Coupling de novo protein folding with subunit exchange into pre-formed oligomeric protein complexes: the ‘heritable template’ hypothesis
-
Michael A. McMurray


Abstract
Despite remarkable advances in synthetic biology, the fact remains that it takes a living cell to make a new living cell. The information encoded in the genome is necessary to direct assembly of all cellular components, but it may not be sufficient. Some components (e.g. mitochondria) cannot be synthesized de novo, and instead require pre-existing templates, creating a fundamental continuity of life: if the template information is ever lost, the genomic code cannot suffice to ensure proper biogenesis. One type of information only incompletely encoded in the genome is the structures of macromolecular assemblies, which emerge from the conformations of the constituent molecules coupled with the ways in which these molecules interact. For many, if not most proteins, gene sequence is not the sole determinant of native conformation, particularly in the crowded cellular milieu. A partial solution to this problem lies in the functions of molecular chaperones, encoded by nearly all cellular genomes. Chaperones effectively restrict the ensemble of conformations sampled by polypeptides, promoting the acquisition of native, functional forms, but multiple proteins have evolved ways to achieve chaperone independence, perhaps by coupling folding with higher-order assembly. Here, I propose the existence of another solution: a novel mechanism of de novo folding in which the folding of specific proteins is templated by pre-folded molecules of a partner protein whose own folding also required similar templating. This hypothesis challenges prevailing paradigms by predicting that, in order to achieve a functional fold, some non-prion proteins require a seed passed down through generations.
Introduction
Even the most minimalist of organisms require an astounding complexity of cellular processes. Proteins, nucleic acids, lipids, ions, and other molecules must interact in highly specific ways to form functional and dynamic complexes. Evolution sculpted the forms adopted by proteins and nucleic acids to optimize these intermolecular associations: some became high-affinity interactions with ultraslow dissociation kinetics, while others maintained looser connections to facilitate dynamics and multiplicity of interaction partners. Here I focus on what I predict is a small subset of protein-protein interactions that are high-affinity but are nonetheless subject to an ‘exchange’ step in which an association is transiently loosened to allow release of a pre-existing partner and switching to a newly synthesized molecule of the same protein.
The tertiary structure of a protein is not a fixed entity dictated directly by the primary sequence. Polypeptides populate an ensemble of conformations, and what fraction of molecules populate which conformations is influenced by a number of factors. Interactions between hydrophobic amino acid side chains, which typically mediate formation of the core of folded proteins wherein solvent molecules are excluded, are intrinsically sensitive to temperature. For nascent polypeptides emerging from the ribosome, C-terminal sequences are unavailable when N-terminal stretches of amino acids begin to fold. Thus, native associations between N- and C-terminal residues are not possible until translation is complete. Finally, and most importantly, with regard to the subject of this piece, interactions between distinct polypeptides promote the population of those conformations that are most compatible with oligomerization, and in many cases the creation of a high-affinity interface involves the exclusion of solvent via association between otherwise exposed hydrophobic residues.
In a complex environment crowded with many other molecules, however, a native oligomerization partner is unlikely to be the first protein encountered by a nascent polypeptide. Particularly when C-terminal sequences are required to bury the hydrophobic core, a nascent polypeptide is inherently vulnerable to non-native associations with other similarly exposed proteins that can form non-functional (even toxic) aggregates, which are often difficult to dissolve and/or destroy. Accordingly, as should be clear from the paragraphs above, proteins destined for high-affinity interactions with a specific partner face considerable challenges during the early stages of maturation. They must remain on the proper pathway toward acquisition of the native conformation, which may ultimately only be achieved in the context of the native oligomer.
Here I describe mechanisms that facilitate, and may even be required for, successful folding and oligomerization by proteins with specific properties. These mechanisms include roles for chaperone proteins in client/substrate protein folding and oligomerization. I also introduce a concept that is, to my knowledge, novel: that successful de novo folding may require a chaperone-mediated templating interaction between a nascent protein and a pre-folded oligomerization partner, even when the partner itself previously required such a templating event for its own de novo folding. The resulting chicken-and-egg scenario presents challenges to the heterologous production of certain kinds of proteins and, more fundamentally, to the field of synthetic biology. Moreover, loss of functional ‘heritable templates’ could underlie diseases whose etiologies remain unknown.
The acquisition of oligomerization- competent conformations by subunits of high-affinity oligomers is not spontaneous
Protein sequence is not the sole determinant of protein structure. An informative illustration of this concept is the inability to reconstitute many simple high-affinity multisubunit complexes from individually synthesized parts. For example, αtubulin cannot adopt the native fold unless it interacts with β tubulin, and vice versa; each protomer requires structural information that its partner provides (1). Moreover, additional factors, in the form of general cytosolic chaperones and tubulin-specific folding cofactors (here considered to be chaperones), are further required to direct the tubulin polypeptides to adopt conformations primed for heterodimerization (2).
One possible explanation for the requirement for chaperones during de novo tubulin folding is that some features of the native tubulin fold (which is highly similar between the α and β subunits) are intrinsically difficult to attain in a spontaneous manner. However, at least one genus of bacteria (Prosthecobacter) obtained, likely via horizontal gene transfer, sequences encoding clear homologs of eukaryotic α and β tubulins, and these bacteria manage to fold their tubulin-like proteins and assemble them into heterodimers without assistance from cofactors (3), (4). Remarkably, each prokaryotic tubulin can even refold successfully in solution following chemical denaturation. High-resolution structures of BtubA and BtubB confirm that these proteins spontaneously attain all the major structural features of their eukaryotic counterparts (3). What purpose, then, does chaperone- and partner-assisted tubulin folding serve for the eukaryotic species that are clearly under strong selection to maintain them?
The answer may be that the ways most eukaryotes evolved to use tubulins require not just a heterodimer but an exceedingly stable one: the BtubA-BtubB heterodimer is significantly lower-affinity than the α-β tubulin heterodimer (3), (4). It would appear that the conformational states acquired by the bacterial proteins are not quite equivalent to those achieved in the context of the exquisitely intimate association between the eukaryotic tubulin pair. Accordingly, it must be easier for a polypeptide to acquire the native BtubA or BtubB fold than it is to acquire the native α or β tubulin fold; no additional folding information needs to be supplied by a chaperone or interaction partner.
