Kinetic studies on cyclopalladation in palladium(II) complexes containing an indole moiety
-
Satoshi Iwatsuki
 ,
Takuya Suzuki
,
Takuya Suzuki

Abstract
Various Pd–C complexes have been developed to date, affording deep insights into the reaction intermediates in useful catalytic reactions in organic syntheses. Cyclopalladation is one of the most famous Pd–C bond formation reactions to generate the palladacycles. Indole is an electron-rich aromatic ring involved in the side chain of an essential amino acid, tryptophan (Trp), and Trp and its derivatives are important in biological systems, such as electron transfer in protein, cofactors for conversion of biological molecules and so on. Pd catalysts are also useful for syntheses of such indole derivatives, and the mechanisms are considered to be through the Pd–C intermediates. However, the detailed properties and formation mechanisms of Pd–indole species are still unclear. With these points in mind, we focus on Pd(II)–indole-C2 carbon bond formations using various Pd(II) complexes having an indole moiety, especially on the recent studies on the kinetic analyses for these cyclopalladation reactions and their detailed mechanisms.
Introduction
Cyclopalladation has been intensely studied not only for the coordination and organometallic chemistry but also for the importance of Pd–C intermediates in catalytic organic syntheses. Many Pd–C bond formations including cyclopalladation have been widely developed to date, affording deep insights into the reaction mechanisms for many useful homogeneous catalytic reactions [1–10]. Especially Pd–C binding complexes, which are becoming important intermediates in catalytic systems, have been formed in general by the oxidative insertion of the organic substrates or by the reaction with the C–H activated species [1–10].
On the other hand, the indole ring of the tryptophyl side chain residue having the highest hydrophobicity among natural α-amino acids [11] is known to provide a hydrophobic microenvironment for enzyme catalysis, protein stability, specific binding of molecules, etc. [12–15]. It is involved in electron-transfer pathways, and formation of the indolyl radical at a tryptophyl residue has been established for the intermediate, compound I, formed in the catalytic cycle of cytochrome c peroxidase (CcP) [15–19]. It is known to form covalent bonds with Pd(II), etc. through the nitrogen and carbon atoms [20–30] besides π–π interactions with aromatic nitrogen donors in metal complexes [30–34]. The pendent indole ring in metal complexes of tripodal ligands was found to bind to Cu(I) by η2 coordination of the C2=C3 bond to form a tetrahedral structure [20]. Similar η2 coordination has been recently detected in biological systems for a Cu chaperone CusF [21, 22]. On the other hand, Yamauchi et al. reported Pd(II)–C3 bond formation in complexes with indole derivatives [23–25]. Kostić et al. developed unique artificial peptidases using Pd(II) and Pt(II), which recognize the indole moiety and cleave the adjacent amide or peptide bond specifically [26–29].
With these points in mind, we have synthesized and characterized Pd(II)–indole C2 bonding complexes and analyzed their Pd–indole carbon bond formation through cyclopalladation [30, 35–38]. We present here our recent studies on reversible Pd–indole C2 carbon bond formations in complexes of 2N1O-donor ligands with an indole and phenol moiety [35–37] and on a low concentration-favored Pd–C bond formation of the complex with a 2N-donor ligand with an indole moiety (Scheme 1) [38]. These results may be important for deeper understanding of the reaction mechanism of the catalytic conversion for the organic substrates having Pd–C intermediate as well as the significance of metal–indole complexes in biological systems.
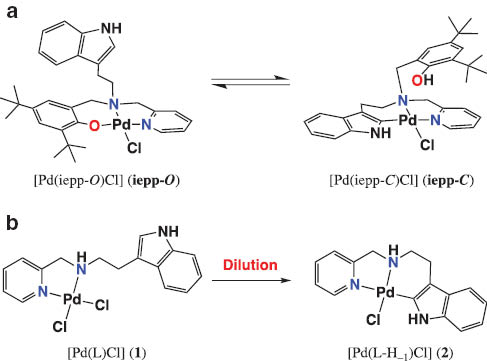
Reversible Pd–indole C2 carbon bond formations: (a) a complex of 2N1O-donor ligand with an indole and phenol moiety; (b) low concentration-favored Pd–C bond formation of the complex with a 2N-donor ligand with an indole moiety.
