from: https://www.bmj.com/content/348/bmj.f766110.1016/j.jvs.2014.03.264Search in Google Scholar
5 Anjum A, von Allmen R, Greenhalgh R, Powell JT. Explaining the decrease in mortality from abdominal aortic aneurysm rupture. Br J Surg. 2012 May;99(5):637–45.10.1002/bjs.8698Search in Google Scholar PubMed
6 Thompson SG, Brown LC, Sweeting MJ, Bown MJ, Kim LG, Glover MJ, et al. Systematic review and meta-analysis of the growth and rupture rates of small abdominal aortic aneurysms: implications for surveillance intervals and their cost-effectiveness. Health Technol Assess. 2013 Sep;17(41):1–118.10.3310/hta17410Search in Google Scholar PubMed PubMed Central
7 Moll FL, Powell JT, Fraedrich G, Verzini F, Haulon S, Waltham M, et al.; European Society for Vascular Surgery. Management of abdominal aortic aneurysms clinical practice guidelines of the European society for vascular surgery. Eur J Vasc Endovasc Surg. 2011 Jan;41 Suppl 1:S1–58.10.1016/j.ejvs.2010.09.011Search in Google Scholar PubMed
8 Merali FS, Anand SS. Immediate repair compared with surveillance of small abdominal aortic aneurysms. Vasc Med. 2002 Aug;7(3):249–50.10.1191/1358863x02vm444xxSearch in Google Scholar PubMed
9 Svensjö S, Björck M, Wanhainen A. Update on screening for abdominal aortic aneurysm: a topical review. Eur J Vasc Endovasc Surg. 2014 Dec;48(6):659–67.10.1016/j.ejvs.2014.08.029Search in Google Scholar PubMed
10 Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. 2019 Apr;16(4):225–42.10.1038/s41569-018-0114-9Search in Google Scholar PubMed
11 Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017 Aug;14(8):457–71.10.1038/nrcardio.2017.52Search in Google Scholar PubMed
12 Sakalihasan N, Michel JB, Katsargyris A, Kuivaniemi H, Defraigne JO, Nchimi A, et al. Abdominal aortic aneurysms. Nat Rev Dis Primers. 2018 Oct;4(1):34.10.1038/s41572-018-0030-7Search in Google Scholar PubMed
13 Golledge J, Shi GP, Norman P. Pathogenesis of Aortic Aneurysms. In: Fitridge R, Thompson M, editors. Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet]. Adelaide (AU): University of Adelaide Press; 2011 [cited 2020 Oct 19]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK534278/10.1017/UPO9781922064004.013Search in Google Scholar
14 Coscas R, Dupont S, Mussot S, Louedec L, Etienne H, Morvan M, et al. Exploring antibody-dependent adaptive immunity against aortic extracellular matrix components in experimental aortic aneurysms. Journal of Vascular Surgery. 2018 Dec 1;68(6):60S–71S.e3. https://doi.org/10.1016/j.jvs.2017.11.090.10.1016/j.jvs.2017.11.090Search in Google Scholar PubMed
15 Lareyre F, Clément M, Raffort J, Pohlod S, Patel M, Esposito B, et al. TGFβ (Transforming Growth Factor-β) Blockade Induces a Human-Like Disease in a Nondissecting Mouse Model of Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2017 Nov;37(11):2171–81.10.1161/ATVBAHA.117.309999Search in Google Scholar PubMed
16 Myasoedova VA, Di Minno A, Songia P, Massaiu I, Alfieri V, Valerio V, et al. Sex-specific differences in age-related aortic valve calcium load: A systematic review and meta-analysis. Ageing Res Rev. 2020 Aug;61:101077.10.1016/j.arr.2020.101077Search in Google Scholar PubMed
17 Forester ND, Cruickshank SM, Scott DJA, Carding SR. Functional characterization of T cells in abdominal aortic aneurysms. Immunology. 2005;115(2):262–70. 131. https://doi.org/10.1111/j.1365-2567.2005.02157.x.10.1111/j.1365-2567.2005.02157.xSearch in Google Scholar PubMed PubMed Central
18 Forester ND, Cruickshank SM, Scott DJ, Carding SR. Increased natural killer cell activity in patients with an abdominal aortic aneurysm. Br J Surg. 2006 Jan;93(1):46–54.10.1002/bjs.5215Search in Google Scholar PubMed
19 Rabkin SW. The Role Matrix Metalloproteinases in the Production of Aortic Aneurysm. Prog Mol Biol Transl Sci. 2017;147:239–65.10.1016/bs.pmbts.2017.02.002Search in Google Scholar PubMed
20 Chew DK, Conte MS, Khalil RA. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J Vasc Surg. 2004 Nov;40(5):1001–10.10.1016/j.jvs.2004.08.035Search in Google Scholar PubMed
21 Chew DK, Orshal JM, Khalil RA. Elastase-induced suppression of endothelin-mediated Ca2+ entry mechanisms of vascular contraction. Hypertension. 2003 Oct;42(4):818–24.10.1161/01.HYP.0000086200.93184.8ESearch in Google Scholar PubMed
22 Jagadesham VP, Scott DJ, Carding SR. Abdominal aortic aneurysms: an autoimmune disease? Trends Mol Med. 2008 Dec;14(12):522–9.10.1016/j.molmed.2008.09.008Search in Google Scholar PubMed
