Synthesis of 2,6-dimethoxy-9-phenyl-1H-phenalen-1-one and structural revision of the benzoindenone from Eichhornia crassipes
-
Xiaowan Li
 and
Albert S.C. Chan
and
Albert S.C. Chan

Abstract
The total synthesis of the natural phytoalexin 2,6-dimethoxy-9-phenyl-1H-phenalen-1-one (3) isolated from Eichhornia crassipes was accomplished in seven steps. The synthetic strategy included a Friedel–Crafts acylation and a Jacobsen–Katsuki epoxidation as the key reactions to obtain the target compound. Based on a comparison of the NMR data and a crystal structure of 3, the previously proposed structure of 2,5-dimethoxy-4-phenyl-benzoindenone (2) isolated from the same plant has been revised.
1 Introduction
Phytoalexins are a group of secondary metabolites produced by plants in response to pathogen infections, pest attacks, and abiotic stresses [1], [2]. They exhibit in vitro or in vivo antimicrobial activity against a variety of phytopathogens. Therefore, it is possible to utilize them as templates for synthesizing new pesticides [3]. Phenylphenalenones, which are known as Musaceae phytoalexins [4], are antifungal substances induced in banana (Masa spp.) at the early stage of Mycosphaerella fijensis infection [5]. Recently, Hölscher and co-workers reported that high concentrations of anigorufone 1 (Fig. 1) and other phenylphenalenones in the roots of nematode-infected Musa resulted in resistance to the nematode Radopholus similis. Compound 1 was identified to function as a strong nematocidal compound by incorporating with lipids into droplets throughout the body of the nematode, causing immobilization and eventual death [4]. Phenylphenalenone-type phytoalexins also show biological activities against various other pathogenic organisms [6], [7], [8].
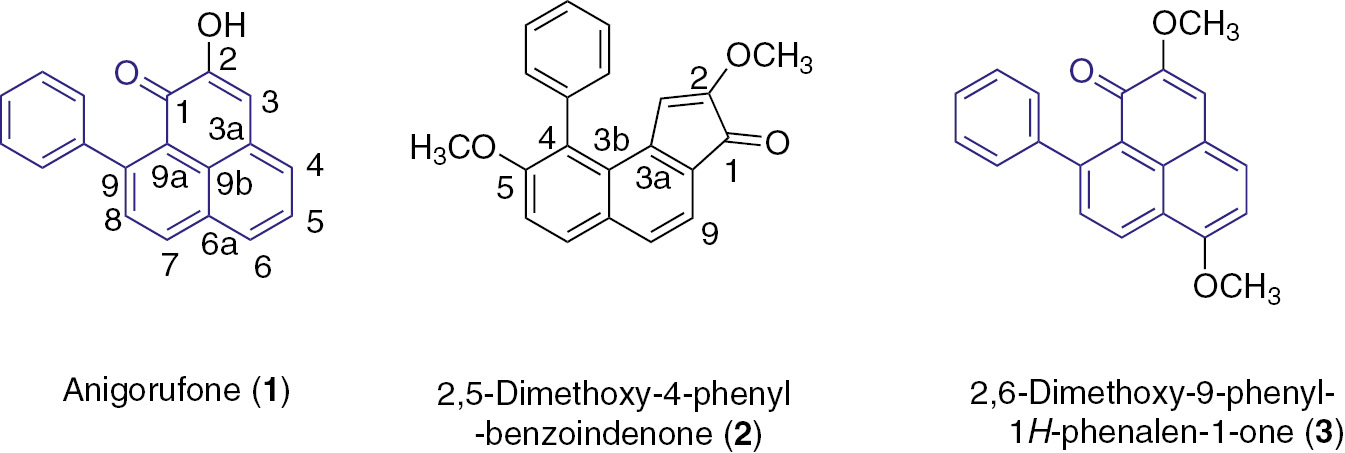
Chemical structures of compounds 1–3.
Eichhornia crassipes (water hyacinth) is a highly problematic, invasive species in freshwater habitats all over the world, but this devastating aquatic weed was found to be a good source of fungicides [9]. 2,5-Dimethoxy-4-phenyl-benzoindenone 2 (Fig. 1), the first benzoindenonic compound to be discovered from E. crassipes, was isolated by Della Greca et al. in 1991, who proposed the structure shown in Fig. 1 [10]. The compound exhibited moderate activity in inhibiting the growth of Candida albicans. In search of potential allelochemical compounds from E. crassipes, compound 3 (Fig. 1) was isolated and its structure determined [11]. Recently, we found that the NMR data of 2 highly matched with those of 3 [11]. The originally assigned structure of 2 was found to be incorrect. Importantly, 3 has the 9-phenyl-phenalenone core structure, which is the same as that of 1. Compounds 3 and 1 occur naturally as phytoalexins in plants, but they are normally present only in very small amounts, which limited their applications in agricultural and clinical uses. Thus, there is the need for an independent synthesis and further structure confirmation of 3.
2 Results and discussion
Here we describe the first total synthesis of 3 and the revised chemical structure of 2 by comparison of its NMR data with that of compound 3. In a retro-synthetic analysis, 3 was considered to be synthesized from 6-methoxy-9-phenyl-1H-phenalen-1-one (4) via a Yang–Finnegan epoxidation and the ring-opening reaction between C-2 and C-3 and a methoxylation reaction. Compound 4 could be synthesized from 6-methoxy-1H-phenalen-1-one (5) by Michael addition with phenylmagnesium bromide and dehydrogenation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). Compound 5 could be prepared from 6-bromo-1H-phenalen-1-one (6) with sodium methoxide through a nucleophilic substitution reaction. Based on our previous work on the synthesis of 2-hydroxy-8-(4-hydroxyphenyl)-1H-phenalen-1-one [12], the intermediate 6 could be synthesized from the aldehyde 7 by a Wittig reaction, saponification, and a Friedel–Crafts reaction (Fig. 2).