Cyclopalladation of Pd(II) complexes with 2N1O-donor ligand with an indole ring: interconversion between phenolate complex and indole-binding complex
The pendent indole ring in the Pd(II) complex of a 2N1O-donor ligand involving a phenolate, a pyridine, and a tertiary amine as metal binding sites ([Pd(iepp-O)Cl] in Scheme 1a) replaced the phenolate group under suitable conditions to form a complex with a direct Pd–C2 bond ([Pd(iepp-C)Cl] in Scheme 1a) [35]. In this Pd–C bond formation mechanism, the proton dissociated from the indole ring binds to the phenolate moiety. The conversion property depends on the nature not only of the O-donor but also of the aromatic N-donor atoms.
Effect of N-donor properties on cyclopalladation
In order to understand the participation of the pendent indole ring of Pd(II) complexes in Pd–C(indole) bond formation, we synthesized the Pd(II) complexes of 2N1O-donor tridentate ligands having a different aromatic N-donor such as imidazole (Hieip) and methyl-substituted imidazole and pyridine derivatives (HMe3-ieip and HMe-iepp, respectively) as a substitute for the pyridine ring in ligand Hiepp (Fig. 1) [35]. Among the methyl-substituted pyridines as aromatic N-donors, 2-methylpyridine has been reported to show weaker donor properties than pyridine itself due to steric requirement [36, 39]. However, the steric effect in 4,5-dimethylimidazole is smaller in comparison to that in pyridine due to a larger N–C–Me angle in the pentagonal ring.

Structures of 2N1O-tridentate ligands with a pendent indole moiety and various aromatic nitrogen donors.
The X-ray structures of the phenolate complexes (Fig. 2) revealed the square-planar four-coordinated Pd(II) complexes. However, the Pd–N bond length and the distortion from the coordination plane depended on the N-donor properties. The Pd–aromatic nitrogen (Narom) bond length in this series was found to be longer for the methyl-substituted complexes, the order being ieip-O (1.986(4) Å) < iepp-O (2.011(4)) < Me3-ieip-O (2.015(7)) < Me-iepp-O (2.050(2)), while the Pd–tertiary amine nitrogen (Namine) bond length showed the opposite characteristics [36]. The results suggest that the methyl-substituted pyridine and imidazole are weaker N-donors than unsubstituted ones. Further, the angles of O–Pd–Narom and Cl–Pd–Namine are affected by the methyl-substituted aromatic nitrogen ligand; for example, the Cl–Pd–Namine angles are distorted from 180° in the order ieip-O (176.9(1)°) ≤ iepp-O (176.2(1)°) ≤ Me3-ieip-O (175.2(2)°) < Me-iepp-O (170.47(6)°). Thus, the methyl substituent on the aromatic N-donor causes distortion from the square plane due to its steric hindrance [36, 39].
![Fig. 2
Molecular structures of Pd(II)–phenolato complexes: (a) [Pd(iepp-O)Cl] (iepp-O), (b) [Pd(ieip-O)Cl] (ieip-O), (c) [Pd(Me3-ieip-O)Cl] (Me3-ieip-O), and (d) [Pd(Me-iepp-O)Cl] (Me-iepp-O).](/document/doi/10.1515/pac-2014-5020/asset/graphic/j_pac-2014-5020_fig_003.jpg)
Molecular structures of Pd(II)–phenolato complexes: (a) [Pd(iepp-O)Cl] (iepp-O), (b) [Pd(ieip-O)Cl] (ieip-O), (c) [Pd(Me3-ieip-O)Cl] (Me3-ieip-O), and (d) [Pd(Me-iepp-O)Cl] (Me-iepp-O).