23 Kobayashi M, Matsubara J, Matsushita M, Nishikimi N, Sakurai T, Nimura Y. Expression of angiogenesis and angiogenic factors in human aortic vascular disease. J Surg Res. 2002 Aug;106(2):239–45.10.1006/jsre.2002.6468Search in Google Scholar PubMed
24 Spin JM, Hsu M, Azuma J, Tedesco MM, Deng A, Dyer JS, et al. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm. Physiol Genomics. 2011 Sep;43(17):993–1003.10.1152/physiolgenomics.00044.2011Search in Google Scholar PubMed PubMed Central
25 Emeto TI, Golledge S-WS and J. Targets for Medical Therapy to Limit Abdominal Aortic Aneurysm Progression [Internet]. Vol. 15, Current Drug Targets. 2014 [cited 2020 Oct 21]. p. 860–73. Available from: https://www.eurekaselect.com/123671/article10.2174/1389450115666140804155036Search in Google Scholar PubMed
26 McCormick ML, Gavrila D, Weintraub NL. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2007 Mar;27(3):461–9.10.1161/01.ATV.0000257552.94483.14Search in Google Scholar PubMed
27 Kaneko H, Anzai T, Morisawa M, Kohno T, Nagai T, Anzai A, et al. Resveratrol prevents the development of abdominal aortic aneurysm through attenuation of inflammation, oxidative stress, and neovascularization. Atherosclerosis. 2011 Aug;217(2):350–7.10.1016/j.atherosclerosis.2011.03.042Search in Google Scholar PubMed
28 Sawada H, Hao H, Naito Y, Oboshi M, Hirotani S, Mitsuno M, et al. Aortic iron overload with oxidative stress and inflammation in human and murine abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015 Jun;35(6):1507–14.10.1161/ATVBAHA.115.305586Search in Google Scholar PubMed
29 Shang T, Liu Z, Liu CJ. Antioxidant Vitamin C attenuates experimental abdominal aortic aneurysm development in an elastase-induced rat model. J Surg Res. 2014 May;188(1):316–25.10.1016/j.jss.2013.11.1105Search in Google Scholar PubMed
30 Emeto TI, Moxon JV, Au M, Golledge J. Oxidative stress and abdominal aortic aneurysm: potential treatment targets. Clin Sci (Lond). 2016 Mar;130(5):301–15.10.1042/CS20150547Search in Google Scholar PubMed
31 Weintraub NL. Understanding abdominal aortic aneurysm. N Engl J Med. 2009 Sep;361(11):1114–6.10.1056/NEJMcibr0905244Search in Google Scholar PubMed PubMed Central
32 Wanhainen A, Mani K, Golledge J. Surrogate Markers of Abdominal Aortic Aneurysm Progression. Arterioscler Thromb Vasc Biol. 2016 Feb;36(2):236–44.10.1161/ATVBAHA.115.306538Search in Google Scholar PubMed
33 Förstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008 Jun;5(6):338–49.10.1038/ncpcardio1211Search in Google Scholar PubMed
34 Summerhill VI, Moschetta D, Orekhov AN, Poggio P, Myasoedova VA. Sex-Specific Features of Calcific Aortic Valve Disease. Int J Mol Sci. 2020 Aug;21(16):5620.10.3390/ijms21165620Search in Google Scholar PubMed PubMed Central
35 Malekzadeh S, Fraga-Silva RA, Trachet B, Montecucco F, Mach F, Stergiopulos N. Role of the renin-angiotensin system on abdominal aortic aneurysms. Eur J Clin Invest. 2013 Dec;43(12):1328–38.10.1111/eci.12173Search in Google Scholar PubMed
36 Griendling KK, Ushio-Fukai M, Lassègue B, Alexander RW, Angiotensin II. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997 Jan;29(1 Pt 2):366–73.10.1161/01.HYP.29.1.366Search in Google Scholar
37 Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009 Apr;302(2):148–58.10.1016/j.mce.2008.11.003Search in Google Scholar PubMed PubMed Central
38 Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008 Feb;102(4):488–96.10.1161/CIRCRESAHA.107.162800Search in Google Scholar PubMed
39 Eguchi S, Kawai T, Scalia R, Rizzo V. Understanding Angiotensin II Type 1 Receptor Signaling in Vascular Pathophysiology. Hypertension. 2018 May;71(5):804–10.10.1161/HYPERTENSIONAHA.118.10266Search in Google Scholar PubMed PubMed Central
40 Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassègue B, Griendling KK, et al. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal. 2014 Jan;20(2):281–94.10.1089/ars.2012.4918Search in Google Scholar PubMed PubMed Central
41 Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Flynn CR, Uzhachenko R, et al. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ Res. 2017 Aug;121(5):564–74.10.1161/CIRCRESAHA.117.310933Search in Google Scholar PubMed PubMed Central
42 Sies H, Berndt C, Jones DP. Oxidative Stress. Annu Rev Biochem. 2017 Jun;86(86):715–48.10.1146/annurev-biochem-061516-045037Search in Google Scholar PubMed
43 Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017 Jul;2(1):1–9.10.1038/sigtrans.2017.23Search in Google Scholar PubMed PubMed Central
44 Lim S, Lee SY, Seo HH, Ham O, Lee C, Park JH, et al. Regulation of mitochondrial morphology by positive feedback interaction between PKCδ and Drp1 in vascular smooth muscle cell. J Cell Biochem. 2015 Apr;116(4):648–60.10.1002/jcb.25016Search in Google Scholar PubMed