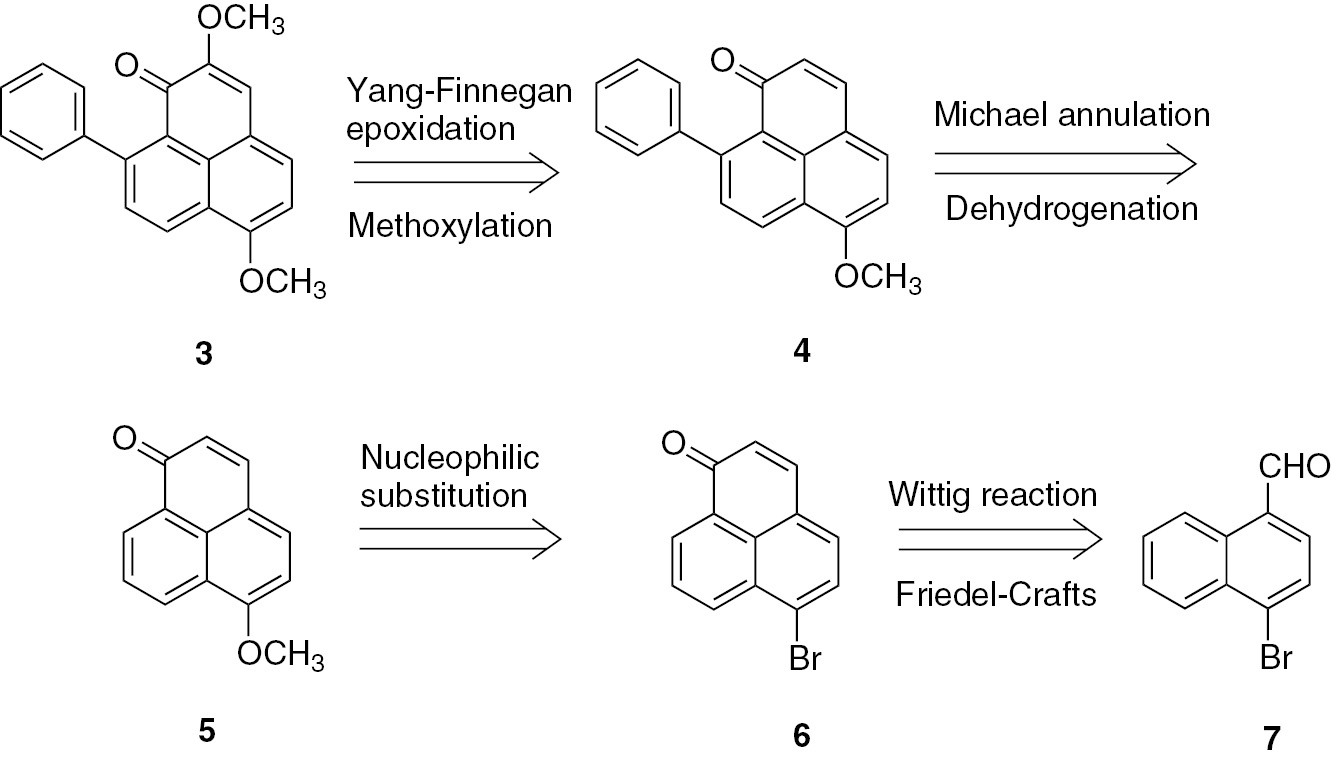
Retro-synthetic analysis toward compound 3.
We thus used commercially available 4-bromo-1-naphthoic acid (7a) as the starting material to synthesize 4-bromo-1-naphthaldehyde (7) by reduction with BH3, followed by oxidation with Dess–Martin periodinane (DMP) in 78% yield. The compound (E)-3-(4-bromonaphthalen-1-yl)acrylic acid (7b) was synthesized by the Wittig reaction of 7 with carbethoxymethyl-triphenylphosphonium bromide (Ph3P+CH2CO2Et Br−), followed by saponification. The acrylic acid 7c was converted to 6 in a one-pot reaction by Friedel–Crafts acylation in 56% yield (Scheme 1).
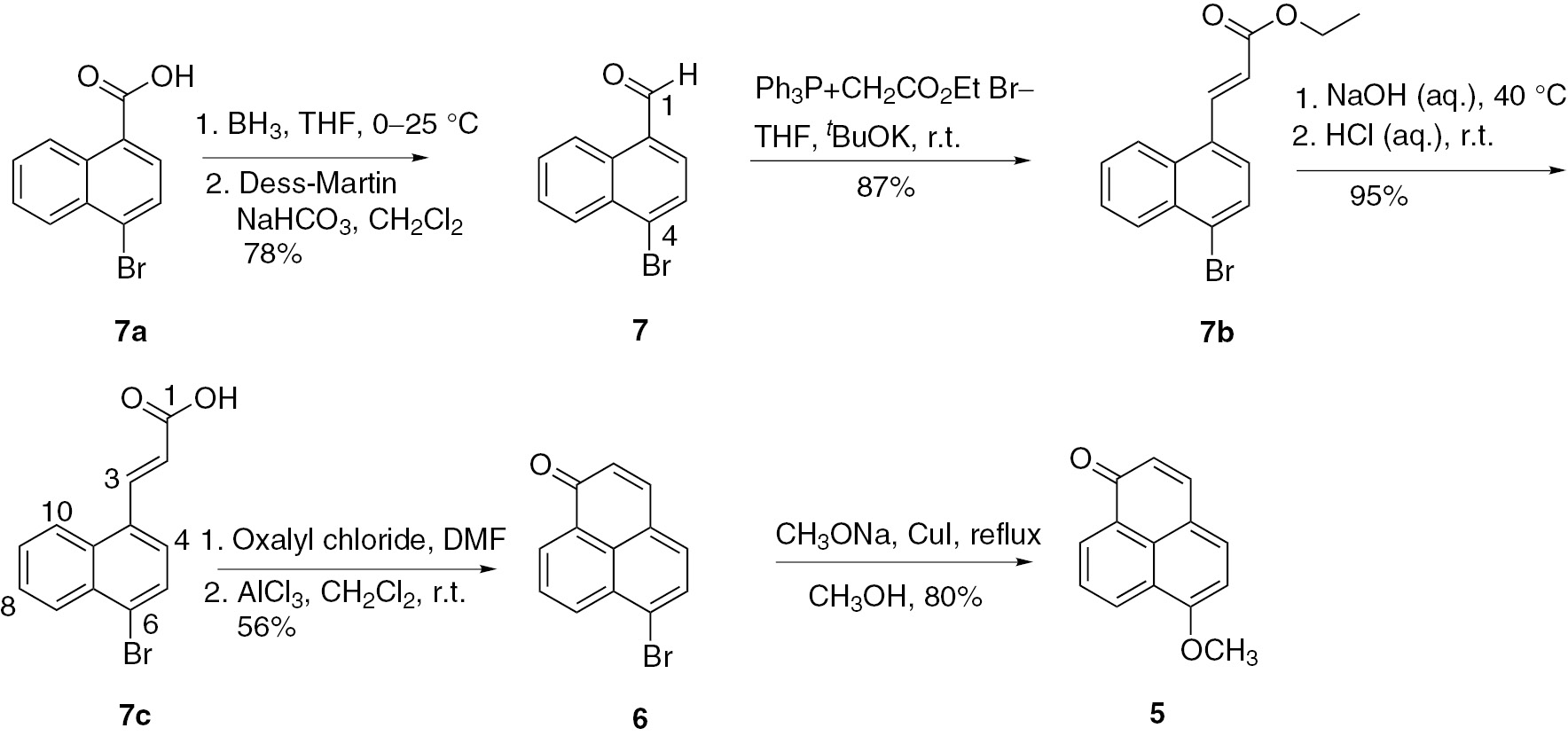
Synthesis of 6-methoxy-1H-phenalen-1-one (5).
