Emulsion-templated macroporous polymer/polymer composites with switchable stiffness
-
Qixiang Jiang
 and
Alexander Bismarck
and
Alexander Bismarck

Abstract
Emulsion templates containing monomers in both emulsion phases were used to manufacture polystyrene-co-divinylbenzene based polymerized high internal phase emulsions (polyHIPEs) which have been reinforced by poly(methacrylic acid) (polyMAA) and poly(dimethyl aminoethyl methacrylate) (polyDMAEMA). The morphology of the hydrogel-filled polyHIPEs is affected by the hydrogels synthesized in the aqueous emulsion phase. The pore structure of polyMAA-filled polyHIPEs is highly interconnected indicating the formation of a methacrylic acid-co-styrene copolymer at the oil/water interface of the emulsion templates during synthesis. However, polyDMAEMA-filled polyHIPEs are predominately closed celled and the pore walls are covered by grafted hydrogel. The ability of the hydrogel-filled polyHIPEs to absorb water decreased with increasing crosslinking density of the hydrogels. The dry hydrogel reinforced the polyHIPE scaffolds possessed higher elastic moduli and crush strengths than the control polyHIPEs. The reinforcing ability of the dry hydrogels was further enhanced by increasing their degree of crosslinking. However, the reinforcement could be “switched off” simply by hydrating the hydrogels. The switchable mechanical properties of the hydrogel-filled polyHIPEs could potentially be utilized in smart humidity sensor technology.
Introduction
Hydrogels are crosslinked hydrophilic polymers or copolymers, which are characterized by their ability to swell in water or other polar solvents. The network, formed after the swelling, can possess many interesting properties, such as inner space for trapping and adsorbing molecules [1], permeability [2], and biocompatibility [3]. Additionally, many hydrogels also exhibit environmental sensitivity, which means their swelling behavior is affected by environmental stimuli, such as pH [4], temperature [5], the presence of certain chemical species [6, 7], electric fields [8], and light [9], making these hydrogels “intelligent” materials. Due to these properties, hydrogels are applicable in many fields, such as drug release, tissue engineering [1], sensors [10], and separation membranes [2, 11–14].
Polymerized high-internal-phase emulsions, also known as polyHIPEs, are macroporous polymers prepared via emulsion templating [15, 16]. Usually, a high-internal-phase emulsion is firstly prepared by dispersing a large volume ratio (more than 74 vol % with respect to the emulsion [15, 16]) of internal phase into a continuous phase, which consists of monomers or is a solution of monomers, emulsifiers, and sometimes initiators. After the polymerization of the emulsion and the removal of the internal phase, the product becomes a macroporous polymer, whose porous structure is a replica of the morphology of the emulsion template. A number of polyHIPEs were prepared from water-in-oil (w/o) emulsions, thus the resulting polyHIPEs are hydrophobic, which limits the applications of these polyHIPEs. One straightforward method to prepare hydrophilic polyHIPEs is using oil/water (o/w) emulsion templates to prepare hydrogel polyHIPEs. Krajnc et al. [17] reported poly(acrylic acid)HIPEs prepared by polymerizing o/w HIPEs. The purpose for the synthesis of the polyHIPEs was to induce acid groups in polyHIPEs in a one-step synthesis. Kulygin and Silverstein [18] have reported the preparation of poly(hydroxyethyl methacrylate)HIPEs from o/w HIPEs. These porous hydrogels are expected to be of interest for tissue engineering and drug release applications. Butler et al. [19] prepared poly(acrylamide)HIPEs by the polymerization of CO2-in-water emulsion templates, in which supercritical CO2 was used as internal phase for the emulsion templates. These polyHIPEs could potentially be used for enzyme immobilization. Grant et al. [20] prepared poly(N-isopropylacrylamide)HIPEs, which possessed temperature-responsive swelling. These polyHIPEs exhibited a temperature-controlled loading and release of polystyrene particles, which may be applicable in drug delivery and in smart coating. Zhou et al. [21] reported dextran-based polyHIPEs, which were synthesized via crosslinking glycidyl methacrylate functionalized dextran within particle-stabilized o/w emulsions. Furthermore, they also prepared thermo-responsive polyHIPEs by physical crosslinking dextran-co-poly(N-isopropylacrylamide) which occurred if the temperature exceeded the lower critical solution temperature of the grafted poly(N-isopropylacrylamide) [22]. More recently, macroporous modified alginates were synthesized by Zhou et al. [23]. Besides chemical crosslinks, these macroporous hydrogels could be further reversibly crosslinked by Ca2+ ions, which introduced an interesting route to control the dimensions and pore structure of the macroporous hydrogel. This type of macroporous hydrogels could be extruded through a hypodermic needle, rendering it suitable to be used as injectable scaffolds for tissue engineering. Moreover, the nature of emulsion templating allows for the preparation of hydrogel-polyHIPE composites via one-step synthesis: if the continuous phase of an emulsion template contains hydrophobic monomers and the internal phase of the emulsion template hydrophilic monomers. After simultaneous polymerization of both phases, the resulting products are hydrophobic polyHIPE scaffolds filled with hydrogels. Ruckenstein et al. [24, 25] reported poly(acrylamide)-filled polyHIPEs and poly(sodium acrylate)-filled polyHIPEs, i.e., polyHIPE-supported hydrogels, which could be used as pervaporation membranes. Gitli and Silverstein [26] have reported the preparation of polyacrylamide-filled polyHIPEs using a similar method and investigated the effect of the locus of initiation of polymerization on resulting pore structures. They reported the loading and release of Eosin Y in polyacrylamide-filled polyHIPEs, aiming to develop drug release systems [27]. More recently, Kovacic et al. [28] reported poly(acrylic acid)-filled polyHIPEs and poly(N-isopropylacrylamide)-filled polyHIPEs. They demonstrated that the water flux through the hydrogel-filled porous polyHIPE monoliths is pH- or temperature-dependent and suggested that the hydrogel-filled polyHIPEs might be of interest for chromatography applications.