45 Lee H, Yoon Y. Mitochondrial fission: regulation and ER connection. Mol Cells. 2014 Feb;37(2):89–94.10.14348/molcells.2014.2329Search in Google Scholar PubMed PubMed Central
46 Cowan JA, Dimick JB, Henke PK, Rectenwald J, Stanley JC, Upchurch GR. Epidemiology of Aortic Aneurysm Repair in the United States from 1993 to 2003. Diabetes mellitus. :10.10.1196/annals.1383.030Search in Google Scholar PubMed
47 Golledge J, Muller J, Daugherty A, Norman P. Abdominal aortic aneurysm: pathogenesis and implications for management. Arterioscler Thromb Vasc Biol. 2006 Dec;26(12):2605–13.10.1161/01.ATV.0000245819.32762.cbSearch in Google Scholar PubMed
48 Cornuz J, Sidoti Pinto C, Tevaearai H, Egger M. Risk factors for asymptomatic abdominal aortic aneurysm: systematic review and meta-analysis of population-based screening studies. Eur J Public Health. 2004 Dec;14(4):343–9.10.1093/eurpub/14.4.343Search in Google Scholar PubMed
49 Toghill BJ, Saratzis A, Bown MJ. Abdominal aortic aneurysm-an independent disease to atherosclerosis? Cardiovasc Pathol. 2017 Mar – Apr;27:71–5.10.1016/j.carpath.2017.01.008Search in Google Scholar PubMed
50 Brady AR, Thompson SG, Fowkes F, Gerald R. Greenhalgh Roger M., Powell Janet T. Abdominal Aortic Aneurysm Expansion. Circulation. 2004 Jul;110(1):16–21.10.1161/01.CIR.0000133279.07468.9FSearch in Google Scholar PubMed
51 Ghosh A, Pechota LV, Upchurch GR Jr, Eliason JL. Cross-talk between macrophages, smooth muscle cells, and endothelial cells in response to cigarette smoke: the effects on MMP2 and 9. Mol Cell Biochem. 2015 Dec;410(1–2):75–84.10.1007/s11010-015-2539-3Search in Google Scholar
52 Ferguson CD, Clancy P, Bourke B, Walker PJ, Dear A, Buckenham T, et al. Association of statin prescription with small abdominal aortic aneurysm progression. Am Heart J. 2010 Feb;159(2):307–13.10.1016/j.ahj.2009.11.016Search in Google Scholar
53 Schlösser FJ, Tangelder MJ, Verhagen HJ, van der Heijden GJ, Muhs BE, van der Graaf Y, et al.; SMART study group. Growth predictors and prognosis of small abdominal aortic aneurysms. J Vasc Surg. 2008 Jun;47(6):1127–33.10.1016/j.jvs.2008.01.041Search in Google Scholar
54 Hackam DG, Thiruchelvam D, Redelmeier DA. Angiotensin-converting enzyme inhibitors and aortic rupture: a population-based case-control study. Lancet. 2006 Aug;368(9536):659–65.10.1016/S0140-6736(06)69250-7Search in Google Scholar
55 López-Lluch G, Santos-Ocaña C, Sánchez-Alcázar JA, Fernández-Ayala DJ, Asencio-Salcedo C, Rodríguez-Aguilera JC, et al. Mitochondrial responsibility in ageing process: innocent, suspect or guilty. Biogerontology. 2015 Oct;16(5):599–620.10.1007/s10522-015-9585-9Search in Google Scholar PubMed
56 Foote K, Reinhold J, Yu EP, Figg NL, Finigan A, Murphy MP, et al. Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell. 2018 Aug;17(4):e12773.10.1111/acel.12773Search in Google Scholar PubMed PubMed Central
57 Li Q, Youn JY, Siu KL, Murugesan P, Zhang Y, Cai H. Knockout of dihydrofolate reductase in mice induces hypertension and abdominal aortic aneurysm via mitochondrial dysfunction. Redox Biol. 2019 Jun;24:101185.10.1016/j.redox.2019.101185Search in Google Scholar PubMed PubMed Central
58 Miyao M, Cicalese S, Cooper HA, Eguchi S. Endoplasmic reticulum stress and mitochondrial biogenesis are potential therapeutic targets for abdominal aortic aneurysm. Clin Sci (Lond). 2019 15;133(19):2023–8. https://doi.org/10.1042/CS20190648.10.1042/CS20190648Search in Google Scholar PubMed
59 Sinha I, Sinha-Hikim AP, Hannawa KK, Henke PK, Eagleton MJ, Stanley JC, et al. Mitochondrial-dependent apoptosis in experimental rodent abdominal aortic aneurysms. Surgery. 2005 Oct;138(4):806–11.10.1016/j.surg.2005.07.011Search in Google Scholar PubMed
60 Yu E, Foote K, Bennett M. Mitochondrial function in thoracic aortic aneurysms. Cardiovasc Res. 2018 01;114(13):1696–8. https://doi.org/10.1093/cvr/cvy180.10.1093/cvr/cvy180Search in Google Scholar PubMed PubMed Central
61 Docherty CK, Carswell A, Friel E, Mercer JR. Impaired mitochondrial respiration in human carotid plaque atherosclerosis: A potential role for Pink1 in vascular smooth muscle cell energetics. Atherosclerosis. 2018 Jan;268:1–11.10.1016/j.atherosclerosis.2017.11.009Search in Google Scholar PubMed PubMed Central
62 Yuan K, Liang W, Zhang J. A comprehensive analysis of differentially expressed genes and pathways in abdominal aortic aneurysm. Mol Med Rep. 2015 Aug;12(2):2707–14.10.3892/mmr.2015.3709Search in Google Scholar PubMed