Compound 6 was subsequently reacted with sodium methoxide and catalytic amounts of copper iodide through a nucleophilic substitution in methanol to give 5 in 80% yield (Scheme 1), which in turn was followed by Michael annulation with phenylmagnesium bromide and dehydrogenation with DDQ to afford 4 in 65% yield. With sufficient amounts of 4 in hand, the focus was initially on a Yang–Finnegan epoxidation and ring-opening reactions as key processes to give 2-hydroxy-6-methoxy-9-phenyl-1H-phenalen-1-one 8 (Scheme 2).
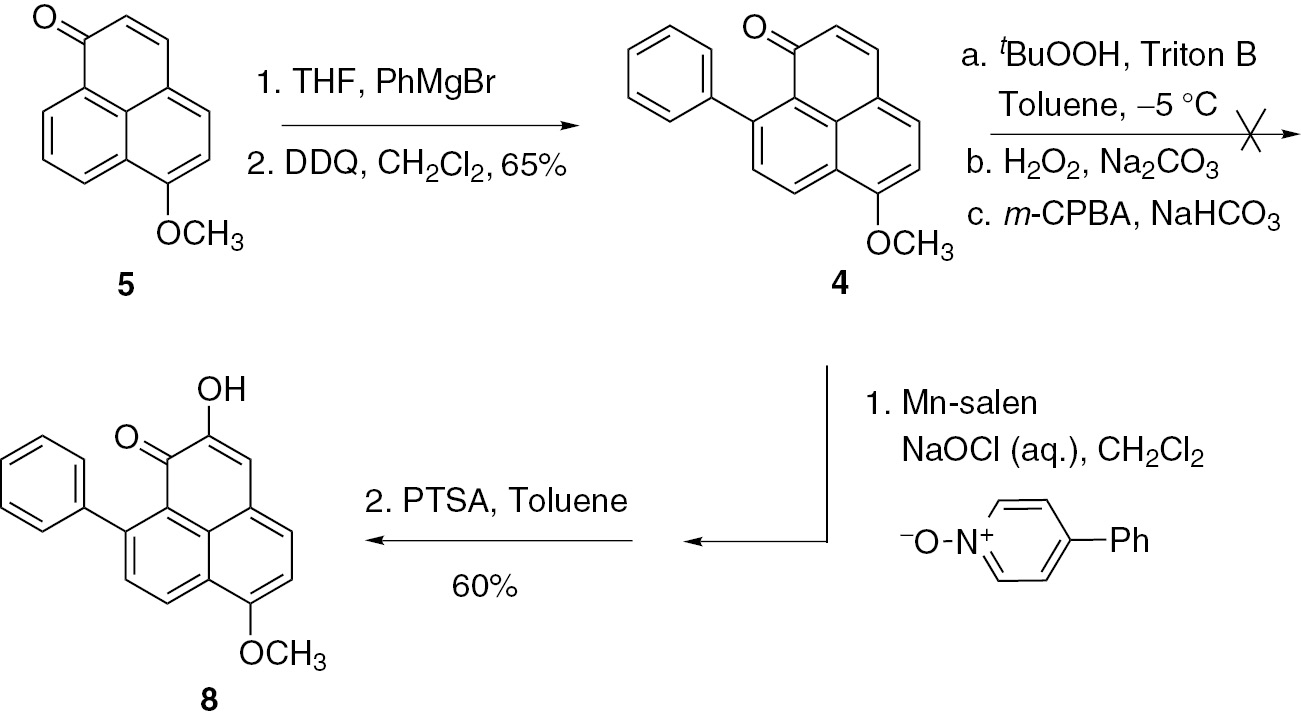
Synthesis of compound 8.
However, we were unable to obtain 8 using this reaction sequence, which previously had been used successfully to prepare hydroxyanigorufone [13]. Similar findings were also reported by Otálvaro et al. in the synthesis of lachnanthocarpone [14]. Subsequently, several other hydroxylation conditions were screened for the formation of 8. Unfortunately, all attempts to effect the epoxidation of 8 under various conditions (H2O2/Na2CO3, m-CPBA/NaHCO3, etc.) failed. Referring to literature procedures [14], finally, 4 was sequentially treated under catalysis with (S,S)-[N,N’-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamine]manganese(II) chloride (Mn-salen) with 4-phenylpyridine N-oxide and sodium hypochlorite (aq.) to promote a Jacobsen–Katsuki epoxidation, followed by a ring-opening process catalyzed by p-toluenesulfonic acid (PTSA) to afford 8 smoothly in moderate yield (60%) (Scheme 2).
Finally, treatment of 8 with potassium carbonate and iodomethane in DMF gave 3 in 90% yield through a nucleophilic substitution reaction (Scheme 3). The physical and spectroscopic data of the synthetic compound 3 were identical to those reported in the literature [11] (Supplementary material).