In contrast to the research discussed above, we aim to make use of the inherently different mechanical properties of dry and wet hydrogels. Wet hydrogels are mechanically weak and disintegrate easily [29], whereas hydrogels with low or zero swelling are usually tough and stiff materials [29, 30]. Our own preliminary experiments with up to 10% crosslinked poly(methacrylic acid) (polyMAA) and poly(dimethyl aminoethyl methacrylate) (polyDMAEMA) show that the dehydrated hydrogels are almost incompressible. Moreover, they are difficult to be cut or to be torn apart while the wet hydrogels can be easily destroyed. We, therefore, decided to create macroporous polymer/polymer composites, whose stiffness can be adjusted simply by hydration of the hydrogel component of the porous polymer composites. Such materials can potentially find applications as humidity sensors. The macroporous polymer/polymer composites, namely, polyMAA- and polyDMAEMA-filled poly(ST-co-DVB) polyHIPEs, were manufactured by polymerizing emulsion templates, which contain monomers in both phases. The influence of the crosslinking degree of the respective hydrogels on the morphology of the macroporous composites as well as their water uptake behavior and, most importantly, on the mechanical performance of the dry and wet macroporous polymer composites was investigated.
Experiments
Materials
Styrene (ST), divinylbenzene (DVB), dimethyl aminoethyl methacrylate (DMAEMA), N,N′-methylenebisacrylamide (MBA), α,α′-azobisisbutyronitrile (AIBN), ammonium peroxodisulfate (APS), NaOH, HCl (37.5%), and CaCl2·2H2O were purchased from Sigma-Aldrich (Gillingham, UK). Methacrylic acid (MAA) was purchased from Acros (Geel, Belgium), and Hypermer B246 was kindly supplied by Croda (Snaith, UK). All the chemicals were used as received.
Preparation of hydrogel-filled ST-co-DVB-based polyHIPEs
The setup for HIPE preparation consisted of a reaction vessel equipped with a glass anchor stirrer connected to an overhead stirrer. The continuous-phase components ST, DVB, Hypermer B246, and AIBN were mixed in the reaction vessel. In order to prepare the internal phases for the HIPEs, 12 vol % MAA or DMAEMA and varying amounts of MBA were dissolved in deionized water. Afterwards, the MAA in water was deprotonated by adjusting the pH of the aqueous solution to 14 by addition of NaOH, while the DMAEMA was protonated by adding HCl until a pH of 1 was reached. This was done to limit the portioning of the water-soluble monomers into organic emulsion phase. The premixed internal phase was placed in a dropping funnel and added dropwise into the continuous phase while stirring at a constant rate of 400 rpm. Afterwards, the HIPEs containing MAA in their internal phase were stirred for 2 min and while those containing DMAEMA for 9 min, respectively, at a speed of 2000 rpm to produce homogeneous emulsions. The initiator APS was dissolved in <1 mL deionized water, and the solution was added into the ready-made emulsions while stirring them for another minute to disperse the APS evenly. Finally, the emulsions were transferred into freestanding polypropylene centrifuge tubes and polymerized in an oven at 70°C for 24 h. Afterwards, the hydrogel-filled polyHIPEs were purified by washing them in mixtures of water and methanol (1:1 by volume) for 48 h. The samples were dried at 70°C initially in air followed by under vacuum for 24 h, respectively.
A poly(ST-co-DVB) polyHIPE (without grafted hydrogel) was prepared as control following the same procedure. In this case, an aqueous CaCl2·2H2O solution with a concentration of 5 g/L was used as internal HIPE phase instead of the monomer salt/APS mixtures discussed above. The formulation of the samples is summarized in Table 1. C stands for the control sample. Sample codes starting with M represent polyMAA-filled poly(ST-co-DVB) polyHIPEs, while sample codes starting with D represent polyDMAEMA-filled poly(ST-co-DVB) polyHIPEs. The numbers behind M or D refer to the molar ratio of MBA (crosslinker) with respect to MAA or DMAEMA.
Formulations of hydrogel filled poly(ST-co-DVB)HIPEs.
| Continuous phasea |
Internal phasea |
||||||
|---|---|---|---|---|---|---|---|
| ST (vol %)b | DVB (vol %)b | B246 (vol %)b | AIBN (mol %)c | Monomer | MBA (mol %)d | APS (mol %)e | |
| C | 60 | 20 | 20 | 1 | – | – | – |
| M0 | 60 | 20 | 20 | 1 | MA | 0 | 1 |
| M1 | 60 | 20 | 20 | 1 | MA | 1 | 1 |
| M2 | 60 | 20 | 20 | 1 | MA | 2 | 1 |
| M5 | 60 | 20 | 20 | 1 | MA | 5 | 1 |
| M10 | 60 | 20 | 20 | 1 | MA | 10 | 1 |
| D0 | 60 | 20 | 20 | 1 | DMAEMA | 0 | 1 |
| D1 | 60 | 20 | 20 | 1 | DMAEMA | 1 | 1 |
| D2 | 60 | 20 | 20 | 1 | DMAEMA | 2 | 1 |
| D5 | 60 | 20 | 20 | 1 | DMAEMA | 5 | 1 |
| D10 | 60 | 20 | 20 | 1 | DMAEMA | 10 | 1 |
aContinuous and internal phase volumes were 20 and 80% of the total volume of emulsions, respectively.Manual Line Break
bPercentage volume of monomers and surfactant with respect to the continuous-phase volume.Manual Line Break
cPercentage molar concentration of AIBN with respect to the molar number of double bonds in the monomers in continuous phase.
dPercentage molar concentration of MBA with respect to the molar numbers of aqueous-phase monomers.
ePercentage molar concentration of APS with respect to the molar number of double bonds in the aqueous-phase monomers.