63 Palmer CS, Osellame LD, Stojanovski D, Ryan MT. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal. 2011 Oct;23(10):1534–45.10.1016/j.cellsig.2011.05.021Search in Google Scholar PubMed
64 Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010 Jul;1201(1):21–5.10.1111/j.1749-6632.2010.05615.xSearch in Google Scholar PubMed
65 Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012 Jul;393(7):547–64.10.1515/hsz-2012-0119Search in Google Scholar PubMed PubMed Central
66 Diot A, Morten K, Poulton J. Mitophagy plays a central role in mitochondrial ageing. Mamm Genome. 2016 Aug;27(7–8):381–95.10.1007/s00335-016-9651-xSearch in Google Scholar PubMed PubMed Central
67 Cooper HA, Cicalese S, Preston KJ, Kawai T, Okuno K, Choi ET, et al. Targeting Mitochondrial Fission as a Potential Therapeutic for Abdominal Aortic Aneurysm. Cardiovasc Res. 2020 May.10.1093/cvr/cvaa133Search in Google Scholar PubMed PubMed Central
68 Cavallini G, Donati A, Taddei M, Bergamini E. Evidence for selective mitochondrial autophagy and failure in aging. Autophagy. 2007 Jan–Feb;3(1):26–7.10.4161/auto.3268Search in Google Scholar PubMed
69 Zamorano-Leon JJ, Serna-Soto M, Moñux G, Freixer G, Zekri-Nechar K, Cabrero-Fernandez M, et al. Factor Xa Inhibition by Rivaroxaban Modified Mitochondrial-Associated Proteins in Human Abdominal Aortic Aneurysms. Ann Vasc Surg. 2020 Aug;67:482–9.10.1016/j.avsg.2020.02.005Search in Google Scholar PubMed
70 Gkikas I, Palikaras K, Tavernarakis N. The Role of Mitophagy in Innate Immunity [Internet]. Front Immunol. 2018 Jun;9:1283. [cited 2020 Nov 25] Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2018.01283/full#B1910.3389/fimmu.2018.01283Search in Google Scholar PubMed PubMed Central
71 Mills EL, Kelly B, O’Neill LA. Mitochondria are the powerhouses of immunity. Nat Immunol. 2017 Apr;18(5):488–98.10.1038/ni.3704Search in Google Scholar PubMed
72 Miller DV, Oderich GS, Aubry MC, Panneton JM, Edwards WD. Surgical pathology of infected aneurysms of the descending thoracic and abdominal aorta: clinicopathologic correlations in 29 cases (1976 to 1999). Hum Pathol. 2004 Sep;35(9):1112–20.10.1016/j.humpath.2004.05.013Search in Google Scholar PubMed
73 Sodeck G, Domanovits H, Khanakah G, Schillinger M, Thalmann M, Bayegan K, et al. The role of Chlamydia pneumoniae in human aortic disease-a hypothesis revisited. Eur J Vasc Endovasc Surg. 2004 Nov;28(5):547–52.10.1016/j.ejvs.2004.07.019Search in Google Scholar PubMed
74 Gredmark-Russ S, Dzabic M, Rahbar A, Wanhainen A, Björck M, Larsson E, et al. Active cytomegalovirus infection in aortic smooth muscle cells from patients with abdominal aortic aneurysm. J Mol Med (Berl). 2009 Apr;87(4):347–56.10.1007/s00109-008-0413-4Search in Google Scholar PubMed
75 Kurihara N, Inoue Y, Iwai T, Umeda M, Huang Y, Ishikawa I. Detection and localization of periodontopathic bacteria in abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. 2004 Nov;28(5):553–8.10.1016/j.ejvs.2004.08.010Search in Google Scholar PubMed
76 Orekhov AN, Nikiforov NN, Ivanova EA, Sobenin IA. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: focus on Atherosclerosis. J Clin Med. 2020 Apr;9(4):978.10.3390/jcm9040978Search in Google Scholar PubMed PubMed Central
77 Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA. 2014 Oct;111(43):15514–9.10.1073/pnas.1414859111Search in Google Scholar PubMed PubMed Central
78 Caruso C, Buffa S, Candore G, Colonna-Romano G, Dunn-Walters D, Kipling D, et al. Mechanisms of immunosenescence. Immun Ageing. 2009 Jul;6(1):10.10.1186/1742-4933-6-10Search in Google Scholar PubMed PubMed Central
79 Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014 Jun;69 Suppl 1:S4–9.10.1093/gerona/glu057Search in Google Scholar PubMed
80 McGuire PJ. Mitochondrial Dysfunction and the Aging Immune System. Biology (Basel). 2019 May;8(2):E26.10.3390/biology8020026Search in Google Scholar PubMed PubMed Central
81 Folsom AR, Yao L, Alonso A, Lutsey PL, Missov E, Lederle FA, et al. Circulating Biomarkers and Abdominal Aortic Aneurysm Incidence: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2015 Aug;132(7):578–85.10.1161/CIRCULATIONAHA.115.016537Search in Google Scholar PubMed PubMed Central
82 Zhang J, Ji Y, Lu Y, Fu R, Xu M, Liu X, et al. Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T > C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum Mol Genet. 2018 01;27(11):1999–2011.10.1093/hmg/ddy107Search in Google Scholar PubMed
83 Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008 Apr;40(4):392–4.10.1038/ng.95Search in Google Scholar PubMed