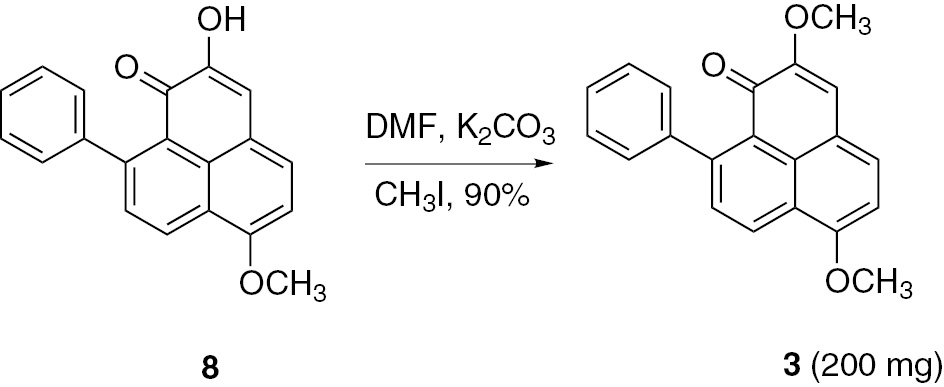
Synthesis of compound 3 from 8.
On comparing the 1H and 13C NMR data of 2 (literature-reported) with those of 3 (synthetic), it was found that all NMR data of 2 were identical to those of 3. The positions of C-4, C-7, C-9, C-10, C-3a, C-3b, C-7a, and C-1′ in 2 were reassigned by 2D NMR (Figs. 3 and 4 , Tables 1 and 2 ). Furthermore, the structure of 3 was firmly established also by X-ray structure determination. Single crystals of the as-synthesized 3 suitable for X-ray diffraction were obtained from methanol/ethyl acetate, and their crystal and molecular structure were determined (Fig. 3; see Experimental for further details), which in turn confirmed that the published structure of 2 was incorrect.
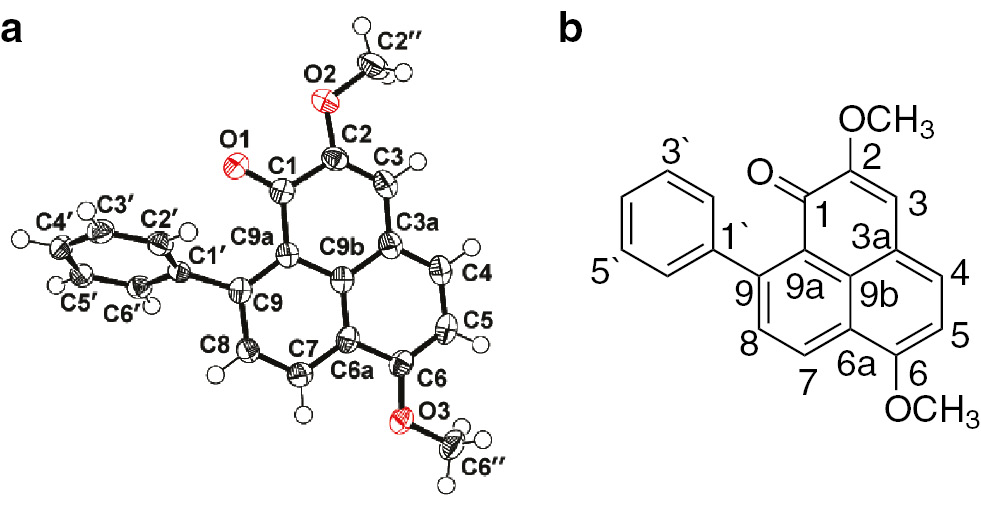
Molecular structure of 3 in the crystal (a) and carbon numbering (b). Displacement ellipsoids are drawn at the 50% probability level, and H atoms as spheres with arbitrary radii.
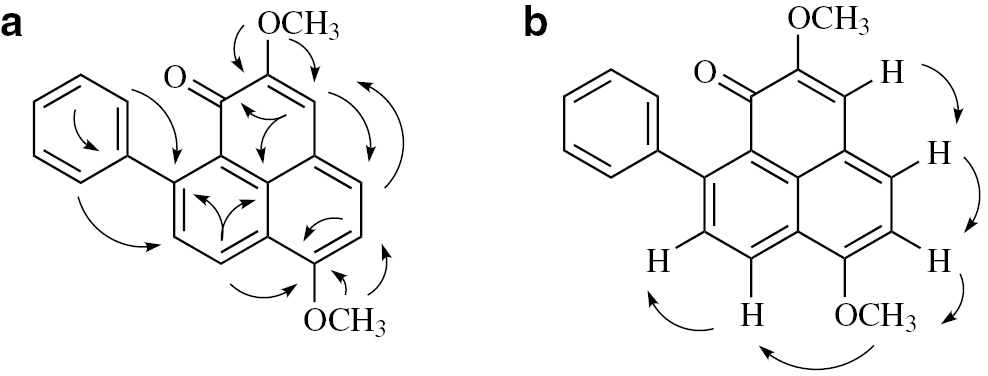
Key HMBC (a) and NOESY (b) correlations of 3.
Comparison of 13C NMR data of synthesized 3 with literature-reported data of 2 and 3 (CDCl3, δ in ppm).