Characterization
Morphology of the polyHIPEs
Fractured surfaces of polyHIPEs were investigated using scanning electron microscopy (SEM, Hitachi S-3400N, Hitachi High Technologies, Germany). Prior to SEM, the polyHIPE fragments were gold-coated using Agar Auto Sputter Coater (Agar Scientific, Essex, UK) to guarantee sufficient electrical conductivity. The SEM was operated in secondary electron mode using a voltage of 15 kV for the electron beam. The images were further analyzed using UTHSCSA ImageTool software. To determine the pore and pore throat sizes, at least 200 pores as well as pore throats were measured.
Density and porosity of the polyHIPEs
The skeletal densities of the samples were determined using a helium replacement pycnometer (Accupyc 1330, Micrometrics Ltd., Dunstable, UK). About 0.5 g sample was crushed into a powder, weighed, and placed into the sample chamber. The foam densities were measured using powder pycnometry (Geopyc 1360, Micrometrics Ltd., Dunstable, UK). About 0.1 g of small polyHIPE fragments was investigated. The porosity (P) was calculated from the measured densities as follows:

where ρf is the foam density and ρs is the skeletal density.
Water uptake behavior of the polyHIPEs
Samples pieces of about (5×5×5) mm were first dried under vacuum and placed in DI water in a sample glass. The polyHIPE samples were kept immersed in water by placing two five-penny coins on top the samples. In order to determine the weight of the wet samples they were taken out of the water and any water drops on the surface were wiped off using filter paper immediately before weighing. Afterwards, the samples were dried again and weighted. The water uptake (S) was calculated using the equation below:

where mw is the weight of the wet sample and md that of the dry sample.
Mechanical properties of wet and dry hydrogel-filled polyHIPEs
Mechanical properties of wet and dry hydrogel-filled polyHIPEs were determined by compression tests using a Lloyds EZ50 (Lloyds Instruments Ltd., Fareham, UK) equipped with a 50 kN load cell. Prior to the compression tests, the samples were cut into disks with a diameter of about 25 mm and a height of about 10 mm. The sample disks were compressed between compression plates to half of their original height at a speed of 1 mm/min while their stress–strain curves were recorded. The elastic modulus was determined from the slope of the initial linear region of the stress–strain curves. The crush strength was defined as the maximum strength at the end of the initial linear elastic region. At least six disks per sample were investigated. It is worth noting that samples, which were purified by extraction but were not dried, are referred to as “wet”.
Results and discussion
Emulsion templates containing monomers in both phases were prepared and polymerized to produce moisture-sensitive polyHIPE composites with adjustable stiffness. The hydrophobic monomers ST and DVB were employed in the organic phase of the templates to form the characteristic macroporous polyHIPE scaffolds. Wet hydrogels are known to be mechanically weak while they turn into tough, stiff materials upon drying [29, 30]. This makes hydrogels ideal candidates to provide a “smart” reinforcement for polyHIPEs, which can be switched on or off as required. Therefore, we introduced deprotonated MAA (M0–M10) or protonated DMAEMA (D0–D10) as hydrophilic monomers into the internal phase. The hydrophilic monomer concentrations were kept constant in all samples, but the concentration of crosslinker MBA was varied, in order to investigate the influence of the crosslinking degree on the properties of the hydrogels and macroporous polymer/polymer composites.
The SEM images of the control sample and polyMAA-filled poly(ST-co-DVB) polyHIPEs are shown in Fig. 1. Their pore sizes, pore throat sizes, densities, and porosities are listed in Table 2. As expected, the control poly(ST-co-DVB) polyHIPE has the morphology of typical polyHIPEs [16] containing spherical pores, which are interconnected by pore throats (Fig. 1a,b). Surprisingly, however, there are only very few morphological changes visible in the SEM images of M0–M10 (Fig. 1c–l) compared to the control (Fig. 1a,b). The MAA in the internal phase of the emulsion templates was deprotonated in order to reduce its solubility in the organic phase. One would, therefore, expect the formation of a hydrogel during polymerization, which fills the pores of the polyHIPE scaffold or at least covers the pore walls in the form of a thin layer. In this case, the pores would appear to be partially or predominately closed cell [26, 28]. However, the pores of M0–M10 are well interconnected by pore throats; only the denser crosslinked hydrogels of M5 and M10 seem to cover a few of the pore throats in their polyHIPE scaffolds as indicated by the arrows in the SEM images (Fig. 1j,l). The pore and pore throat sizes of M0–M10 are with approximately 3–4 μm and 0.8–1.3 μm in diameter, respectively, similar or only slightly smaller than those of the control (Table 2). This seems to indicate that no or very little hydrogel formed during polymerization. However, the measured densities and porosities suggest otherwise. The significant increase in skeletal density observed for M0 (1.24 g/m3) compared to the control (1.14 g/m3) can be attributed to the presence of the denser polyMAA hydrogel. Although, the crosslinking density does not significantly influence the skeletal densities of M0–M10, they are identical within error. The foam densities of M0–M10 are higher than that of the control. This indicates the presence of hydrogels, occupying the pore volume. Furthermore, the foam densities of M0–M10 increased gradually, corresponding to the decrease of their porosities (Table 2). The highest foam densities and lowest porosities possess samples M5 and M10. This is evidence for the presence of hydrogel grafted to the pore walls.

SEM images of poly(MAA)-filled poly(St-co-DVB)HIPEs. (a,b) control sample (c); (c,d) 0% (M0); (e,f) 1% (M1); (g, h) 2% (M2); (i,j) 5% (M5); (k,l) 10% (M10). Arrows point at pore throats covered by hydrogels.