Generally speaking, the conformation achieved just prior to the acquisition of the native state is often the most aggregation-prone (5). It may be the case that protein conformations poised for the highest-affinity protein-protein interactions also have the highest propensity for non-native aggregation. Such a relationship would predict that chaperone involvement is particularly important to prevent aggregation during the folding of proteins destined for high-affinity interactions. Certainly, chaperones play an important role in preventing aggregation, a fact easily illustrated by the widespread protein aggregation observed following depletion of GroEL, a nearly ubiquitous prokaryotic chaperone that is essential in Escherichia coli (6). As with the BtubA-BtubB example, some prokaryotes have found solutions to the aggregation problem that do not require GroEL: the Mycoplasma species Ureaplasma urealyticum lacks any gene encoding a homolog of GroEL (7), yet its homologs of many obligate GroEL substrates are able to fold properly when expressed in GroEL-deficient E. coli (6) and even avoid aggregation upon refolding in solution (8). Single amino acid substitutions in the U. urealyticum homolog of the homotetrameric enzyme MetK render the mutant MetK protein both prone to aggregation and dependent on GroEL for proper folding (8). It would be very interesting to know if the affinity of the MetK subunits for each other is increased by these mutations.
One final conclusion can be drawn from the analysis of other structurally similar proteins that differ in their GroEL dependence. Analysis of the folding pathways of E. coli DapA (GroEL-dependent) and Mycoplasma synoviae NanA (GroEL-independent) revealed that, both with and without GroEL assistance, DapA folds first into a native monomer that subsequently oligomerizes into a functional tetramer. By contrast, NanA folding and tetramerization are coupled, and contacts between non-native monomers precede acquisition of the native conformation (9). These studies did not demonstrate that the act of tetramerization promotes folding of the monomers. However, it is tempting to speculate that the need for chaperone assistance was supplanted in NanA partly by the evolution of intersubunit interactions that accelerate on-pathway folding and disfavor off-pathway conformations.
Templated folding
The notion that a partner subunit of an oligomeric protein complex can provide structural information that guides the folding of a nascent polypeptide toward the native state is further supported by examples of other high-affinity heteromers. Independent refolding following denaturation of the α and β subunits of the bacterial luciferase heterodimer results in proteins that cannot properly interact, but refolding of the two subunits together reconstitutes the functional enzyme (10), (11) [and addition of GroEL further improves yield (12)]. Similarly, when septin family proteins, which form high-affinity heteromers in their native eukaryotic hosts, are expressed individually in bacteria, they commonly purify as non-native homodimers (13), (14), (15), (16) or engage in non-native heteromeric interactions when subsequently mixed with other individually purified subunits (17). Upon heterologous co-expression, on the other hand, septins assemble into complexes with the native organization (13), (18). Although it remains to be determined whether the subunit-subunit interactions that guide native folding occur co-translationally or post-translationally, these examples reinforce the idea that primary sequence is often insufficient to direct native folding in cis, but the missing information can in some cases be provided in trans. From a kinetic perspective, for some proteins the lowest-energy state appears to be a non-native one (perhaps an aggregate), and interactions with an oligomerization partner during the folding process avoids a kinetic trap and allows the protein to remain in the higher-energy native conformation required for function.
Release from ‘late-stage’ chaperones is triggered by client protein oligomerization
From the examples introduced above it should be clear that nascent proteins destined for high-affinity protein-protein interactions often require assistance to stay on pathway toward the native conformation and that chaperones and partner proteins can both provide such assistance. In the case of most chaperones, assistance comes primarily in the form of transient shielding or sequestration of exposed hydrophobic patches away from other hydrophobic sequences (in the same or a distinct polypeptide) to prevent non-native associations at early steps in the folding pathway. In the case of the partner protein(s), non-transient contacts that are not limited to burial of hydrophobic interactions provide an endpoint to an otherwise protracted series of bind-and-release chaperone associations. Additionally, a special class of chaperones (broadly defined) may play a conceptually distinct role at a late step in the folding pathway that facilitates the formation of high-affinity protein-protein partnerships.
Chaperonins function as nanocages inside which single client polypeptides fold without interference from other proteins (recently reviewed in Ref. 19). The hydrolysis of adenosine triphosphate (ATP) triggers release of the client, whereupon the client re-binds if it is not yet properly folded. Prior to its implication in protein folding per se, GroEL, the founding member of the chaperonin family, was first found to be required for the assembly of oligmeric complexes of bacteriophage head and tail proteins, and its plant homolog was found to be required for assembly of the multimeric chloroplast enzyme Rubisco (20). In contrast to other classes of chaperones (e.g. Hsp70), in which folding by the client polypeptide occurs in solution in between cycles of chaperone binding, each client binding event by a chaperonin provides the opportunity for the client to make extensive folding progress; unfolded α tubulin can even fold a functional GTP-binding pocket during a single voyage through the chaperonin chamber (21). For GroEL, ATP hydrolysis appears to follow a simple timer mechanism, giving the client protein ~2 s of folding time before it is released and can attempt to oligomerize (22). If an oligomerization-competent conformation has not yet been achieved, the client protein is re-bound by GroEL and provided with another folding opportunity. Similarly, successful acquisition of an oligomerization-competent conformation appears to be a prerequisite for the permanent release of many clients from the chaperonin of the eukaryotic cytosol, called CCT or TRiC (23), (24). An idea emerges that for a client that is a subunit of a multiprotein complex, another subunit that interacts directly with the client (a partner) often provides an interaction interface that promotes the client’s acquisition of the native conformation, i.e. a template. Consequently, a client that cannot fold without a partner remains trapped in futile cycles of release and re-engagement by the chaperone. Chaperonins thus maintain such clients in a quasi-native conformation, protecting them from alternative ‘dead-end’ conformations, until hetero-oligomerization with the partner induces acquisition of the native state and thereby puts an end to the folding process.