84 Nicholls TJ, Minczuk M. In D-loop: 40 years of mitochondrial 7S DNA. Exp Gerontol. 2014 Aug;56:175–81.10.1016/j.exger.2014.03.027Search in Google Scholar PubMed
85 Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet. 2009 Mar;18(6):1028–36.10.1093/hmg/ddn437Search in Google Scholar PubMed PubMed Central
86 Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79(1):683–706.10.1146/annurev-biochem-060408-093701Search in Google Scholar PubMed
87 Li H, Shen L, Hu P, Huang R, Cao Y, Deng J, et al. Aging-associated mitochondrial DNA mutations alter oxidative phosphorylation machinery and cause mitochondrial dysfunctions. Biochim Biophys Acta Mol Basis Dis. 2017 Sep;1863(9):2266–73.10.1016/j.bbadis.2017.05.022Search in Google Scholar PubMed
88 Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010 Jun;51(5):440–50.10.1002/em.20586Search in Google Scholar PubMed
89 Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci USA. 2001 Mar;98(7):4038–43.10.1073/pnas.061038798Search in Google Scholar PubMed PubMed Central
90 Weakley SM, Jiang J, Kougias P, Lin PH, Yao Q, Brunicardi FC, et al. Role of somatic mutations in vascular disease formation. Expert Rev Mol Diagn. 2010 Mar;10(2):173–85.10.1586/erm.10.1Search in Google Scholar PubMed PubMed Central
91 Sazonova MA, Sinyov VV, Barinova VA, Ryzhkova AI, Zhelankin AV, Postnov AY, et al. Mosaicism of Mitochondrial Genetic Variation in Atherosclerotic Lesions of the Human Aorta [Internet]. Vol. 2015, BioMed Research International. Hindawi; 2015 [cited 2020 Nov 19]. p. e825468. Available from: https://www.hindawi.com/journals/bmri/2015/825468/10.1155/2015/825468Search in Google Scholar PubMed PubMed Central
92 Matsunaga H, Tanaka Y, Tanaka M, Gong JS, Zhang J, Nomiyama T, et al. Antiatherogenic mitochondrial genotype in patients with type 2 diabetes. Diabetes Care. 2001 Mar;24(3):500–3.10.2337/diacare.24.3.500Search in Google Scholar PubMed
93 Mukae S, Aoki S, Itoh S, Sato R, Nishio K, Iwata T, et al. Mitochondrial 5178A/C genotype is associated with acute myocardial infarction. Circ J. 2003 Jan;67(1):16–20.10.1253/circj.67.16Search in Google Scholar PubMed
94 Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012 Oct;12(10):685–98.10.1038/nrc3365Search in Google Scholar PubMed PubMed Central
95 Park CB, Larsson NG. Mitochondrial DNA mutations in disease and aging. J Cell Biol. 2011 May;193(5):809–18.10.1083/jcb.201010024Search in Google Scholar PubMed PubMed Central
96 Krishnan KJ, Greaves LC, Reeve AK, Turnbull D. The ageing mitochondrial genome. Nucleic Acids Res. 2007;35(22):7399–405.10.1093/nar/gkm635Search in Google Scholar PubMed PubMed Central
97 Khrapko K, Turnbull D. Mitochondrial DNA mutations in aging. Prog Mol Biol Transl Sci. 2014;127:29–62.10.1016/B978-0-12-394625-6.00002-7Search in Google Scholar PubMed
98 Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9(9):e1003794.10.1371/journal.pgen.1003794Search in Google Scholar PubMed PubMed Central
99 Lagouge M, Larsson NG. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med. 2013 Jun;273(6):529–43.10.1111/joim.12055Search in Google Scholar PubMed PubMed Central
100 Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging. Free Radic Biol Med. 2015 Aug;85:250–8.10.1016/j.freeradbiomed.2015.05.005Search in Google Scholar PubMed PubMed Central
101 Dasouki M, Markova D, Garola R, Sasaki T, Charbonneau NL, Sakai LY, et al. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007 Nov;143A(22):2635–41.10.1002/ajmg.a.31980Search in Google Scholar PubMed
102 Hanada K, Vermeij M, Garinis GA, de Waard MC, Kunen MG, Myers L, et al. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ Res. 2007 Mar;100(5):738–46.10.1161/01.RES.0000260181.19449.95Search in Google Scholar PubMed
103 Davis EC. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest. 1993 Jan;68(1):89–99.Search in Google Scholar
104 van der Pluijm I, Burger J, van Heijningen PM, IJpma A, van Vliet N, Milanese C, et al. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc Res. 2018 Nov;114(13):1776–93.10.1093/cvr/cvy150Search in Google Scholar PubMed PubMed Central
105 Burger J, van Vliet N, van Heijningen P, Kumra H, Kremers GJ, Alves M, et al. Fibulin-4 deficiency differentially affects cytoskeleton structure and dynamics as well as TGFβ signaling. Cell Signal. 2019 Jun;58:65–78.10.1016/j.cellsig.2019.02.008Search in Google Scholar PubMed
106 Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006 Mar;116(3):615–22.10.1172/JCI27794Search in Google Scholar PubMed PubMed Central