| Position | 3a | 3b | 2c | Δδ1 | Δδ2 |
|---|---|---|---|---|---|
| 1 | 179.8, C | 179.8, C | 179.8, C | ||
| 2 | 152.1, C | 152.1, C | 152.1, C | ||
| 3 | 111.9, CH | 112.0, CH | 112.0, CH | 0.1 | 0.1 |
| 3a (4)d | 121.4, C | 121.5, C | 121.4, C | 0.1 | |
| 4 (7) | 130.6, CH | 130.7, CH | 130.7, CH | 0.1 | 0.1 |
| 5 (6) | 104.6, CH | 104.7, CH | 104.7, CH | 0.1 | 0.1 |
| 6 (5) | 157.4, C | 157.4, C | 157.4, C | ||
| 6a (10) | 124.3, C | 124.3, C | 124.3, C | ||
| 7 (9) | 128.3, CH | 128.3, CH | 128.3, CH | ||
| 8 | 130.6, CH | 130.6, CH | 130.7, CH | ||
| 9 (1′) | 148.4, C | 148.4, C | 148.4, C | ||
| 9a (7a) | 126.3, C | 126.3, C | 126.3, C | ||
| 9b (3b) | 125.7, C | 125.8, C | 125.7, C | 0.1 | |
| 1′(3a) | 142.9, C | 143.0, C | 143.0, C | 0.1 | 0.1 |
| 2′ | 127.8, CH | 127.9, CH | 127.9, CH | 0.1 | 0.1 |
| 3′ | 128.1, CH | 128.1, CH | 128.1, CH | ||
| 4′ | 126.8, CH | 126.9, CH | 126.9, CH | 0.1 | 0.1 |
| 5′ | 128.1, CH | 128.1, CH | 128.1, CH | ||
| 6′ | 127.8, CH | 127.9, CH | 127.9, CH | 0.1 | 0.1 |
| MeO-2 | 55.4, CH3 | 55.4, CH3 | 55.4, CH3 | ||
| MeO-6 (5) | 55.8, CH3 | 55.9, CH3 | 55.9, CH3 | 0.1 | 0.1 |
Comparison of 1H NMR data of synthesized 3 with literature-reported data of 2 and 3 (CDCl3, δ in ppm, J in Hz).
| Position | 3a | 3b | 2c | Δδ1 | Δδ2 |
|---|---|---|---|---|---|
| 3 | 6.82, s | 6.80, s | 6.83, s | –0.02 | 0.01 |
| 4 (7)d | 7.60, d (8.0) | 7.58, d (8.0) | 7.61, d (8.4) | –0.02 | 0.01 |
| 5 (6) | 6.89, d (8.0) | 6.87, d (8.0) | 6.90, d (8.4) | –0.02 | 0.01 |
| 7 (9) | 8.63, d (8.4) | 8.61, d (8.4) | 8.63, d (8.0) | –0.02 | |
| 8 | 7.57, d (8.4) | 7.55, d (8.4) | 7.57, d (8.0) | –0.02 | |
| 2′ | 7.37, d (7.6) | 7.35, d (7.6) | 7.34–7.46, m | –0.02 | |
| 3′ | 7.43, d (7.6) | 7.41, d (7.6) | 7.34–7.46, m | –0.02 | |
| 4′ | 7.39, d (7.6) | 7.38, d (7.6) | 7.34–7.46, m | –0.01 | |
| 5′ | 7.43, d (7.6) | 7.41, d (7.6) | 7.34–7.46, m | –0.02 | |
| 6′ | 7.37, d (7.6) | 7.35, d (7.6) | 7.34–7.46, m | –0.02 | |
| MeO-2 | 3.86, s | 3.84, s | 3.86, s | –0.02 | |
| MeO-6 (5) | 4.08, s | 4.06, s | 4.08, s | –0.02 |
In summary, a concise and efficient strategy was developed for the synthesis of 3 utilizing Friedel–Crafts acylation and Jacobsen–Katsuki epoxidation as the key reactions. The observed overall yield of 3 (seven steps, starting from 7a) was 10.0%. This is the first report of the synthesis and X-ray crystal structure of 3. Based on a comparison of the NMR and crystal structural data of 3, the previously proposed structure of 2 [10] isolated from E. crassipes was revised.
3 Experimental section
3.1 Materials and measurements
Commercially available reagents were used without further purification. All reactions were conducted in oven-dried (T=120°C) or flame-dried glassware under N2 atmosphere and at ambient temperature (T=20–25°C) unless otherwise stated. All solvents were distilled prior to use: THF, benzene, and toluene were distilled from Na/benzophenone, and DMF, Et3N, and CH2Cl2 from CaH2. All non-aqueous reactions were performed under an atmosphere of nitrogen or argon using oven-dried glassware and standard syringe-in-septa techniques. Concentration under reduced pressure was performed at 5–350 mbar. Melting points were measured with a Stanford Research Systems apparatus (OptiMelt-100). 1H NMR spectra were recorded in CDCl3 (unless stated otherwise) on a Bruker Avance 400 or 500 instrument at 400 or 500 MHz (13C NMR spectra at 100 or 125 MHz). Chemical shifts are reported as δ values referenced to TMS. Mass spectra were measured on ABI Q-star Elite mass spectrometer. Thin-layer chromatography (TLC) was carried out using precoated sheets (Qingdao silica gel 60-F250, 0.2 mm) which, after development, were visualized under UV light at 254 nm. Column chromatography was performed on Qingdao silica gel 60 (230–400 mesh ASTM). Yields refer to chromatographically purified compounds.
3.2 General procedure for the synthesis of 3–8
3.2.1 4-Bromo-1-naphthaldehyde (7)
BH3 (40.0 mL, 1 mol L−1) was added to a solution of 7a (5.0 g, 20.0 mmol) in dry THF (50.0 mL) at 0°C, and the mixture stirred at room temperature for 5 h. The reaction was quenched with a saturated aqueous solution of NH4Cl and concentrated under vacuum. The aqueous residue was extracted with EtOAc (3×200.0 mL). The combined organic layers were dried over Na2SO4 and concentrated under vacuum. The residue was used in the next step without purification. Dess–Martin reagent (8.5 g, 20.0 mmol) and NaHCO3 (5.0 g, 60.0 mmol) were added to a solution of the residue in CH2Cl2 (150.0 mL) at 0°C. The reaction mixture was stirred at 0°C for 4 h, H2O (200.0 mL) was added, and the mixture was extracted with CH2Cl2 (3×200.0 mL). The combined organic layer was concentrated under vacuum. The residue was purified by chromatography (EtOAc/hexanes=1:20) to afford the compound 7 (yield 3.6 g, 78%) as a colorless solid; m.p. 79–81°C. 1H NMR (400 MHz, CDCl3): δ=10.36 (s, 1H, –CHO), 9.27 (d, J=8.4 Hz, 1H, Ar-H), 8.35 (d, J=8.4 Hz, 1H, Ar-H), 7.95 (d, J=7.6 Hz, 1H, Ar-H), 7.79 (d, J=7.6 Hz, 1H, Ar-H), 7.69–7.75 (m, 2H, Ar-H). 13C NMR (100 MHz, CDCl3): δ=192.6, 136.1, 132.1, 131.4, 131.3, 130.9, 129.8, 129.3, 128.3, 127.7, 125.1. HRMS ((+)-ESI): m/z=234.9731. (calcd. 234.9753 for C11H7BrO, [M+H]+).