The indole-binding complexes (Fig. 3) revealed different structural features from the phenolate complexes (Fig. 2); the Pd–C bond length in these complexes is affected by the methyl substituent of the aromatic N-donor, as observed for the Pd–C bond length of ieip-C (1.972(3) Å) and of Me3-ieip-C (2.005(6) Å). In comparison to the phenolate complexes, all of the indole–binding complexes have a longer Pd–N bond, suggesting that the carbanion donor of the indole moiety is more donating to the Pd ion. On the other hand, the differences in the angles of C–Pd–Narom and Cl–Pd–Namine in indole-binding complexes are rather small in comparison to the phenolate complexes, indicating that the Pd–indole bond requires a “flat” geometry [39]. It is interesting to note in Fig. 3 that the OH group of the phenol moiety in complex iepp-C is directed toward the center of the Pd–C bond with weak interaction, while that in Me3-ieip-C is directed toward the outside of the molecule. Complexes ieip-C and Me-iepp-C were revealed to be a 1:1 mixture of the structures with these different OH group directions, and as a result, the phenol OH group was disordered. These structural differences suggest that the electron density of the Pd–C bond may be ordered as Me3-ieip-C < ieip-C = Me-iepp-C < iepp-C, which may correlate with the Pd–O to Pd–C conversion rate and the stability of the indole-binding complexes.
![Fig. 3
Molecular structures of Pd–indole binding complexes: (A) [Pd(iepp-C)Cl] (iepp-C), (B) [Pd(ieip-C)Cl] (ieip-C), (C) [Pd(Me3-ieip-C)Cl] (Me3-ieip-C), and (D) [Pd(Me-iepp-C)Cl] (Me-iepp-C). (a) and (b) are the structures of two isomers with a different OH direction and observed as disordered.](/document/doi/10.1515/pac-2014-5020/asset/graphic/j_pac-2014-5020_fig_004.jpg)
Molecular structures of Pd–indole binding complexes: (A) [Pd(iepp-C)Cl] (iepp-C), (B) [Pd(ieip-C)Cl] (ieip-C), (C) [Pd(Me3-ieip-C)Cl] (Me3-ieip-C), and (D) [Pd(Me-iepp-C)Cl] (Me-iepp-C). (a) and (b) are the structures of two isomers with a different OH direction and observed as disordered.
All of the phenolate complexes convert to the indole-binding complexes with a color change from orange to pale yellow, in which typical absorption bands of the phenolato complexes gradually decreased with a new band appearing with isosbestic points [36]. Such spectral changes were also observed in the 1H NMR spectra; the signals of the phenolate complexes gradually disappeared, and those of the indole-binding complexes appeared without proton signals of any other products. Further, the formation ratio from the phenolate to indole-binding species depends on the temperature. These results suggest reversible conversion and an intramolecular process, which is expressed by the following equation:

where k1 and k–1 denote the rate constants of the forward and backward processes, respectively, and the equilibrium constant is expressed by K = k1/k–1 = [indole-binding complex]/[phenolate complex]. All the reversible conversions could be analyzed by the first-order kinetics, and the observed first-order rate constant kobs is expressed by kobs = k1 + k–1 [40, 41]. From the dependences of K and kobs on temperature, the thermodynamic parameters (ΔH° and ΔS°) and activation parameters (ΔH1‡ and ΔS1‡ for k1 path, and ΔH–1‡ and ΔS–1‡ for k–1 path, respectively) for all the complexes in this study were determined as shown in Table 1.
Thermodynamic and activation parameters for interconversion between phenolate and indole-binding species [36].
| Complex |
K
|
k
1 path |
K
–
1 path |
Temperature range | |||
|---|---|---|---|---|---|---|---|
| ΔH°/kJ mol–1 | ΔS°/J mol–1K–1 | ΔH1‡/kJ mol–1 | ΔS1‡/J mol–1 K–1 | ΔH–1‡/kJ mol–1 | ΔS–1‡/J mol–1 K–1 | ||
| iepp | 1 | 1 | 99 ± 2 | –47 ± 7 | 98 ± 2 | –47 ± 6 | 60–90 °C |
| ieip | 12 | 23 | 100 ± 5 | –51 ± 15 | 88 ± 6 | –74 ± 16 | 60–90 °C |
| Me3-ieip | 11 | 23 | 104 ± 1 | –39 ± 1 | 94 ± 1 | –61 ± 1 | 60–90 °C |
| Me-iepp | 10 | 27 | 102 ± 2 | –26 ± 7 | 92 ± 1 | –54 ± 4 | 50–80 °C |
In comparison to the iepp complex system, all the other complexes showed large ΔH° and ΔS° values, which indicates that the formation ratio of the indole-binding species increases with the increase of the reaction temperature. The iepp and Me-iepp complex systems showed that the thermodynamic parameters are significantly different due to the effect of steric hindrance, while the ieip and Me3-ieip systems showed very similar thermodynamic parameters.