A second example of chaperones in this category of late-acting facilitators of oligomerization can be found in the tubulin cofactors. Tubulin polypeptides pass through CCT yet emerge in conformations that are not competent for efficient heterodimerization (21), (25). The cofactors engage the tubulin subunits at this point and promote a series of additional conformational changes, after which the tubulins are in a high-energy state primed for heterodimerization (1). It has been suggested by others (26) that the tubulin cofactors evolved from ancestral proteins that simply bound the primordial tubulin monomers and/or heterodimer, and it is easy from this perspective to consider the cofactors themselves as templates that are required for de novo tubulin folding, albeit via interactions that do not involve the native tubulin dimerization interface. Release from the cofactors into the heterodimer allows the tubulin subunits to achieve the native state only found in the heterodimer context. Mutant tubulins that cannot achieve the early quasi-native states remain trapped in futile cycles of CCT binding, with some degradation (27), (28); mutant tubulins that progress further but cannot reach the primed state remain stuck on the cofactors (29). Crucially, a wild-type tubulin also remains stuck on the co-factors if its partner tubulin is missing (30). Thus, one can imagine scenarios in which a perfectly normal protein is rendered effectively non-functional by the inability of a partner to achieve the oligomerization-competent state.
A final example comes from the mechanism of bacterial pili/fimbria fiber assembly, in which nascent, non-native fiber subunits are engaged by a periplasmic chaperone in a stable chaperone-client complex. In a process called donor strand complementation, the chaperone provides in trans a small amount of structural information, in the form of a single β strand, that is ‘missing’ from the subunit polypeptide; the β strand occupies a hydrophobic groove that, if left exposed, triggers degradation (reviewed in Ref. 31). When the chaperone-fiber-subunit heterodimer arrives at the growing end of the pre-assembled fiber, it encounters a similar chaperone-client heterodimer occupying the end position, and a sequence at the N-terminus of the newest fiber subunit displaces from the previously incorporated fiber subunit the β strand provided by the chaperone associated with that older subunit, liberating that chaperone molecule and incorporating the new chaperone-fiber-subunit heterodimer. Remarkably, if the N-terminal donor β strand from one fiber subunit is simply fused to the C-terminus of another, it occupies the groove in cis and allows the subunit to fold stably, independent of the chaperone (32). Transient donor strand complementation by the chaperone acts as a kinetic trap to prevent the inappropriate assembly of non-functional, low-energy oligomers (33). Moreover, as with the tubulin cofactors, the chaperones acting in pilus fiber assembly maintain their clients in high-energy states, priming them for the oligomerization event that drives acquisition of the native conformation (31).
Subunit exchanges within high-affinity complexes
High-affinity complexes are characterized by ultra-slow kinetics of spontaneous dissociation, and one would predict that once the subunits of a high-affinity complex associate, they will rarely dissociate, and pre-existing subunits should not exchange with newly synthesized subunits. Metaphorically, the constituent molecules have entered into a monogamous relationship in which all future events during their lifetimes should be undertaken as a single functional unit.
However, even high-affinity complexes display evidence of subunit exchange. For example, the same tubulin cofactor complex that first introduces the nascent α and β tubulin polypeptides to each other is also able to mediate dissociation of a pre-formed heterodimer and exchange of an old (previously folded) subunit for a new (nascent) one (34). Histone-specific chaperones similarly mediate exchange of subunits within pre-formed histone complexes (35). In this scenario, the release from late-stage chaperone engagement that is triggered by oligomerization is in fact not so permanent, because the same chaperone can in fact re-intervene in the relationship.
By definition, the subunits within preformed high-affinity complexes are already in the native conformations. Hence, unless the exchange reaction involves considerable unfolding of the constituent subunits, it follows that subunit exchange within such complexes provides opportunities for a different kind of templating event, in which a protein that itself previously required folding information from a partner (e.g. nascent β tubulin interacting for the first time with nascent α tubulin) could store that information and transmit it, via exchange, to a new partner. Extending the metaphor of the monogamous relationship, match-maker becomes home-breaker as an age-matched partner is swapped for a new/improved one. The structural dynamics occurring during exchange within high-affinity hetero-oligmers remain largely unknown. Is extensive unfolding of the pre-existing subunits required, or can at least one of the protein-protein interaction interfaces remain mostly native, and therefore primed for interaction with a new partner without the need for significant refolding? In the absence of evidence to the contrary, I assume for the purposes of this piece that in at least some cases exchange requires negligible refolding.
The heritable template hypothesis
The discussion of established biological phenomena that I presented above is intended to provide the basis for what is, to my knowledge, a novel hypothesis, which I call the heritable template hypothesis. Let us consider two hypothetical partner proteins, A and B, that form an A–B heterodimer (Figure 1). Each possesses a low-energy, non-functional folding state that is the default in the absence of chaperone assistance. For simplicity, I will consider this state to be misfolded, but that is a purely functional assignment. Chaperone assistance promotes folding into a quasi-native, higher-energy conformation. The native A fold (relatively high-energy) is only achieved in the context of the A–B dimer and the same is true of B. Formation of the A–B heterodimer triggers folding and permanent release of A and B from chaperone sequestration; quasi-native A and B remain chaperone-bound. These properties would be shared with α and β tubulin, with the co-factors as the chaperone. Unlike other proteins that have been studied, however, chaperone-bound, quasi-native A cannot heterodimerize with quasi-native, chaperone-bound B (Figure 1). Instead, in order to fold and escape chaperone sequestration, A requires interaction with a fully folded, native molecule of B, and vice versa (Figure 1). Accordingly, the subunits within a preformed A–B heterodimer must be exchangeable: if A never dissociates from B, pre-folded B will never be available to template the folding of new A molecules. It follows that A–B heterodimers must persist and be distributed through cell divisions; otherwise, daughter cells would be unable to fold new molecules of A or B. We define the A–B partner pair as a heritable template.
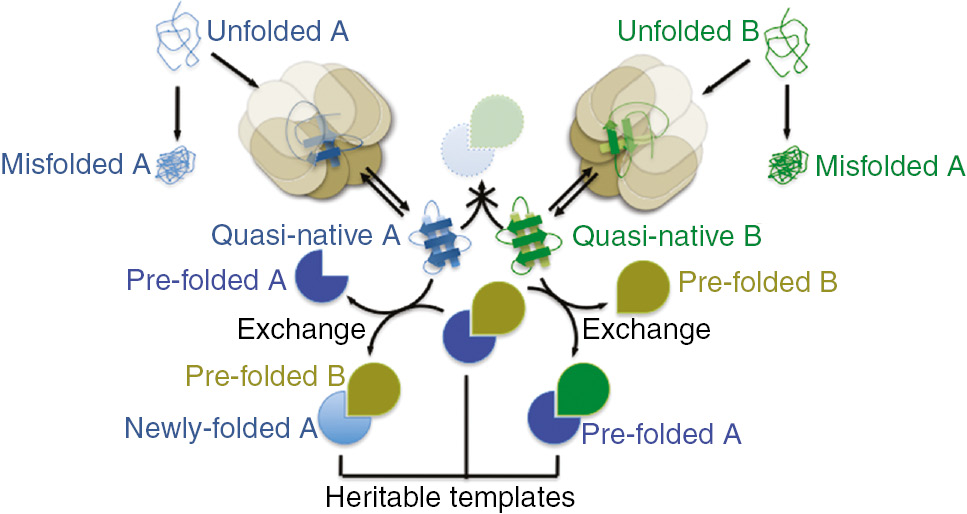
An illustration of a heterodimeric example of the heritable template hypothesis.