3.2.2 (E)-Ethyl 3-(4-bromonaphthalen-1-yl)acrylate (7b)
To a solution of (carbethoxymethy1)triphenylphosphonium bromide (9.2 g, 15.0 mmol) in 100 mL THF, tBuOK (2.3 g, 15.0 mmol) was added. The solution was stirred for 2 h, compound 7 (3.5 g, 15.0 mmol) was added, and stirring continued for 15 h. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc (2×100.0 mL) and H2O (200.0 mL). The organic layer was concentrated and purified by chromatography (EtOAc/hexanes=1:10) to afford (E)-ethyl 3-(4-bromonaphthalen-1-yl)acrylate as a colorless oil (yield 4.0 g, 87%). 1H NMR (400 MHz, CHCl3): δ=8.33 (d, J=15.6 Hz, 1H), 8.21–8.26 (m, 2H), 7.93 (d, J=8.0 Hz, 1H), 7.83 (d, J=6.8 Hz, 1H), 7.72–7.75 (m, 2H), 6.63 (d, J=15.6 Hz, 1H). 13C NMR (100 MHz, CHCl3): δ=167.2, 139.3, 131.8, 131.5, 131.1, 129.9, 128.1, 128.0, 127.1, 125.7, 124.1, 123.9, 122.9. HRMS ((+)-ESI): m/z=305.0159 (calcd. 305.0172 for C15H14BrO2, [M+H]+).
3.2.3 (E)-3-(4-Bromonaphthalen-1-yl)acrylic acid (7c)
NaOH (50.0 mL, 1 mol L−1) was added to (E)-ethyl 3-(4-bromonaphthalen-1-yl)acrylate (3.6 g, 12.0 mmol) that was diluted with THF (50.0 mL). The mixture was stirred at 40°C for 15 h. Then THF was removed under vacuum, and the residue was extracted with CH2Cl2 (100.0 mL) and H2O (200.0 mL). The aqueous phase was acidified with concentrated HCl to give 7c (yield 3.1 g, 95%) as a white solid, which was collected by filtration and dried under reduced pressure; m.p. 255–257°C. 1H NMR (400 MHz, [D6]DMSO): δ=8.33 (d, J=15.6 Hz, 1H), 8.21–8.26 (m, 2H), 7.93 (d, J=8.0 Hz, 1H), 7.83 (d, J=6.8 Hz, 1H), 7.72–7.75 (m, 2H), 6.63 (d, J=15.6 Hz, 1H). 13C NMR (100 MHz, [D6]DMSO): δ=167.2, 139.3, 131.8, 131.5, 131.1, 129.9, 128.1, 128.0, 127.1, 125.7, 124.1, 123.9, 122.9. HRMS ((+)-ESI): m/z=274.9713 (calcd. 274.9713 for C13H8BrO2, [M – H]+).
3.2.4 6-Bromo-1H-phenalen-1-one (6)
To a solution of compound 7c (3.0 g, 11.5 mmol) in dry CH2Cl2 (100.0 mL), oxalyl chloride (7.0 mL, 80.0 mmol) was added and the mixture stirred for 20 min. Then, two drops of dry DMF were added, and the mixture was stirred overnight at room temperature. The light yellow solution was evaporated under vacuum to afford (E)-3-(4-bromonaphthalen-1-yl)acryloyl chloride, which was rapidly dissolved in dry CH2Cl2 (100.0 mL). AlCl3 (2.4 g, 18.0 mmol) was added at 0°C, and stirred overnight at 25°C. The mixture was poured slowly onto ice, and the aqueous phase was extracted with CH2Cl2 (2×200.0 mL). The organic phase was combined and concentrated under vacuum, and the residue was purified by chromatography on silica gel (petroleum ether/EtOAc=9:1) to afford 6 as an orange solid (yield 1.7 g, 56%); m.p. 177–179°C. 1H NMR (400 MHz, CDCl3): δ=8.64 (dd, J=7.2, 1.2 Hz, 1H), 8.56 (dd, J=8.4, 1.2 Hz, 1H), 7.84 (t, J=8.4 Hz, 2H), 7.67 (d, J=9.6 Hz, 1H), 7.52 (d, J=7.6 Hz, 1H), 6.72 (d, J=9.6 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ=184.9, 141.3, 134.1, 131.1, 131.1, 131.1, 130.7, 129.6, 129.2, 128.7, 128.2, 128.0, 127.6. HRMS ((+)-ESI): m/z=280.9574 (calcd. 280.9572 for C13H7BrONa, [M+Na]+).
3.2.5 6-Methoxy-9-phenyl-1H-phenalen-1-one (4)
A solution of 5 (840.0 mg, 4.0 mmol) in dry THF (50.0 mL) was added slowly to the Grignard reagent (6.0 mmol, prepared from 1-bromo-4-methoxybenzene and Mg in dry THF) under nitrogen atmosphere. After stirring for 30 min at –10°C, the mixture was allowed to warm up to 25°C and stirred for 40 min. The mixture was quenched with HCl (aq., 5.0%) and extracted with EtOAc (2×100.0 mL), and the combined organic phase was washed with H2O and evaporated to dryness. The crude material was diluted with CH2Cl2 (100.0 mL) and treated with DDQ (2.0 equiv.), refluxed for 2 h, and cooled to 25°C. The mixture was concentrated and purified by chromatography on silica gel (petroleum ether/EtOAc=9:1) to afford 4 as an orange solid (yield 740.0 mg, two steps, 65%); m.p. 183–185°C. 1H NMR (400 MHz, CDCl3): δ=8.58 (dd, J=8.4, 1.2 Hz, 1H), 7.66 (d, J=8.0 Hz, 1H), 7.56 (ddd, J=9.6, 8.0, 1.2 Hz, 2H), 7.51–7.39 (m, 2H), 7.37 (td, J=6.0, 2.8 Hz, 3H), 6.88 (d, J=8.0 Hz, 1H), 6.47 (dd, J=9.6, 1.2 Hz, 1H), 4.08 (s, 3H). 13C NMR (100 MHz, CDCl3): δ=185.8, 159.5, 147.9, 143.2, 140.4, 133.1, 130.8, 129.4, 128.2, 127.9, 127.9, 127.0, 126.2, 124.5, 121.6, 104.4, 56.0. HRMS ((+)-ESI): m/z=287.1063 (calcd. 287.1067 for C20H15O3, [M+H]+).