Pore sizes, pore throat sizes, densities, and porosities of polyMAA-filled polyHIPEs.
| Pore size (μm) | Pore throat size (μm) | Skeletal density (g/cm3) | Foam density (g/cm3) | Porosity (%) | |
|---|---|---|---|---|---|
| Control | 4±1 | 1.5±0.4 | 1.14±0.05 | 0.15±0.01 | 87±1 |
| M0 | 4±1 | 1.3±0.6 | 1.25±0.01 | 0.24±0.03 | 81±2 |
| M1 | 4±1 | 1.2±0.3 | 1.25±0.01 | 0.27±0.03 | 78±2 |
| M2 | 3±1 | 1.2±0.3 | 1.25±0.01 | 0.25±0.02 | 80±1 |
| M5 | 3±1 | 0.8±0.3 | 1.24±0.01 | 0.35±0.01 | 71±1 |
| M10 | 3±1 | 1.0±0.3 | 1.24±0.01 | 0.37±0.01 | 70±1 |
A possible explanation for the seemingly contradicting density and porosity data on the one side and the observed morphology on the other might be the formation of ST-MAA copolymer at what used to be the o/w interface in the emulsion template. The reactivity ratios of ST (1) and MAA (2) are approximately r1=0.2~0.3 and r2=0.4~0.6 depending on the polymerization conditions [31]. This means it is more likely that these monomers are consumed in a copolymerization than in homopolymerization. However, the ST-MAA copolymerization competes against the crosslinking reaction of the hydrogel. As long as the crosslinking degree of the hydrogel is low (or zero), MAA or the growing polyMAA network can be incorporated into the pore walls, which explains the absence of hydrogel-covered pore throats in the highly interconnected pore structure of M0–M2 (Fig. 1c–h). However, with increasing the MBA concentration in the internal phase of the emulsions from 5 to 10%, the crosslinking density of the hydrogels increased during polymerization so dramatically that an incorporation of the formed hydrogel network into the pore walls is hindered. Grafting of the hydrogel onto the pore walls occurred in accordance to the mechanism suggested by Mika et al. [32] and Susanto and Ulbricht [33]. This leads to the observed coverage of some of M5’s and M10’s pore throats by hydrogel, which reduces the free pore volume and, therefore, causes the significant further increase in foam density of M5–M10 compared to M0–M2.
The porosities, skeletal and foam densities of the polyDMAEMA-filled polyHIPEs D0–D10 (Table 3) show the same trend as observed for M0–M10. The skeletal densities of D0–D10 are identical within errors but significantly larger than those of the control. The foam density increased and the porosity decreased upon introduction of polyDMAEMA in to the polyHIPEs (control vs. D0) as well as with increasing crosslinking density of the hydrogel within the group D0–D10. This is evidence for the successful incorporation of polyDMAEMA and an increasing amount of hydrogel grafting onto the polyHIPE scaffold. However, the morphology of polyDMAEMA-filled poly(ST-co-DVB) polyHIPEs differs significantly from that of the polyMAA-containing polyHIPEs. D0–D10 possess hierarchical pore structures consisting of large pores of about 30 μm in diameter, which are surrounded by relatively small pores (Fig. 2). This indicates that the emulsion templates used to manufacture D0–D10 experienced droplet coalescence during polymerization. One possible explanation might be the slow decomposition rate of the initiator APS at low pH values [34]; the pH had to be adjusted to 1 to promote the protonation of DMAEMA to reduce its solubility in the organic phase. The reduced decomposition rate of APS resulted in a lower polymerization rate so that the emulsion templates were exposed to elevated temperature for longer. Furthermore, only D0 appears to possess an interconnected pore structure although its average pore throat size and number of pore throats per pore was dramatically reduced compared to the control (Table 3, Fig. 2a–d). PolyHIPEs D1–D10 have a mainly closed cell pore structure as the pore walls especially of the large pores seem to be covered by hydrogel layers. The thickness of these layers increased with increasing crosslinking density.
Pore sizes, pore throat sizes, densities, and porosities of polyDMAEMA-filled polyHIPEs.
| Pore size (μm) | Pore throat size (μm) | Skeletal density (g/cm3) | Foam density (g/cm3) | Porosity (%) | |
|---|---|---|---|---|---|
| C | 4±1 | 1.5±0.4 | 1.14±0.05 | 0.15±0.01 | 87±1 |
| D0 | 4–30 | 0.8±0.2 | 1.2±0.1 | 0.19±0.03 | 84±2 |
| D1 | 3–28 | – | 1.20±0.06 | 0.21±0.03 | 83±3 |
| D2 | 3–30 | – | 1.2±0.1 | 0.27±0.01 | 78±1 |
| D5 | 2–46 | – | 1.21±0.09 | 0.32±0.03 | 73±2 |
| D10 | 1–50 | – | 1.2±0.1 | 0.36±0.01 | 70±1 |

SEM images of poly(DMAEMA) -filled poly(St- co-DVB)HIPEs. (a,b) control sample (c); (c,d) 0% (D0); (e, f) 1% (D1); (g,h) 2% (D2); (i,j) 5% (D5); (k,l) 10% (D10).
The changed morphology of D0–D10 compared to M0–M10 suggests a different mechanism for the DMAEMA consumption during polymerization. Looking at the copolymerization reactivity ratios of ST (1) and DMAEMA (2) (r1=1.74 and r2=0.43 [35]), it becomes clear that ST as well as DMAEMA favors the reaction with ST; copolymerization at the o/w interface is, therefore, unlikely. The formed hydrogel, however, is directly grafted to the pore walls, which explains the presence of hydrogel layers covering the pore walls and pore throats and, therefore, the reduced interconnectivity of D0–D10 compared to the control.