For two proteins, A and B, four distinct conformational states are illustrated in the context of a simplified folding pathway. Nascent polypeptides are ‘unfolded’, and without the assistance of a chaperone (beige octameric barrel) tend to populate a low-energy conformational state that is non-functional and thus considered ‘misfolded’. Chaperone engagement promotes a ‘quasi-native’ conformation that shares many structural features with the native state (Pacman shape for A, teardrop shape for B), but the native state is difficult for two quasi-native molecules to achieve. Quasi-native molecules continue to cycle through rounds of chaperone binding and release. An exchange reaction between a quasi-native protein A or B and a pre-formed, native A–B heterodimer is the only efficient route to the native state. Exchange liberates a pre-folded A or B molecule that may be able to itself template the folding of other quasi-native B or A molecules without requiring exchange, but this ability is not a necessary component of the hypothesis. Exchange produces a native heterodimer in which one subunit is newly folded. All native heterodimers can act as heritable templates if they persist through cell division and/or differentiation.
Importantly, this form of molecular memory provided by heritable templates could actually protect cells from otherwise deleterious effects of mutations. Imagine that, like A and B, two hypothetical proteins C and D template each other’s folding, but, unlike A–B, the C–D heterodimer can assemble de novo from newly synthesized molecules. The D-encoding gene is able to accumulate mutations (*) that prevent spontaneous D* folding, for example by strongly favoring an inactive, low-energy conformation if D* tries to fold in the absence of C. So long as molecules of C are present during de novo D* folding, these mutations are phenotypically silent. If C and D* are (like tubulins) long-lived proteins, then subsequent mutations in the C-encoding gene could also be tolerated, because pre-existing D* molecules template the folding of newly made mutant C* proteins. Folded C* molecules can then template the folding of new D* proteins, and thus the C*–D* dimer persists despite the fact that neither C* nor D* could correctly fold in isolation. C* and D* are, according to our definition, heritable templates, and natural selection was powerless to prevent their evolution.
Some experimental evidence for heritable templating allowing mutant proteins to tolerate otherwise catastrophic folding defects may be found in unexpected behaviors of mutants studied in the laboratory setting. My lab has noticed that certain mutant septin proteins with folding difficulties display a longer-than-expected ability to assemble properly under conditions in which a pre-formed mutant-containing septin complex may be acting as a heritable template (unpublished results). Somewhat similarly, a temperature-sensitive mutant of the prokaryotic tubulin homolog FtsZ in Caulobacter crescentus (EGR142AGA) may retain partial function at low temperatures via templating off of FtsZ polymers initially produced by wild-type FtsZ that was depleted from the cells at the start of the experiment (36). It is unsurprising that few examples exist, as one would predict that mutants for which de novo folding defects are masked by heritable templating would be almost always overlooked.
Alternatively, imagine that in most organisms the ability of proteins C and D to fold into conformations that are capable of de novo heterodimerization requires the action of a series of chaperones, but that exchange of subunits within pre-assembled C–D heterodimers is either spontaneous or involves a distinct chaperone. No heritable templates are required for C–D biogenesis in most organisms. On the other hand, if during evolution mutations arose not in C or D but in an early acting D-folding chaperone, then in some organisms de novo C–D biogenesis becomes impossible, yet these organisms persist thanks to the existence of heritable C–D templates first assembled in the ‘chicken’ that produced a mutant ‘egg’. According to this logic, I believe the existence of heritable folding templates may, in fact, be an evolutionary inevitability.
It is worth noting that in the simplest hypothetical example of heritable templating, I assume that functional folding of the participant proteins fails entirely in the absence of a pre-folded partner. In fact, even a slight reduction in the efficiency of folding could, for proteins whose high-efficiency folding is of crucial importance to the cell, impose a fitness cost, particularly under stressful conditions (e.g. heat shock). Indeed, even a conformation that under normal conditions is very unlikely to be achieved without a template, such as that of classical yeast prions (see below), will arise spontaneously in about one cell in a million (37), so when, regarding spontaneous folding, I say ‘cannot’, the reader should understand ‘almost never under normal circumstances’. I used heterodimers to illustrate the heritable template hypothesis, but the concept also applies to homo-oligomers, although the C*–D* evolutionary argument described above would only hold in organisms in which the protein in question is encoded by more than one genetic locus (e.g. diploids). Finally, from the description I provided above, incorporating a new subunit into the polymer during pilus fiber assembly pathway would seem to preclude efficient de novo pilus assembly without some sort of pre-existing seed. However, a pore-shaped usher factor inserted in the outer membrane first (via kinetic partitioning) incorporates at the future tip of the fiber a distinct adhesin-chaperone heterodimer that alters usher conformation to accept fiber-subunit-chaperone heterodimers and thereby initiate fiber polymerization (38). Thus, the evolution of nucleating factors can drive local assembly to overcome barriers to spontaneous oligomerization.
The catalytic potential of exchange-mediated templating
If the exchange reaction does not involve significant unfolding, then each heterodimer exchange event liberates a pre-folded molecule of the other subunit that may be capable of templating de novo folding of a new partner. By analogy to simple chemical reactions, this phenomenon could initiate a chain reaction of templated folding events including obvious initiation, propagation, and termination events (Figure 2). In this way, the information that is transmitted via the templating reaction is not only never lost but might actually be multiplied. However, chain reaction propagation would require that the liberated, pre-folded subunit persist in its high-energy (free-radical-like) native conformation long enough to engage in a productive encounter with a quasi-native nascent partner. Unless exchange-mediating chaperones are capable of kinetically storing such activated monomers, information multiplication may be limited.