3.2.6 2-Hydroxy-6-methoxy-9-phenyl-1H-phenalen-1-one (8)
A solution of compound 4 (500.0 mg, 1.7 mmol), (S,S)-[N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamine]manganese(II) chloride (55.0 mg, 0.08 mmol), and 4-phenylpyridine-N-oxide (72.0 mg, 0.4 mmol) in dichloromethane (100.0 mL) was cooled to 0°C and treated by precooled sodium hypochlorite (40.0 mL, 14.3 wt%). The mixture was stirred for 4 h at 0°C. Formation of the epoxide was observed by TLC. The reaction was quenched with saturated Na2S2O3 solution, the mixture extracted with DCM (2×100.0 mL) and H2O (200.0 mL), and the organic layer concentrated to give a residue. The residue was diluted in toluene (100.0 mL), and p-toluenesulfonic acid monohydrate (0.65 g, 3.4 mmol) was added, and the mixture was stirred overnight. The mixture was concentrated and extracted with EtOAc (2×100.0 mL) and H2O (200.0 mL). The organic layer was concentrated, and the residue was purified by column chromatography on silica gel (petroleum ether/EtOAc=7:1) to afford 8 as a red solid (yield 300.0 mg, 60%); m.p. 183–185°C. 1H NMR (400 MHz, CDCl3): δ=8.70 (dd, J=8.4, 1.2 Hz, 1H), 7.64 (d, J=8.0 Hz, 1H), 7.58 (dd, J=8.4, 1.2 Hz, 1H), 7.48 (ddd, J=10.4, 7.2, 3.6 Hz, 3H), 7.37 (dt, J=8.0, 1.2 Hz, 2H), 7.08 (d, J=1.2 Hz, 1H), 6.94–6.86 (m, 2H), 4.08 (s, 3H). 13C NMR (100 MHz, CDCl3): δ=179.9, 158.1, 149.2, 148.4, 142.6, 132.4, 130.3, 129.6, 128.2, 127.8, 127.5, 125.8, 124.5, 123.6, 121.5, 113.2, 105.1, 56.0. HRMS ((+)-ESI): m/z=303.1013 (calcd. 303.1016 for C20H15O3, [M+H]+).
3.2.7 2,6-Dimethoxy-9-phenyl-1H-phenalen-1-one (3)
To a solution of 8 (210.0 mg, 0.7 mmol) and K2CO3 (960.0 mg, 7.0 mmol) in DMF (15.0 mL) stirred for 1 h, CH3I (0.2 mL, 3.5 mmol) was added. The mixture was kept stirred for 10 h at 25°C. Then the reaction was quenched with H2O (100.0 mL), extracted with EtOAc (2×100.0 mL), and the combined organic extract evaporated to dryness. The residue was separated and purified by chromatography on silica gel (petroleum ether/EtOAc=9:1) to afford 3 as an orange solid (yield 200.0 mg, 90%); m.p. 183–185°C. 1H and 13C NMR (Tables 1 and 2). HRMS ((+)-ESI): m/z=317.1164 (calcd. 317.1172 for C21H17O3, [M+H]+).
3.3 Crystal structure determination of 3
The crystal and molecular structure of 3 were determined by single-crystal X-ray diffraction. A suitable single crystal was obtained from methanol/ethyl acetate and investigated on an Xcalibur Onyx Nova diffractometer with the temperature kept at T=100(2) K during data collection. The structure was solved and refined using the programs XS and Olex2 [15], respectively. The program X-Seed [16] was used as an interface to the program Shelx [17], [18] to produce the molecular graphics.
Crystal data for 2,6-dimethoxy-9-phenyl-1H-phenalen-1-one (3): C21H16O3 (Mr=316.3), tetragonal space group I41/a (no. 88), a=b=32.6613(5), c=5.6505(1) Å, V=6027.7(2) Å3, Z=16, T=100(2) K, Cu Kα radiation (λ=1.54184 Å), μ(Cu Kα)=0.746 mm−1, dcalcd=1.394 mg m−3, 5403 measured reflections of which 2693 were unique (Rint=0.0195, Rsigma=0.0221) and were used in all calculations. The final refinement gave R1=0.0350 (I>2 σ(I)) and wR2=0.0933 (all data), Δρfin (max/min=–0.20/0.16 e Å−3).
CCDC 1812926 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Acknowledgment
This work was financially supported by the Research Foundation for Advanced Talents (36000-18821101), Guangdong Natural Science Foundation (2017A030313751), The Science and Technology Planning Project of Guangzhou City (201707010189), The China Postdoctoral Science Foundation (2018M640942), and the Interdisciplinary Research Matching Scheme (RC-IRMS/15-16/02) of Hong Kong Baptist University, Hong Kong SAR, China.