107 Tiano JP, Springer DA, Rane SG. SMAD3 negatively regulates serum irisin and skeletal muscle FNDC5 and peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) during exercise. J Biol Chem. 2015 May;290(18):11431.10.1074/jbc.A114.617399Search in Google Scholar PubMed PubMed Central
108 Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. 2011 Jul;14(1):67–79.10.1016/j.cmet.2011.04.013Search in Google Scholar PubMed PubMed Central
109 Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, et al. PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis [Internet]. PLoS Biol. 2005 Apr;3(4):e101. [cited 2020 Nov 11] Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1064854/10.1371/journal.pbio.0030101Search in Google Scholar PubMed PubMed Central
110 Gabrielson M, Vorkapic E, Folkesson M, Welander M, Matussek A, Dimberg J, et al. Altered PPARγ Coactivator-1 Alpha Expression in Abdominal Aortic Aneurysm: Possible Effects on Mitochondrial Biogenesis. J Vasc Res. 2016;53(1–2):17–26.10.1159/000446653Search in Google Scholar PubMed
111 Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008 Nov;8(5):347–58.10.1016/j.cmet.2008.08.017Search in Google Scholar PubMed
112 Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, et al. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab. 2011 Nov;14(5):623–34.10.1016/j.cmet.2011.09.013Search in Google Scholar PubMed PubMed Central
113 Ramnath NW, Hawinkels LJ, van Heijningen PM, te Riet L, Paauwe M, Vermeij M, et al. Fibulin-4 deficiency increases TGF-β signalling in aortic smooth muscle cells due to elevated TGF-β2 levels. Sci Rep. 2015 Nov;5(1):16872.10.1038/srep16872Search in Google Scholar PubMed PubMed Central
114 Gao F, Chambon P, Offermanns S, Tellides G, Kong W, Zhang X, et al. Disruption of TGF-β signaling in smooth muscle cell prevents elastase-induced abdominal aortic aneurysm. Biochem Biophys Res Commun. 2014 Nov;454(1):137–43.10.1016/j.bbrc.2014.10.053Search in Google Scholar PubMed
115 Yu E, Foote K, Bennett M. Mitochondrial function in thoracic aortic aneurysms. Cardiovasc Res. 2018 01;114(13):1696–8. https://doi.org/10.1093/cvr/cvy180.10.1093/cvr/cvy180Search in Google Scholar PubMed PubMed Central
116 Kent KC, Zwolak RM, Egorova NN, Riles TS, Manganaro A, Moskowitz AJ, et al. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg. 2010 Sep;52(3):539–48.10.1016/j.jvs.2010.05.090Search in Google Scholar PubMed
117 Wahlgren CM, Larsson E, Magnusson PK, Hultgren R, Swedenborg J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J Vasc Surg. 2010 Jan;51(1):3–7.10.1016/j.jvs.2009.08.036Search in Google Scholar PubMed
118 Sakalihasan N, Defraigne JO, Kerstenne MA, Cheramy-Bien JP, Smelser DT, Tromp G, et al. Family members of patients with abdominal aortic aneurysms are at increased risk for aneurysms: analysis of 618 probands and their families from the Liège AAA Family Study. Ann Vasc Surg. 2014 May;28(4):787–97.10.1016/j.avsg.2013.11.005Search in Google Scholar PubMed PubMed Central
119 Ogata T, MacKean GL, Cole CW, Arthur C, Andreou P, Tromp G, et al. The lifetime prevalence of abdominal aortic aneurysms among siblings of aneurysm patients is eightfold higher than among siblings of spouses: an analysis of 187 aneurysm families in Nova Scotia, Canada. J Vasc Surg. 2005 Nov;42(5):891–7.10.1016/j.jvs.2005.08.002Search in Google Scholar PubMed PubMed Central
120 Shibamura H, Olson JM, van Vlijmen-Van Keulen C, Buxbaum SG, Dudek DM, Tromp G, et al. Genome scan for familial abdominal aortic aneurysm using sex and family history as covariates suggests genetic heterogeneity and identifies linkage to chromosome 19q13. Circulation. 2004 May;109(17):2103–8.10.1161/01.CIR.0000127857.77161.A1Search in Google Scholar PubMed
121 Van Vlijmen-Van Keulen CJ, Rauwerda JA, Pals G. Genome-wide linkage in three Dutch families maps a locus for abdominal aortic aneurysms to chromosome 19q13.3. Eur J Vasc Endovasc Surg. 2005 Jul;30(1):29–35.10.1016/j.ejvs.2004.12.029Search in Google Scholar PubMed
122 Hinterseher I, Tromp G, Kuivaniemi H. Genes and abdominal aortic aneurysm. Ann Vasc Surg. 2011 Apr;25(3):388–412.10.1016/j.avsg.2010.09.004Search in Google Scholar PubMed PubMed Central
123 Jones GT, Tromp G, Kuivaniemi H, Gretarsdottir S, Baas AF, Giusti B, et al. Meta-Analysis of Genome-Wide Association Studies for Abdominal Aortic Aneurysm Identifies Four New Disease-Specific Risk Loci. Circ Res. 2017 Jan;120(2):341–53.10.1161/CIRCRESAHA.116.308765Search in Google Scholar PubMed PubMed Central
124 Frontini MJ, Nong Z, Gros R, Drangova M, O’Neil C, Rahman MN, et al. Fibroblast growth factor 9 delivery during angiogenesis produces durable, vasoresponsive microvessels wrapped by smooth muscle cells. Nat Biotechnol. 2011 May;29(5):421–7.10.1038/nbt.1845Search in Google Scholar PubMed