Pre-folded proteins liberated from a native heterodimer via subunit exchange may initiate a templating ‘chain reaction’.
The folding states for the hypothetical A–B heterodimer introduced in Figure 1 are distinguished here by font formatting, with quasi-native molecules in plain font and native molecules in bold. Initiation of the chain reaction is represented by an exchange reaction by which a pre-existing A–B heterodimer templates the native folding of a quasi-native A molecule and liberates a native, pre-existing A molecule. These liberated pre-existing proteins are assumed in this example to retain a very nearly native conformation long enough to allow direct (exchange-independent) templating of the folding of a newly made, quasi-native partner molecule. This step propagates the native state and generates a new native A–B heterodimer capable of templating, via exchange, the native folding of another B molecule. The propagation phase effectively multiplies the amount of native A and B molecules, and is terminated only by heterodimerization between pre-folded, native A and B.
Prions: the archetypal heritable templates?
Notably, the protein folding field already espouses the notion that particular protein conformations are unlikely to occur spontaneously, and instead are templated by molecules whose templating ability was itself a product of prior templating. This is, in essence, the prion hypothesis (39). Prions are proteins that can adopt (at least) two distinct conformations, one soluble and one aggregation-prone. The aggregation-prone (prion) conformation can recruit formerly soluble versions of the protein into the prion form. The prion form arises rarely in cells in which all the pre-existing molecules are in the soluble form, which is favored during spontaneous de novo folding. If, however, prion-form molecules are present in a cell in which a new molecule of the protein is produced, the new protein is very likely to be converted into the prion form. This mechanism for perpetuating the prion form allows it to be maintained through cell divisions, so long as prion-form templates are inherited by each daughter cell. Progeny cells can be cured of prions by various methods that prevent template inheritance (40). Thus, prion propagation requires a heritable template that shares many fundamental properties with the heritable folding templates proposed in my hypothesis.
The critical difference, in my view, between prions and the kinds of heritable templates I discuss here is the nature of the subunit-subunit interactions. In the prion field, this concept is mostly limited to prions with highly specific glutamine/asparagine-rich structural motifs that promote amyloid-like folds, which associate in stereotypical ways (e.g. cross-β sheet) (41). Essentially all prions rely on such domains to direct folding into the prion form and eliminating these sequences abolishes prion propagation. I propose that templated folding is not necessarily limited to any particular structural motif, and merely requires a protein-protein interaction interface involving high-energy conformations that are difficult to achieve spontaneously.
Moreover, the prion form is often considered to be dysfunctional relative to the cellular function of the soluble form. There is some evidence from yeast that prion-induced ‘dysfunction’ can be beneficial, for example by facilitating a kind of functional diversity within a population of cells that might prove advantageous in certain situations, sampling a wider swath of phenotypic space, in effect (42), (43). Additionally, some proteins appear to exploit a prion-like state to propagate signals (41), such as a factor involved in making memories in neurons (44). Overall, however, due to their propensity for aggregation, prions are usually thought to be harmful; in fact, it has been proposed that nearly every protein-aggregate-associated disease (Parkinson’s, Alzheimer’s, etc.) is prion-based (45). By contrast, I envision that non-prion heritable templates are critical to normal, native folding of proteins into conformations that are important for healthy cellular functions. Rather than ‘curing’ a cell and restoring normal function, to lose a heritable template of the sort I envision could easily result in death.
If heritable templates exist, how would we find them?
To my knowledge, there is yet no clear evidence that supports the identification of any non-prion (see below) heritable template in wild-type cells of any species. Indeed, in order to demonstrate heritable templating, a number of non-trivial experimental approaches must be employed and focused on proteins that meet a few basic criteria. For example, pulse-chase analysis could first be used to demonstrate protein half-life longer than a cell division cycle, or else a burst of short-lived complex assembly just prior to cell division. Next, exchange with a pre-formed complex containing a pre-folded partner could be shown using affinity co-purification, size exclusion chromatography, or non-denaturing gel electrophoresis, combined with pulse-chase labeling or pre-existing complexes in which the constituent subunits are modified in other ways that allow discrimination from newly synthesized forms (e.g. addition of distinct epitope tags). If a late-stage chaperone is suspected to mediate the exchange reaction, then a further prediction is that, in the absence of a pre-folded complex, newly made subunit molecules might remain trapped in association with the chaperone, but then be released upon addition of purified, pre-folded complex.
A useful comparison can be made with observations found in the early literature surrounding the discovery of the tubulin-folding chaperones. Purified CCT was found to act on unfolded, purified α or β tubulin to produce quasi-native subunits that are resistant to limited proteolysis, but are not competent to undergo exchange into a pre-existing α–β heterodimer (mediated by the ‘latest-acting’ tubulin cofactor) (25). Only upon addition of the earlier-acting cofactors could α or β tubulin fold into a conformation capable of exchange; in the absence of exchange, the exchange-incompetent protein remained stuck in CTT (25). Because in these experiments only a single tubulin subunit was synthesized, there was no newly synthesized partner protein with which to interact, and exchange-mediated contact with a pre-folded partner was the only route to the native conformation. Therefore, these results included all the hallmarks of a requirement for a heritable template, but they were artifacts of an in vitro system that lacked important components of living cells. Along these same lines, it is also worth noting here that polypeptide refolding following denaturation, and the chaperone roles therein, are in many cases fundamentally different from what occurs in vivo, particularly in the context of the act of ribosomal translation itself (19), (46), (47), (48).
Ultimately, then, the most direct demonstration of a heritable template would involve transient depletion from living cells of all pre-existing molecules of candidate template protein (for example, using an inducible degron system), and detection of clear defects in de novo complex assembly. Ideally, such defects would be non-lethal, allowing a rescue of normal assembly by introduction into the defective cells of purified, pre-folded complex. Analogous experiments have already been performed for prion proteins (49). Notably, chaperones themselves could masquerade as heritable templates by virtue of their folding functions. For example, in a classic chicken-and-egg paradox, de novo folding of the mitochondrial chaperonin Hsp60 requires the function of the Hsp60 chaperonin (50), but we can assume that this requirement reflects chaperone-client interactions between pre-existing and nascent Hsp60 molecules, not the kinds of subunit-subunit interface interactions that pertain to the heritable template hypothesis.