References
[1] M. H. Cho, S. W. Lee, Int. J. Mol. Sci.2015, 16, 29120.10.3390/ijms161226152Search in Google Scholar
[2] I. Ahuja, R. Kissen, A. M. Bones, Trends Plant Sci.2012, 17, 73.10.1016/j.tplants.2011.11.002Search in Google Scholar
[3] G. F. Yang, X. H. Jiang, H. Z. Yang, Pest Manage. Sci.2002, 58, 1063.10.1002/ps.584Search in Google Scholar
[4] D. Hölscher, S. Dhakshinamoorthy, T. Alexandrov, M. Becker, T. Bretschneider, A. Buerkert, A. C. Crecelius, D. De Waele, A. Elsen, D. G. Heckel, H. Heklau, C. Hertweck, M. Kai, K. Knop, C. Krafft, R. K. Maddula, C. Matthäus, J. Popp, B. Schneider, U. S. Schubert, R. A. Sikora, A. Svatoš, R. L. Swennen, Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 105.10.1073/pnas.1314168110Search in Google Scholar
[5] F. Echeverri, F. Torres, W. Quiñones, G. Escobar, R. Archbold, Phytochem. Rev.2012, 11, 1.10.1007/s11101-010-9205-xSearch in Google Scholar
[6] K. Jitsaeng, B. Schneider, Phytochem. Lett.2010, 3, 84.10.1016/j.phytol.2010.02.001Search in Google Scholar
[7] F. Otálvaro, J. Nanclares, L. E. Vásquez, W. Quiñones, F. Echeverri, R. Arango, B. Schneider, J. Nat. Prod.2007, 70, 887.10.1021/np070091eSearch in Google Scholar
[8] J. G. Luis, W. Q. Fletcher, F. Echeverri, T. A. Grillo, Tetrahedron1994, 50, 10963.10.1016/S0040-4020(01)85707-0Search in Google Scholar
[9] L. M. Lenora, N. Senthilkumar, Asian J. Plant Sci. Res.2017, 7, 1.Search in Google Scholar
[10] M. Della Greta, R. Lanzetta, L. Mangoni, P. Monaco, L. Previtera, Bioorg. Med. Chem. Lett.1991, 1, 599.10.1016/S0960-894X(01)81159-8Search in Google Scholar
[11] M. Z. Wang, X. H. Cai, X. D. Luo, Helv. Chim. Acta2011, 94, 61.10.1002/hlca.200900428Search in Google Scholar
[12] M. Z. Wang, C. F. Ku, T. X. Si, S. W. Tsang, X. M. Lv, X. W. Li, Z. M. Li, H. J. Zhang, A. S. C. Chan, J. Nat. Prod.2018, 81, 98.10.1021/acs.jnatprod.7b00709Search in Google Scholar
[13] W. Quiñones, G. Escobar, F. Echeverri, F. Torres, Y. Rosero, V. Arango, G. Cardona, A. Gallego, Molecules2000, 5, 974.10.3390/50700974Search in Google Scholar
[14] F. Otálvaro, W. Quiñones, F. Echeverri, B. Schneider, J. Labelled Compd. Radiopharm. 2004, 47, 147.10.1002/jlcr.808Search in Google Scholar
[15] O. V. Dolomanov, L. J. Bourhis, R. J.Gildea, J. A. K. Howard, H. Puschmann, J. Appl. Crystallogr. 2009, 42, 339.10.1107/S0021889808042726Search in Google Scholar
[16] L. J. Barbour, J. Supramol. Chem.2001, 1, 189.10.1016/S1472-7862(02)00030-8Search in Google Scholar
[17] G. M. Sheldrick, Acta Crystallogr. 2008, A64, 112.10.1107/S0108767307043930Search in Google Scholar PubMed
[18] G. M. Sheldrick, Acta Crystallogr. 2015, C71, 3.Search in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/znb-2018-0181).
©2019 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this Issue
- Synthesis and structure of the donor-free potassium silanide K[SiPh3]
- A series of Keggin- and Wells-Dawson-polyoxometalate-based compounds constructed from oxygen-functional imidazole derivatives
- Synthesis, crystal structure, photoluminescence and photochemistry of bis(triphenylphosphine)silver(I) flavonolate
- Facile synthesis of new pyrazolo[4′,3′:5,6]pyrano[2,3-d]pyrimidin-5(1H)-ones via the tandem intramolecular Pinner–Dimroth rearrangement and their antibacterial evaluation
- Synthesis of 2,6-dimethoxy-9-phenyl-1H-phenalen-1-one and structural revision of the benzoindenone from Eichhornia crassipes
- Addition of some 6-amino-4-aryl-2(1H)-pyridones to phenylisocyanate and related reactions
- Study on the chemical constituents of Dacrydium elatum and their cytotoxic activity
- RhSn3 and the Modifications of RhSn4 – Structure and 119Sn Mössbauer spectroscopic characterization
- Equiatomic iron-based tetrelides TFeSi and TFeGe (T = Zr, Nb, Hf, Ta) – A 57Fe Mössbauer-spectroscopic study
- The reaction of a particularly electrophilic acyclic diaminocarbene with carbon monoxide: formation of β- and γ-lactams
- Zinc-lead ordering in equiatomic rare earth plumbides REZnPb (RE=La–Nd and Sm–Tb)
- Ni(II) complexes with thioether-functionalized silylamide ligands. Synthesis and crystal structures of [Ni{Me2Si(N-C6H4-2-S-t-Bu)2}], [Ni{Ph2Si(N-C6H4-2-SMe)2}] and [Ni{Ph2Si(N-C6H4-2-SPh)2}]
Articles in the same Issue
- Frontmatter
- In this Issue
- Synthesis and structure of the donor-free potassium silanide K[SiPh3]
- A series of Keggin- and Wells-Dawson-polyoxometalate-based compounds constructed from oxygen-functional imidazole derivatives
- Synthesis, crystal structure, photoluminescence and photochemistry of bis(triphenylphosphine)silver(I) flavonolate
- Facile synthesis of new pyrazolo[4′,3′:5,6]pyrano[2,3-d]pyrimidin-5(1H)-ones via the tandem intramolecular Pinner–Dimroth rearrangement and their antibacterial evaluation
- Synthesis of 2,6-dimethoxy-9-phenyl-1H-phenalen-1-one and structural revision of the benzoindenone from Eichhornia crassipes
- Addition of some 6-amino-4-aryl-2(1H)-pyridones to phenylisocyanate and related reactions
- Study on the chemical constituents of Dacrydium elatum and their cytotoxic activity
- RhSn3 and the Modifications of RhSn4 – Structure and 119Sn Mössbauer spectroscopic characterization
- Equiatomic iron-based tetrelides TFeSi and TFeGe (T = Zr, Nb, Hf, Ta) – A 57Fe Mössbauer-spectroscopic study
- The reaction of a particularly electrophilic acyclic diaminocarbene with carbon monoxide: formation of β- and γ-lactams
- Zinc-lead ordering in equiatomic rare earth plumbides REZnPb (RE=La–Nd and Sm–Tb)
- Ni(II) complexes with thioether-functionalized silylamide ligands. Synthesis and crystal structures of [Ni{Me2Si(N-C6H4-2-S-t-Bu)2}], [Ni{Ph2Si(N-C6H4-2-SMe)2}] and [Ni{Ph2Si(N-C6H4-2-SPh)2}]