The morphology of the polyHIPEs M0–M10 and D0–D10 differs significantly. This might have a profound impact on the ability of their hydrogel components to swell in water or other polar solvents and subsequently on the switchability of the stiffness of polymer/polymer composites. We, therefore, investigated the water uptake behavior of polyHIPEs M0–M10 and D0–D10. All samples were able to absorb water, however, the amount varied depending on the hydrogel and degree of crosslinking of the hydrogel (Fig. 3). The differences in the water uptake can be explained by the processes occurring during water uptake. At the beginning, water does enter the polyHIPEs relatively undisturbed via two routes: passing through uncovered pore throats (main process for the highly interconnected polyHIPEs M0–M10 and D0) and diffuse through the hydrogel layers (main process for the D1–D10). During this stage, air can leave the pores through pore throats. However, while the hydrogels swell, the pore throats either become increasingly blocked by the expanding hydrogel network covering the pore throats (main process for the D1–D10) or their size will be reduced to almost zero due to the swelling of the pore walls in which hydrogel is incorporated (main process for M0–M10 and D0). This means water can still enter pores by diffusion. However, it becomes increasingly difficult for the still entrapped air to leave the pores due to its poor permeability through the wet expanding hydrogel. Ultimately, the pressure of the entrapped air is in balance with the back pressure of water and an equilibrium state reached. Pore throats are much easier routes for water to penetrate the pores and for air to escape compared to diffusion through hydrogels. This explains why, with the exception of M0 and D0, the water uptake of the highly interconnected samples of the M series was considerably larger than that of the corresponding samples of the D series. However, it is worth noting, that in case of the equally well-interconnected samples M0 and D0 the water uptake of D0 exceeds that of M0, which indicates that the polyDMAEMA network renders the polyHIPE surface more efficiently hydrophilic than polyMAA. Furthermore, as expected, the water uptake decreased with increasing crosslinking degrees of the polyMAA and polyDMAEMA hydrogels, respectively, for both groups of the hydrogel-filled polyHIPEs. This is caused by the increasing density of the highly crosslinked hydrogel layers, which hinder more efficiently the water diffusion [36]. Similarly, the hydrogels covering the pore throats in D1–D10 may contribute to their lower water uptake compared to D0.
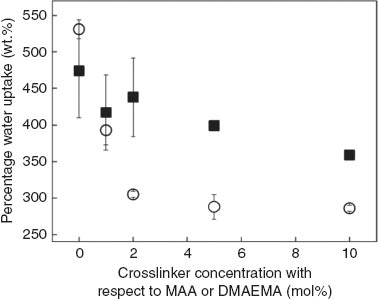
Percentage water uptake of polyMAA-filled poly(ST-co-DVB)HIPEs (solid squares) and polyDMAEMA-filled poly(ST-co-DVB)HIPEs (open circles).
Compression tests were conducted in order to investigate the mechanical performance of dry and wet polyMAA and polyDMAEMA-filled poly(ST-co-DVB) polyHIPEs, respectively; elastic moduli and crush strengths of the polymer/polymer composites were determined and compared to those of the control. However, cracks developed in the samples M10, D5, and D10 during drying as the polyHIPE scaffolds cannot withstand the rising stress levels in the samples caused by the shrinkage of the hydrogels. Therefore, the mechanical properties of those dry samples could not be measured. The dry control sample has an elastic modulus of 14±1 MPa and crush strength of 1.00±0.08 MPa (Fig. 4), which is similar to results previously reported by our group [37]. Both, the elastic modulus (22 MPa, Fig. 4a) as well as the crush strength (1.6 MPa, Fig. 4b) of dry M0 was significantly higher than those of the control indicating a successful reinforcement of the polyHIPE scaffold by dry polyMAA. This behavior can be explained by two effects: Firstly, the presence of dry polyMAA in M0 leads to an increase in the foam densities, which is known to result in improved mechanical properties [38]; secondly, the incorporation of dry and, therefore, tough, rigid polyMAA into and its grafting to the pore walls of the polyHIPE scaffold results in a direct increase of the mechanical performance of the macroporous polymer/polymer composite. Furthermore, an increase in elastic moduli and crush strengths of M1, M2, and M5 can be observed (Fig. 4a,b): the elastic moduli increased from 23 MPa (M1) to 36 MPa (M5), and the crush strengths increased from 1.5 MPa (M1) to 2.1 MPa (M5). This indicates that increasing the crosslinking degree of the hydrogel resulted in an increased stiffness of the dry hydrogels, which in turn provided a better reinforcement to the polyHIPE scaffolds. On the other hand, the elastic moduli and crush strengths of the wet polyMAA-filled poly(ST-co-DVB) polyHIPEs were dramatically reduced compared to their corresponding dry samples. The swollen hydrogels are mechanically weak and cannot, therefore, reinforce the polyHIPE scaffolds. Furthermore, the fact that the mechanical properties of the wet samples M0–M2 are even lower than those of the wet control sample (elastic modulus of 14 MPa and crush strength of 1.0 MPa) indicates that a certain amount of ST-DVB-MAA copolymers formed during the synthesis at the o/w interface of the emulsion templates. Similar observations were made by Gitli and Silverstein [26]. Such copolymers are hydrophilic and can absorb a small amount of water, which weakens their mechanical integrity. Although all wet samples of the M series exhibited similar crush strengths, the elastic moduli of wet M5 and M10 were higher than those of wet M0–M2. As revealed by the SEM images, some of the pore throats of M5 and M10 are covered by hydrogel, indicating that less MAA was incorporated into the ST-MAA-DVB copolymer than into samples M0–M2, which would certainly improve the mechanical integrity of the polyHIPE scaffold. However, it is also possible that the swollen hydrogel layers, which were grafted to the pore walls of wet M5 and M10, effectively cover the pore throats and lock the water in the pores during the compression. This might contribute to the observed slight increase of the elastic moduli of wet M5 and M10.