Finally, given the examples I provided above of homologous proteins from different species that either can or cannot fold spontaneously without chaperone assistance, one would predict that a protein capable of spontaneous native folding in one species could have a homolog in another species that requires a heritable template, and thus, the gene encoding the template-requiring homolog might be unable to complement a null mutation of the spontaneously folding homolog. Indeed, a recent study found that less than half of essential budding yeast genes could be functionally replaced by their human counterparts (51). Those authors noted that, according to computer simulations, as protein sequences drift during evolution, selection for interaction between two partner proteins is sufficient to drive retention of interaction between a divergent protein and its ancestral partner (51). However, these simulations assume that each protein folds independently into the interaction-competent conformation. Potential heritable templates may be found by investigating homologous proteins for which cross-species complementation fails despite strong conservation of interaction interfaces.
Expert opinion
In summary, I have attempted in this piece to synthesize from established concepts in protein folding a new hypothesis regarding how the three-dimensional structures of proteins might be propagated via a mechanism that relies on, and may therefore reinforce, the continuity of life. The core concepts are that the native states of many proteins are not the lowest-energy states spontaneously achieved in solution and that contributions by chaperones and partner proteins can act separately or in concert to promote the native state. By simply considering a plausible scenario in which a pre-folded partner protein holds the information that a newly made polypeptide needs in order to achieve the native conformation, I propose a mechanism of folding that, as the genome’s code offers no assistance, is intrinsically vulnerable to failure upon accident or environmental insult, but which could nonetheless persist despite the watchful eye of natural selection. Although here I offer precedents and clues that may help to uncover examples of heritable templates in nature, I have no evidence, direct or indirect, that they in fact exist.
Outlook
However, as the field of synthetic biology develops to the point of being able to synthesize from scratch in the lab more and more of the molecules found in living cells, I predict that in 5–10 years we will know if indeed there are fundamental barriers that prevent the initiation of life from purely synthetic materials. Of course, from my perspective, the information present in a living cell and missing in biomaterials assembled from simple building blocks is not life per se, but structural complexity, which in principle can be extracted from materials that are not currently (but once were) alive. Hence, in a more modest sense, my hypothesis merely proposes that certain molecular assemblies cannot be synthesized from building blocks below a certain threshold of structural complexity, i.e. quaternary structure.
As the prion field expands, it may naturally encompass the heritable template hypothesis, if prion-like phenomena are observed for molecules that lack the structural properties that currently define the widely accepted prion definition. It is my hope that this piece may encourage researchers to look for such phenomena in places they might not otherwise have considered.
Highlights
High-affinity protein-protein interactions often involve protein partners in high-energy conformations that are not the default during spontaneous folding and are only achieved in the context of a native oligomer.
Some proteins require assistance from molecular chaperones to achieve the native fold, whereas others, even close homologs, fold independently of chaperones, perhaps by exploiting ‘early’ interactions between subunits destined to form stable oligomers.
Interactions between subunits of an oligomer during the folding process (i.e. when folding and assembly are coupled) represent a form of structural templating, in which each partner provides information that the other needs in order to achieve the native fold.
Late-stage chaperones often compete with native oligomerization partners for binding to nascent polypeptides that are approaching the native fold, allowing oligomerization to trigger release from chaperone sequestration.
Despite their high affinity, many very stable protein-protein complexes can undergo exchange, often mediated by the late-stage chaperones that assembled them in the first place.
The heritable template hypothesis proposes that some proteins may only be able to achieve the native conformation via exchange-mediated interaction with a pre-folded partner protein that itself required prior templating for its folding.
Although a requirement for heritable templates seems biologically precarious, in the appropriate circumstances it may have evolved without significant fitness costs.
The subunit exchange reaction in a heritable template scenario could propagate as a chain reaction if the high-energy native conformation is sufficiently long-lived.
Prions are a type of heritable template with specific structural properties and distinct concepts regarding the functionality of the ‘native’ and ‘non-native’ forms.
The experimental approaches used to identify chaperones and prions may allow the identification of natural heritable templates, if applied to the appropriate proteins.
Acknowledgments
The author would like to thank Jeff Moore, Jasper Rine, Jeremy Thorner, and members of the McMurray lab for helpful discussions and an anonymous reviewer for suggesting that I consider pili/fimbria fiber subunit assembly. Funding was provided by the National Institute of General Medical Sciences of the National Institutes of Health under Award R00GM086603 and by the Alzheimer’s Association under Award NIRGD-12-241119. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health. The author declares that there is no conflict of financial or other interest.