125 Klarin D, Verma SS, Judy R, Dikilitas O, Wolford BN, Paranjpe I, et al.; Veterans Affairs Million Veteran Program†. Genetic Architecture of Abdominal Aortic Aneurysm in the Million Veteran Program. Circulation. 2020 Oct;142(17):1633–46.10.1161/CIRCULATIONAHA.120.047544Search in Google Scholar PubMed PubMed Central
126 Marsman J, Gimenez G, Day RC, Horsfield JA, Jones GT. A non-coding genetic variant associated with abdominal aortic aneurysm alters ERG gene regulation. Hum Mol Genet. 2020 Mar;29(4):554–65.10.1093/hmg/ddz256Search in Google Scholar PubMed PubMed Central
127 Shah AV, Birdsey GM, Randi AM. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul Pharmacol. 2016 Nov;86:3–13.10.1016/j.vph.2016.05.003Search in Google Scholar PubMed PubMed Central
128 Kaneko H, Anzai T, Takahashi T, Kohno T, Shimoda M, Sasaki A, et al. Role of vascular endothelial growth factor-A in development of abdominal aortic aneurysm. Cardiovasc Res. 2011 Jul;91(2):358–67.10.1093/cvr/cvr080Search in Google Scholar PubMed
129 Baas AF, Medic J, van ’t Slot R, de Kovel CG, Zhernakova A, Geelkerken RH, et al. Association of the TGF-β receptor genes with abdominal aortic aneurysm. Eur J Hum Genet. 2010 Feb;18(2):240–4.10.1038/ejhg.2009.141Search in Google Scholar PubMed PubMed Central
130 Gretarsdottir S, Baas AF, Thorleifsson G, Holm H, den Heijer M, de Vries JP, et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010 Aug;42(8):692–7.10.1038/ng.622Search in Google Scholar PubMed PubMed Central
131 Elmore JR, Obmann MA, Kuivaniemi H, Tromp G, Gerhard GS, Franklin DP, et al. Identification of a genetic variant associated with abdominal aortic aneurysms on chromosome 3p12.3 by genome wide association. J Vasc Surg. 2009 Jun;49(6):1525–31.10.1016/j.jvs.2009.01.041Search in Google Scholar PubMed
132 Saracini C, Bolli P, Sticchi E, Pratesi G, Pulli R, Sofi F, et al. Polymorphisms of genes involved in extracellular matrix remodeling and abdominal aortic aneurysm. J Vasc Surg. 2012 Jan;55(1):171–179.e2.10.1016/j.jvs.2011.07.051Search in Google Scholar PubMed
133 Wang F, Quan QQ, Zhang CL, Li YB, Jiang TB. Association between polymorphisms in the interleukin-10 gene and risk of abdominal aortic aneurysm. Genet Mol Res. 2015 Dec;14(4):17599–604.10.4238/2015.December.21.32Search in Google Scholar PubMed
134 Helgadottir A, Thorleifsson G, Magnusson KP, Grétarsdottir S, Steinthorsdottir V, Manolescu A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008 Feb;40(2):217–24.10.1038/ng.72Search in Google Scholar PubMed
135 Bown MJ. Genomic insights into abdominal aortic aneurysms. Ann R Coll Surg Engl. 2014 Sep;96(6):405–14.10.1308/003588414X13946184901560Search in Google Scholar PubMed PubMed Central
136 Galora S, Saracini C, Pratesi G, Sticchi E, Pulli R, Pratesi C, et al. Association of rs1466535 LRP1 but not rs3019885 SLC30A8 and rs6674171 TDRD10 gene polymorphisms with abdominal aortic aneurysm in Italian patients. J Vasc Surg. 2015 Mar;61(3):787–92.10.1016/j.jvs.2013.10.090Search in Google Scholar PubMed
137 Jones GT, Bown MJ, Gretarsdottir S, Romaine SP, Helgadottir A, Yu G, et al. A sequence variant associated with sortilin-1 (SORT1) on 1p13.3 is independently associated with abdominal aortic aneurysm. Hum Mol Genet. 2013 Jul;22(14):2941–7.10.1093/hmg/ddt141Search in Google Scholar PubMed PubMed Central
138 Bradley DT, Hughes AE, Badger SA, Jones GT, Harrison SC, Wright BJ, et al. A variant in LDLR is associated with abdominal aortic aneurysm. Circ Cardiovasc Genet. 2013 Oct;6(5):498–504.10.1161/CIRCGENETICS.113.000165Search in Google Scholar PubMed
139 Jones GT, Thompson AR, van Bockxmeer FM, Hafez H, Cooper JA, Golledge J, et al. Angiotensin II type 1 receptor 1166C polymorphism is associated with abdominal aortic aneurysm in three independent cohorts. Arterioscler Thromb Vasc Biol. 2008 Apr;28(4):764–70.10.1161/ATVBAHA.107.155564Search in Google Scholar PubMed PubMed Central
140 McColgan P, Peck GE, Greenhalgh RM, Sharma P. The genetics of abdominal aortic aneurysms: a comprehensive meta-analysis involving eight candidate genes in over 16,700 patients. Int Surg. 2009 Oct–Dec;94(4):350–8.Search in Google Scholar
© 2021 Volha I. Summerhill et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- In Our Image: The Ethics of CRISPR Genome Editing
- Doxorubicin loaded magnetism nanoparticles based on cyclodextrin dendritic-graphene oxide inhibited MCF-7 cell proliferation
- The Effects of Oral Liposomal Glutathione and In Vitro Everolimus in Altering the Immune Responses against Mycobacterium bovis BCG Strain in Individuals with Type 2 Diabetes