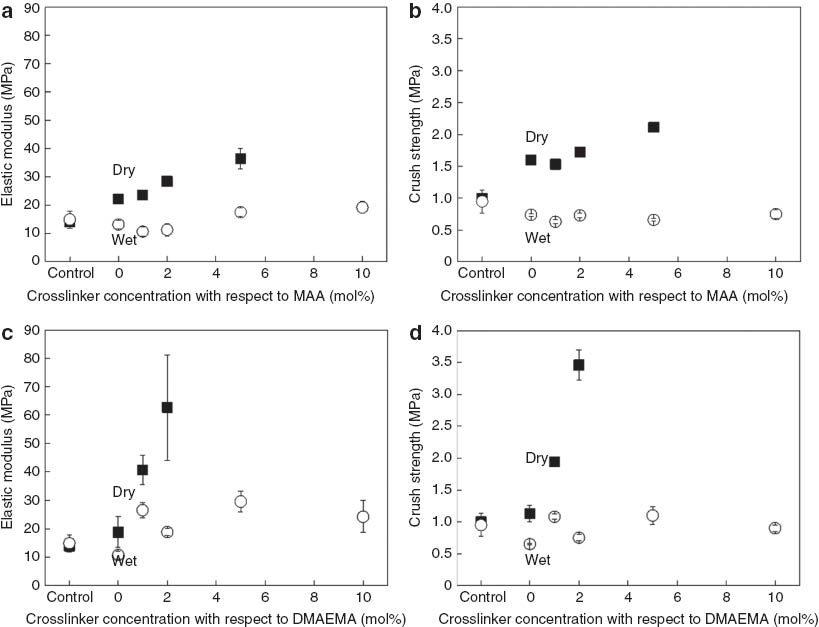
Mechanical properties of polyMAA-filled poly(ST-co-DVB)HIPEs and polyDMAEMA-filled poly(ST-co-DVB)HIPEs. (a) Elastic moduli of polyMAA-filled polyHIPEs. (b) Crush strengths of polyMAA-filled polyHIPEs. (c) Elastic moduli of polyDMAEMA-filled polyHIPEs. (d) Crush strengths of polyDMAEMA-filled polyHIPEs. Solid squares represent the mechanical properties of the dry samples, while open circles represent the mechanical properties of the wet samples.
The polyDMAEMA-reinforced polyHIPEs of the D series show the same trend as the samples of the M series with respect to their mechanical properties (Fig. 4c,d). The elastic modulus of D0 improved from 14 MPa (control) to 20 MPa, while its crush strength increased to 1.1 MPa due to the successful reinforcement of the polyHIPE scaffold by the dry polyDMAEMA (Fig. 4c,d). The mechanical properties improved further with increasing crosslinking degree of the hydrogels. However the dramatic increase in elastic modulus and crush strength observed for D1 (40 MPa, 2 MPa) and D2 (63 MPa, 3.5 MPa) cannot solely be explained by the increasing crosslinking degree of the hydrogels (Fig. 4c,d). The morphology of the samples has also to be considered. D0 possesses an interconnected pore structure while D1 and D2 were closed celled as their pore walls were covered by hydrogel. We previously demonstrated that, if the chemical composition is similar, closed celled macroporous polymers have better mechanical properties than interconnected ones as the pore throats weaken the structural integrity of polyHIPEs [39]. Furthermore, they possess a hierarchical pore structure, which was reported to contribute to the mechanical performance of polyHIPEs as the load is more effectively distributed in such structures [37, 40].
Hydrating the samples of the D series results, as expected, in a dramatic reduction in both, elastic moduli and crush strengths (Fig. 4c,d). The reinforcement was effectively switched off, as the wet hydrogel layers lost their mechanical properties. It is worth noting that only the mechanical properties of D0 dropped below those of the control poly(ST-co-DVB)HIPE, which indicates that polyDMAEMA in D0 was as least partially incorporated into the pore walls of the that only the mechanical properties of D0 dropped below those of the control poly(ST-co-DVB)HIPE scaffold. However, the elastic moduli and crush strengths of D1–D10 are at about 24 and 1 MPa, respectively, similar within the series but, in case of the modulus, considerably higher than that of the control (Fig. 4c). As discussed above, the hydrogel of D1–D10 was grafted to the pore walls of their polyHIPEs scaffolds, leaving the mechanical properties of the scaffold itself unaffected by the hydration of the hydrogel. Furthermore, the entrapped water seems to provide some reinforcement, which causes the elastic moduli of D1–D10 to exceed the modulus of the unreinforced control.
Conclusions
Emulsion templates with monomers in both phases were employed to manufacture macroporous polymer/polymer composites with stiffness that can be controlled by adsorption of water, namely, polyMAA and polyDMAEMA-filled polyHIPEs, respectively. The presence of hydrogel could be proven by density and porosity measurements, as the polymer/polymer composites possess significantly higher densities and lower porosities than the hydrogel free control. However, the morphology differs dramatically depending on the hydrogels employed. The pore structure of polyMAA-filled polyHIPEs is highly interconnected, indicating the formation of an MAA-ST copolymer at the o/w interface of the emulsion templates during synthesis. However, polyDMAEMA-filled polyHIPEs appear to be predominately closed celled; the pore walls are covered by hydrogel, which was grafted to but not copolymerized with the styrene-based matrix. Water uptake measurements showed that the water absorption decreased with increasing crosslinking density of the hydrogel because diffusion through a highly crosslinked hydrogel is restricted. Moreover, evidence was found that the hydrogel covering the pore throats also decreased the water uptake of the samples. Both types of dry hydrogel reinforced the polyHIPE scaffolds; the mechanical properties, namely, the elastic moduli and crush strengths, were significantly higher than those of the hydrogel free control. The reinforcing ability of the incorporated (dried) hydrogels was further enhanced by increasing their crosslinking degree. However, the reinforcement could be “switched off” simply by hydrating the hydrogels. The switchability of mechanical properties might possibly be utilized in applications such as smart humidity sensor technology, where environmental changes can be indicated by variations in stiffness.