List of abbreviations
- CCT
chaperonin containing TCP1 complex
- TRiC
TCP-1 ring complex
References
1. Tian G, Lewis SA, Feierbach B, Stearns T, Rommelaere H, Ampe C, Cowan NJ. Tubulin subunits exist in an activated conformational state generated and maintained by protein cofactors. J Cell Biol 1997; 138: 821–32.10.1083/jcb.138.4.821Search in Google Scholar
2. Lundin VF, Leroux MR, Stirling PC. Quality control of cytoskeletal proteins and human disease. Trends Biochem Sci 2010; 35: 288–97.10.1016/j.tibs.2009.12.007Search in Google Scholar
3. Schlieper D, Oliva MA, Andreu JM, Löwe J. Structure of bacterial tubulin BtubA/B: evidence for horizontal gene transfer. Proc Natl Acad Sci USA 2005; 102: 9170–5.10.1073/pnas.0502859102Search in Google Scholar
4. Martin-Galiano AJ, Oliva MA, Sanz L, Bhattacharyya A, Serna M, Yebenes H, Valpuesta JM, Andreu JM. Bacterial tubulin distinct loop sequences and primitive assembly properties support its origin from a eukaryotic tubulin ancestor. J Biol Chem 2011; 286: 19789–803.10.1074/jbc.M111.230094Search in Google Scholar
5. Landry SJ, Gierasch LM. Polypeptide interactions with molecular chaperones and their relationship to in vivo protein folding. Annu Rev Biophys Biomol Struct 1994; 23: 645–69.10.1146/annurev.bb.23.060194.003241Search in Google Scholar
6. Fujiwara K, Ishihama Y, Nakahigashi K, Soga T, Taguchi H. A systematic survey of in vivo obligate chaperonin-dependent substrates. EMBO J 2010; 29: 1552–64.10.1038/emboj.2010.52Search in Google Scholar
7. Glass JI, Lefkowitz EJ, Glass JS, Heiner CR, Chen EY, Cassell GH. The complete sequence of the mucosal pathogen Ureaplasma urealyticum. Nature 2000;407: 757–62.10.1038/35037619Search in Google Scholar
8. Ishimoto T, Fujiwara K, Niwa T, Taguchi H. Conversion of a chaperonin GroEL-independent protein into an obligate substrate. J Biol Chem 2014; 289: 32073–80.10.1074/jbc.M114.610444Search in Google Scholar
9. Georgescauld F, Popova K, Gupta AJ, Bracher A, Engen JR, Hayer-Hartl M, Hartl FU. GroEL/ES chaperonin modulates the mechanism and accelerates the rate of TIM-barrel domain folding. Cell 2014; 157: 922–34.10.1016/j.cell.2014.03.038Search in Google Scholar
10. Baldwin TO, Ziegler MM, Chaffotte AF, Goldberg ME. Contribution of folding steps involving the individual subunits of bacterial luciferase to the assembly of the active heterodimeric enzyme. J Biol Chem 1993; 268: 10766–72.10.1016/S0021-9258(18)82051-8Search in Google Scholar
11. Clark AC, Sinclair JF, Baldwin TO. Folding of bacterial luciferase involves a non-native heterodimeric intermediate in equilibrium with the native enzyme and the unfolded subunits. J Biol Chem 1993; 268: 10773–9.10.1016/S0021-9258(18)82052-XSearch in Google Scholar
12. Fedorov AN, Baldwin TO. GroE modulates kinetic partitioning of folding intermediates between alternative states to maximize the yield of biologically active protein. J Mol Biol 1997; 268: 712–23.10.1006/jmbi.1997.1007Search in Google Scholar PubMed
13. Sirajuddin M, Farkasovsky M, Hauer F, Kühlmann D, Macara IG, Weyand M, Stark H, Wittinghofer A. Structural insight into filament formation by mammalian septins. Nature 2007; 449: 311–5.10.1038/nature06052Search in Google Scholar PubMed
14. Low C, Macara IG. Structural analysis of septin 2, 6, and 7 complexes. J Biol Chem 2006; 281: 30697–706.10.1074/jbc.M605179200Search in Google Scholar PubMed
15. Macedo JNA, Valadares NF, Marques IA, Ferreira FM, Damalio JCP, Pereira HM, Garratt RC, Araujo AP. The structure and properties of septin 3: a possible missing link in septin filament formation. Biochem J 2013; 450: 95–105.10.1042/BJ20120851Search in Google Scholar PubMed
16. Zeraik AE, Pereira HM, Santos YV, Brandão-Neto J, Spoerner M, Santos MS, Colnago LA, Garratt RC, Araújo AP, DeMarco R. Crystal structure of a Schistosoma mansoni septin reveals the phenomenon of strand slippage in septins dependent on the nature of the bound nucleotide. J Biol Chem 2014; 289: 7799–811.10.1074/jbc.M113.525352Search in Google Scholar PubMed PubMed Central
17. Versele M, Gullbrand B, Shulewitz MJ, Cid VJ, Bahmanyar S, Chen RE, Barth P, Alber T, Thorner J. Protein-protein interactions governing septin heteropentamer assembly and septin filament organization in Saccharomyces cerevisiae. Mol Biol Cell 2004; 15: 4568–83.10.1091/mbc.e04-04-0330Search in Google Scholar PubMed PubMed Central
18. Bertin A, McMurray MA, Grob P, Park S-S, Garcia G, Patanwala I, Ng HL, Alber T, Thorner J, Nogales E. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proc Natl Acad Sci USA 2008; 105: 8274–9.10.1073/pnas.0803330105Search in Google Scholar PubMed PubMed Central
19. Balchin D, Hayer-Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science 2016; 353: aac4354.10.1126/science.aac4354Search in Google Scholar PubMed
20. Hemmingsen SM, Woolford C, van der Vies SM, Tilly K, Dennis DT, Georgopoulos CP, Hendrix RW, Ellis RJ. Homologous plant and bacterial proteins chaperone oligomeric protein assembly. Nature 1988; 333: 330–4.10.1038/333330a0Search in Google Scholar PubMed
21. Tian G, Vainberg IE, Tap WD, Lewis SA, Cowan NJ. Quasi-native chaperonin-bound intermediates in facilitated protein folding. J Biol Chem 1995; 270: 23910–3.10.1074/jbc.270.41.23910Search in Google Scholar
22. Gupta AJ, Haldar S, Miličić G, Hartl FU, Hayer-Hartl M. Active cage mechanism of chaperonin-assisted protein folding demonstrated at single-molecule level. J Mol Biol 2014; 426: 2739–54.10.1016/j.jmb.2014.04.018Search in Google Scholar
23. Yam AY, Xia Y, Lin H-TJ, Burlingame A, Gerstein M, Frydman J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol 2008; 15: 1255–62.10.1038/nsmb.1515Search in Google Scholar
24. Feldman DE, Thulasiraman V, Ferreyra RG, Frydman J. Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol Cell 1999; 4: 1051–61.10.1016/S1097-2765(00)80233-6Search in Google Scholar
25. Gao Y, Vainberg IE, Chow RL, Cowan NJ. Two cofactors and cytoplasmic chaperonin are required for the folding of α- and β-tubulin. Mol Cell Biol 1993; 13: 2478–85.10.1128/MCB.13.4.2478Search in Google Scholar
26. Ludueña RF. A hypothesis on the origin and evolution of tubulin. Int Rev Cell Mol Biol 2013; 302: 41–185.10.1016/B978-0-12-407699-0.00002-9Search in Google Scholar