- Myosteatosis in NAFLD patients correlates with plasma Cathepsin D
- Polyphenols from Passiflora ligularis Regulate Inflammatory Markers and Weight Gain
- Everolimus-induced effector mechanism in macrophages and survivability of Erdman, CDC1551 and HN878 strains of Mycobacterium tuberculosis infection
- Pathophysiological Aspects of the Development of Abdominal Aortic Aneurysm with a Special Focus on Mitochondrial Dysfunction and Genetic Associations
- Size-based Degradation of Therapeutic Proteins - Mechanisms, Modelling and Control
- Incidence and characterisation of Bacillus cereus bacteriophages from Thua Nao, a Thai fermented soybean product
- Bcl2 negatively regulates Protective Immune Responses During Mycobacterial Infection
- Insulin resistance is positively associated with plasma cathepsin D activity in NAFLD patients
- 3D host cell and pathogen-based bioassay development for testing anti-tuberculosis (TB) drug response and modeling immunodeficiency
- Universal scanning-free initiation of eukaryote protein translation–a new normal
- Characterization of Differentially Expressed miRNAs by CXCL12/SDF-1 in Human Bone Marrow Stromal Cells
- Combination therapy using TGF-β1 and STI-571 can induce apoptosis in BCR-ABL oncogene-expressing cells
- SIT1 transporter as a potential novel target in treatment of COVID-19
- Primary Erlotinib Resistance in a Patient with Non-Small Cell Lung Cancer Carrying Simultaneous Compound EGFR L718A, Q849H, and L858R Mutations
- Plant growth-promoting properties of bacterial endophytes isolated from roots of Thymus vulgaris L. and investigate their role as biofertilizers to enhance the essential oil contents
- Implication of plant growth-promoting rhizobacteria of Bacillus spp. as biocontrol agents against wilt disease caused by Fusarium oxysporum Schlecht. in Vicia faba L.
- Antibodies targeting enzyme inhibition as potential tools for research and drug development
- Retraction Note
- Retraction of: Alzheimer's and Danish dementia peptides induce cataract and perturb retinal architecture in rats
Articles in the same Issue
- In Our Image: The Ethics of CRISPR Genome Editing
- Doxorubicin loaded magnetism nanoparticles based on cyclodextrin dendritic-graphene oxide inhibited MCF-7 cell proliferation
- The Effects of Oral Liposomal Glutathione and In Vitro Everolimus in Altering the Immune Responses against Mycobacterium bovis BCG Strain in Individuals with Type 2 Diabetes
- Myosteatosis in NAFLD patients correlates with plasma Cathepsin D
- Polyphenols from Passiflora ligularis Regulate Inflammatory Markers and Weight Gain
- Everolimus-induced effector mechanism in macrophages and survivability of Erdman, CDC1551 and HN878 strains of Mycobacterium tuberculosis infection
- Pathophysiological Aspects of the Development of Abdominal Aortic Aneurysm with a Special Focus on Mitochondrial Dysfunction and Genetic Associations
- Size-based Degradation of Therapeutic Proteins - Mechanisms, Modelling and Control
- Incidence and characterisation of Bacillus cereus bacteriophages from Thua Nao, a Thai fermented soybean product
- Bcl2 negatively regulates Protective Immune Responses During Mycobacterial Infection
- Insulin resistance is positively associated with plasma cathepsin D activity in NAFLD patients
- 3D host cell and pathogen-based bioassay development for testing anti-tuberculosis (TB) drug response and modeling immunodeficiency
- Universal scanning-free initiation of eukaryote protein translation–a new normal
- Characterization of Differentially Expressed miRNAs by CXCL12/SDF-1 in Human Bone Marrow Stromal Cells
- Combination therapy using TGF-β1 and STI-571 can induce apoptosis in BCR-ABL oncogene-expressing cells
- SIT1 transporter as a potential novel target in treatment of COVID-19
- Primary Erlotinib Resistance in a Patient with Non-Small Cell Lung Cancer Carrying Simultaneous Compound EGFR L718A, Q849H, and L858R Mutations
- Plant growth-promoting properties of bacterial endophytes isolated from roots of Thymus vulgaris L. and investigate their role as biofertilizers to enhance the essential oil contents
- Implication of plant growth-promoting rhizobacteria of Bacillus spp. as biocontrol agents against wilt disease caused by Fusarium oxysporum Schlecht. in Vicia faba L.
- Antibodies targeting enzyme inhibition as potential tools for research and drug development
- Retraction Note
- Retraction of: Alzheimer's and Danish dementia peptides induce cataract and perturb retinal architecture in rats