A collection of invited papers based on presentations at the 33rd International Conference on Solution Chemistry (ICSC-33), Kyoto, Japan, 7–12 July 2013.
The authors greatly acknowledge the funding provided by the UK Engineering and Physical Science Research Council (Grant: EP/D068851/1) and further funding by the University of Vienna.
References
[1] N. A. Peppas, Y. Huang, M. Torres-Lugo, J. H. Ward, J. Zhang. Ann. Rev. Biomed. Eng. 2, 9 (2000).10.1146/annurev.bioeng.2.1.9Search in Google Scholar
[2] L. Liu, A. Chakma, X. S. Feng. J. Membr. Sci. 310, 66 (2008).10.1016/j.memsci.2007.10.032Search in Google Scholar
[3] N. A. Peppas, P. Bures, W. Leobandung, H. Ichikawa. Eur. J. Pharm. Biopharm. 50, 27 (2000).Search in Google Scholar
[4] L. Brannon-Peppas, N. A. Peppas. Chem. Eng. Sci. 46, 715 (1991).10.1016/0009-2509(91)80177-ZSearch in Google Scholar
[5] R. Yoshida, K. Uchida, Y. Kaneko, K. Sakai, A. Kikuchi, Y. Sakurai, T. Okano. Nature 374, 240 (1995).10.1038/374240a0Search in Google Scholar
[6] H. Inomata, S. Goto, K. Otake, S. Saito. Langmuir 8, 6870 (1992).10.1021/la00038a064Search in Google Scholar
[7] I. Ohmine, T. Tanaka. J. Chem. Phys. 77, 5725 (1982).10.1063/1.443780Search in Google Scholar
[8] D. De Rossi, M. Suzuki, Y. Osada, P. Morasso. J. Intel. Mater. Syst. Struct. 3, 75 (1992).10.1177/1045389X9200300105Search in Google Scholar
[9] F. M. Andreopoulos, C. R. Deible, M. T. Stauffer, S. G. Weber, W. R. Wagner, E. J. Beckman, A. J. Russell. J. Am. Chem. Soc. 118, 6235 (1996).10.1021/ja954117cSearch in Google Scholar
[10] C. Santulli, S. I. Patel, G. Jeronimidis, F. J. Davis, G. R. Mitchell. Smart Mater. Struct. 14, 434 (2005).10.1088/0964-1726/14/2/018Search in Google Scholar
[11] J. Yang, M. Yang, W. Su, T. Leu. J. Membr. Sci. 236, 39 (2004).10.1016/j.memsci.2004.02.005Search in Google Scholar
[12] H. Matsuyama, M. Teramoto, H. Urano. J. Membr. Sci. 126, 151 (1997).10.1016/S0376-7388(96)00287-6Search in Google Scholar
[13] L. Xu, L. Zhang, H. Chen. Desalination 148, 309 (2002).10.1016/S0011-9164(02)00722-1Search in Google Scholar
[14] S. K. Samal, E. G. Fernandes, F. Chiellini, E. Chiellini. J. Therm. Anal. Calorim. 97, 859 (2009).10.1007/s10973-009-0337-4Search in Google Scholar
[15] N. R. Cameron, D. C. Sherrington. Adv. Polym. Sci. 126, 163 (1996).10.1007/3-540-60484-7_4Search in Google Scholar
[16] N. R. Cameron. Polymer 46, 1439 (2005).10.1016/j.polymer.2004.11.097Search in Google Scholar
[17] P. Krajnc, D. Stefanec, I. Pulko. Macromol. Rapid Commun. 26, 1289 (2005).10.1002/marc.200500353Search in Google Scholar
[18] O. Kulygin, M. S. Silverstein. Soft Matter 3, 1525 (2007).10.1039/b711610aSearch in Google Scholar PubMed
[19] R. Butler, I. Hopkinson, A. I. Cooper. J. Am. Chem. Soc. 125, 14473 (2003).10.1021/ja037570uSearch in Google Scholar
[20] N. C. Grant, A. I. Cooper, H. Zhang. ACS Appl. Mater. Interfaces 2, 1400 (2010).10.1021/am100049rSearch in Google Scholar PubMed
[21] S. Zhou, A. Bismarck, J. H. G. Steinke. J. Mater. Chem. 22, 18824 (2012).10.1039/c2jm33294aSearch in Google Scholar
[22] S. Zhou, A. Bismarck, J. H. G. Steinke. Macromol. Rapid Comm. 33, 1833 (2012).10.1002/marc.201200336Search in Google Scholar
[23] S. Zhou, A. Bismarck, J. H. G. Steinke. J. Mater. Chem. B 1, 4736 (2013).10.1039/c3tb20888eSearch in Google Scholar PubMed
[24] E. Ruckenstein, J. S. Park. J. Appl. Polym. Sci. 40, 213 (1990).10.1002/app.1990.070400117Search in Google Scholar
[25] E. Ruckenstein, H. H. Chen. J. Appl. Polym. Sci. 42, 2429 (1991).10.1002/app.1991.070420908Search in Google Scholar
[26] T. Gitli, M. S. Silverstein. Soft Matter 4, 2475 (2008).10.1039/b809346fSearch in Google Scholar
[27] T. Gitli, M. S. Silverstein. Polymer 52, 107 (2011).10.1016/j.polymer.2010.11.006Search in Google Scholar
[28] S. Kovačič, K. Jeřabek, P. Krajnc. Macromol. Chem. Phys. 212, 2151 (2011).10.1002/macp.201100229Search in Google Scholar
[29] E. Vallés, D. Durando, I. Katime, E. Mendizábal, J. E. Puig. Polym. Bull. 44, 109 (2000).10.1007/s002890050580Search in Google Scholar
[30] S. Kim, G. Iyer, A. Nadarajah, J. M. Frantz, A. L. Spongberg. Int. J. Polym. Anal. Chem. 15, 307 (2010).10.1080/1023666X.2010.493271Search in Google Scholar
[31] N. Sahloul, A. Penlidis. Polym.-Plast. Technol. Eng. 44, 771 (2005).10.1081/PTE-200060379Search in Google Scholar
[32] A. M. Mika, R. F. Childs, J. M. Dickson, B. E. McCarry, D. R. Gagnon. J. Membr. Sci. 135, 81 (1997).10.1016/S0376-7388(97)00122-1Search in Google Scholar
[33] H. Susanto, M. Ulbricht. Langmuir 23, 7818 (2007).10.1021/la700579xSearch in Google Scholar PubMed
[34] I. M. Kolthoff, I. K. Miller. J. Am. Chem. Soc. 73, 3055 (1951).10.1021/ja01151a024Search in Google Scholar