27. Wang Y, Tian G, Cowan NJ, Cabral F. Mutations affecting beta-tubulin folding and degradation. J Biol Chem 2006; 281: 13628–35.10.1074/jbc.M513730200Search in Google Scholar
28. Tian G, Kong X-P, Jaglin XH, Chelly J, Keays D, Cowan NJ. A pachygyria-causing α-tubulin mutation results in inefficient cycling with CCT and a deficient interaction with TBCB. Mol Biol Cell 2008; 19: 1152–61.10.1091/mbc.e07-09-0861Search in Google Scholar
29. Zabala JC, Fontalba A, Avila J. Tubulin folding is altered by mutations in a putative GTP binding motif. J Cell Sci 1996; 109(Pt 6): 1471–8.10.1242/jcs.109.6.1471Search in Google Scholar
30. Tian G, Huang Y, Rommelaere H, Vandekerckhove J, Ampe C, Cowan NJ. Pathway leading to correctly folded β-tubulin. Cell 1996; 86: 287–96.10.1016/S0092-8674(00)80100-2Search in Google Scholar
31. Sauer FG, Remaut H, Hultgren SJ, Waksman G. Fiber assembly by the chaperone-usher pathway. Biochim Biophys Acta 2004; 1694: 259–67.10.1016/j.bbamcr.2004.02.010Search in Google Scholar PubMed
32. Barnhart MM, Pinkner JS, Soto GE, Sauer FG, Langermann S, Waksman G, Frieden C, Hultgren SJ. PapD-like chaperones provide the missing information for folding of pilin proteins. Proc Natl Acad Sci USA 2000; 97: 7709–14.10.1073/pnas.130183897Search in Google Scholar PubMed PubMed Central
33. Bao R, Liu Y, Savarino SJ, Xia D. Off-Pathway assembly of fimbria subunits is prevented by chaperone CfaA of CFA/I fimbriae from enterotoxigenic E. coli. Mol Microbiol 2016. [Epub ahead of print].10.1111/mmi.13530Search in Google Scholar PubMed PubMed Central
34. Tian G, Bhamidipati A, Cowan NJ, Lewis SA. Tubulin folding cofactors as GTPase-activating proteins. GTP hydrolysis and the assembly of the α/β-tubulin heterodimer. J Biol Chem 1999; 274: 24054–8.10.1074/jbc.274.34.24054Search in Google Scholar PubMed
35. Das C, Tyler JK, Churchill MEA. The histone shuffle: histone chaperones in an energetic dance. Trends Biochem Sci 2010; 35: 476–89.10.1016/j.tibs.2010.04.001Search in Google Scholar PubMed PubMed Central
36. Wang Y, Jones BD, Brun YV. A set of ftsZ mutants blocked at different stages of cell division in Caulobacter. Mol Microbiol 2001; 40: 347–60.10.1046/j.1365-2958.2001.02395.xSearch in Google Scholar PubMed
37. Lancaster AK, Bardill JP, True HL, Masel J. The spontaneous appearance rate of the yeast prion [PSI+] and its implications for the evolution of the evolvability properties of the [PSI+] system. Genetics 2010; 184: 393–400.10.1534/genetics.109.110213Search in Google Scholar PubMed PubMed Central
38. Saulino ET, Thanassi DG, Pinkner JS, Hultgren SJ. Ramifications of kinetic partitioning on usher-mediated pilus biogenesis. EMBO J 1998; 17: 2177–85.10.1093/emboj/17.8.2177Search in Google Scholar PubMed PubMed Central
39. Soto C. Prion hypothesis: the end of the controversy? Trends Biochem Sci 2011; 36: 151–8.10.1016/j.tibs.2010.11.001Search in Google Scholar PubMed PubMed Central
40. Wickner RB, Taylor KL, Edskes HK, Maddelein ML, Moriyama H, Roberts BT. Prions of yeast as heritable amyloidoses. J Struct Biol 2000; 130: 310–22.10.1006/jsbi.2000.4250Search in Google Scholar PubMed
41. Sanders DW, Kaufman SK, Holmes BB, Diamond MI. Prions and protein assemblies that convey biological information in health and disease. Neuron 2016; 89: 433–48.10.1016/j.neuron.2016.01.026Search in Google Scholar PubMed PubMed Central
42. Jarosz DF, Taipale M, Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet 2010; 44: 189–216.10.1146/annurev.genet.40.110405.090412Search in Google Scholar PubMed
43. Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012; 482: 363–8.10.1038/nature10875Search in Google Scholar PubMed PubMed Central
44. Si K, Kandel ER. The role of functional prion-like proteins in the persistence of memory. Cold Spring Harb Perspect Biol 2016; 8: a021774.10.1101/cshperspect.a021774Search in Google Scholar PubMed PubMed Central
45. Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012; 336: 1511–3.10.1126/science.1222951Search in Google Scholar PubMed PubMed Central
46. Frydman J, Hartl FU. Principles of chaperone-assisted protein folding: differences between in vitro and in vivo mechanisms. Science 1996; 272: 1497–502.10.1126/science.272.5267.1497Search in Google Scholar PubMed
47. Ellis RJ, Hartl FU. Protein folding in the cell: competing models of chaperonin function. FASEB J 1996; 10: 20–6.10.1096/fasebj.10.1.8566542Search in Google Scholar PubMed
48. Ellis RJ. The “bio” in biochemistry: protein folding inside and outside the cell. Science 1996; 272: 1448–9.10.1126/science.272.5267.1448Search in Google Scholar PubMed
49. Wang F, Wang X, Yuan C-G, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327: 1132–5.10.1126/science.1183748Search in Google Scholar PubMed PubMed Central
50. Cheng MY, Hartl FU, Horwich AL. The mitochondrial chaperonin hsp60 is required for its own assembly. Nature 1990; 348: 455–8.10.1038/348455a0Search in Google Scholar PubMed
51. Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, Marcotte EM. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015; 348: 921–5.10.1126/science.aaa0769Search in Google Scholar PubMed PubMed Central
©2016 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Reviews
- Coupling de novo protein folding with subunit exchange into pre-formed oligomeric protein complexes: the ‘heritable template’ hypothesis
- Role of non-motile microtubule-associated proteins in virus trafficking
- Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy
- Short Conceptual Overviews
- The microRNA-200 family: still much to discover
- Matrix metalloproteinases: an emerging role in regulation of actin microfilament system
- Unusual structures of CCTG repeats and their participation in repeat expansion
Articles in the same Issue
- Frontmatter
- Reviews
- Coupling de novo protein folding with subunit exchange into pre-formed oligomeric protein complexes: the ‘heritable template’ hypothesis
- Role of non-motile microtubule-associated proteins in virus trafficking
- Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy
- Short Conceptual Overviews
- The microRNA-200 family: still much to discover
- Matrix metalloproteinases: an emerging role in regulation of actin microfilament system
- Unusual structures of CCTG repeats and their participation in repeat expansion