[35] P. Hainey, I. M. Huxham, B. Rowatt, D. C. Sherrington, L. Tetley. Macromolecules 24, 117 (1991).10.1021/ma00001a019Search in Google Scholar
[36] Y. Wu, S. Joseph, N. R. Aluru. J. Phys. Chem. B 113, 3512 (2009).10.1021/jp808145xSearch in Google Scholar PubMed
[37] L. L. C. Wong, V. O. Ikem, A. Menner, A. Bismarck. Macromol. Rapid Comm. 32, 1563 (2011).10.1002/marc.201100382Search in Google Scholar
[38] S. S. Manley, N. Graeber, Z. Grof, A. Menner, G. F. Hewitt, F. Stepanek, A. Bismarck. Soft Matter 5, 4780 (2009).10.1039/b900426bSearch in Google Scholar
[39] V. O. Ikem, A. Menner, T. S. Horozov, A. Bismarck. Adv. Mater. 22, 3588 (2010).10.1002/adma.201000729Search in Google Scholar
[40] L. L. C. Wong, P. M. B. Villafranca, A. Menner, A. Bismarck. Langmuir 29, 5952 (2013).10.1021/la3047643Search in Google Scholar PubMed
©2014 IUPAC & De Gruyter Berlin Boston
Articles in the same Issue
- Masthead
- Masthead
- Preface
- International Union of Pure and Applied Chemistry
- Conference paper
- Optimization of superamphiphobic layers based on candle soot
- Probing “ambivalent” snug-fit sites in the KcsA potassium channel using three-dimensional reference interaction site model (3D-RISM) theory
- Perspectives for hybrid ab initio/molecular mechanical simulations of solutions: from complex chemistry to proton-transfer reactions and interfaces
- The complex structure of ionic liquids at an atomistic level: from “red-and-greens” to charge templates
- Amide I IR probing of core and shell hydrogen-bond structures in reverse micelles
- Kinetic studies on cyclopalladation in palladium(II) complexes containing an indole moiety
- Properties of the one-electron oxidized copper(II) salen-type complexes: relationship between electronic structures and reactivities
- Solvation of a sponge-like geometry
- What is “hypermobile” water?: detected in alkali halide, adenosine phosphate, and F-actin solutions by high-resolution microwave dielectric spectroscopy
- Reentrant condensation, liquid–liquid phase separation and crystallization in protein solutions induced by multivalent metal ions
- Emulsion-templated macroporous polymer/polymer composites with switchable stiffness
- Effective interaction between small unilamellar vesicles as probed by coarse-grained molecular dynamics simulations
- Enthalpies of solution, limiting solubilities, and partial molar heat capacities of n-alcohols in water and in trehalose crowded media
- Protonation of alkanolamines and cyclic amines in water at temperatures from 293.15 to 373.15 K
- IUPAC Technical Report
- Defining the transfer coefficient in electrochemistry: An assessment (IUPAC Technical Report)
- IUPAC Recommendations
- Definition of the transfer coefficient in electrochemistry (IUPAC Recommendations 2014)
Articles in the same Issue
- Masthead
- Masthead
- Preface
- International Union of Pure and Applied Chemistry
- Conference paper
- Optimization of superamphiphobic layers based on candle soot
- Probing “ambivalent” snug-fit sites in the KcsA potassium channel using three-dimensional reference interaction site model (3D-RISM) theory
- Perspectives for hybrid ab initio/molecular mechanical simulations of solutions: from complex chemistry to proton-transfer reactions and interfaces
- The complex structure of ionic liquids at an atomistic level: from “red-and-greens” to charge templates
- Amide I IR probing of core and shell hydrogen-bond structures in reverse micelles
- Kinetic studies on cyclopalladation in palladium(II) complexes containing an indole moiety
- Properties of the one-electron oxidized copper(II) salen-type complexes: relationship between electronic structures and reactivities
- Solvation of a sponge-like geometry
- What is “hypermobile” water?: detected in alkali halide, adenosine phosphate, and F-actin solutions by high-resolution microwave dielectric spectroscopy
- Reentrant condensation, liquid–liquid phase separation and crystallization in protein solutions induced by multivalent metal ions
- Emulsion-templated macroporous polymer/polymer composites with switchable stiffness
- Effective interaction between small unilamellar vesicles as probed by coarse-grained molecular dynamics simulations
- Enthalpies of solution, limiting solubilities, and partial molar heat capacities of n-alcohols in water and in trehalose crowded media
- Protonation of alkanolamines and cyclic amines in water at temperatures from 293.15 to 373.15 K
- IUPAC Technical Report
- Defining the transfer coefficient in electrochemistry: An assessment (IUPAC Technical Report)
- IUPAC Recommendations
- Definition of the transfer coefficient in electrochemistry (IUPAC Recommendations 2014)