The activation enthalpies (ΔH1‡) of all the complexes are estimated to be ca. 100 kJ mol–1, which suggest that the sum of the binding energy changes with the conversion in all the complexes is similar. On the other hand, the negative activation entropies (ΔS1‡) indicate that the mechanism of the conversion upon achieving the transition state is associative, that is, the five-coordinated intermediate is preferred in the conversion [42–44]. Further, the order of the ΔS1‡ value is as follows: Me-iepp (ΔS1‡ = –26 J mol–1 K–1) > Me3-ieip (–39) > iepp (–47) > ieip (–51), which agrees well with the order of the distortion from the square plane of the Pd coordination sphere in the phenolate complexes. The ΔS1‡ indicates the changes of the freedom of the structural change upon achieving the transition state [41], and therefore the ΔS1‡ is affected by the distortion of the Pd coordination sphere. Thus, the steric hindrance is more effective in comparison to the basicity of the nitrogen donors in the conversion to the indole-binding species.
Effect of O-donor properties on cyclopalladation
For further understanding of the interconversion between the phenolate and the indole-binding complexes having a 2N1O-donor ligand, it is important to investigate the effect of the O-donor property on the reactivity. We have prepared a series of indole-containing ligands with various substituted phenol moieties or carboxylic acid having a different acidity (Fig. 4).

Structures of 2N1O-tridentate ligands with a pendent indole moiety for investigating the effect of O-donor properties.
X-ray crystal structure analysis revealed that these structures (Fig. 5) are very similar to those of complex iepp-O and its derivatives (Fig. 2) [35, 36]. The Pd–O bond length increased in the order iepp-MeO-O (1.989(2) Å) < iepp-NO2-O (2.009(8) Å) < iepc-O (2.026(2) Å). This tendency is in agreement with the order of the pKa value of the O-donor [45, 46].
![Fig. 5
Structures of Pd(II)–phenolato complexes: (a) [Pd(iepp-MeO-O)Cl] (iepp-MeO-O), (b) [Pd(iepp-NO2-O)Cl] (iepp-NO2-O), and (c) [Pd(iepc-O)Cl] (iepc-O).](/document/doi/10.1515/pac-2014-5020/asset/graphic/j_pac-2014-5020_fig_007.jpg)
Structures of Pd(II)–phenolato complexes: (a) [Pd(iepp-MeO-O)Cl] (iepp-MeO-O), (b) [Pd(iepp-NO2-O)Cl] (iepp-NO2-O), and (c) [Pd(iepc-O)Cl] (iepc-O).
When complex iepp-MeO-O in DMSO and DMF was kept at 50 °C for a few days, a color change from orange to yellow occurred to give the indole-binding complexes [37]. The formation ratio of the indole-binding complex to the phenolate complex was higher for the complexes of the ligands with a higher pKa value of the conjugate acid of O-donor in the order, iepp-MeO > iepp > iepp-NO2 > iepc [37]. The results indicate that the complex of the ligand having a more basic O-donor shows higher formation ratio. The ratio of the phenolate complex to the indole-binding complex was temperature-independent in the range from 40 to 80 °C, i.e., ΔH° ~ 0 kJ mol–1.
All the conversions from the phenolate complex to the indole-binding complex could be analyzed by the first-order kinetics, suggesting a reversible reaction and an intramolecular process. Therefore, the activation parameters for the conversion were determined from the dependence of the observed first-order rate constants (kobs) on temperature, and listed in Table 2.
Activation parameters for formation of the indole-binding Pd(II) complexes of 2N1O-donor ligand having a different O-donor moiety in DMF [37].
| Complex | ΔH‡/kJ mol–1 | ΔS‡/J mol–1 K–1 | 104kobs333/s–1a | Temperature range |
|---|---|---|---|---|
| iepp | 92 ± 4 | –64 ± 10 | 0.122 | 40–70 °C |
| MeO-iepp | 96 ± 2 | –51 ± 6 | 0.122 | 40–70 °C |
| NO2-iepp | 72 ± 1 | –94 ± 3 | 3.64 | 40–70 °C |
| iepc | 38 ± 2 | –170 ± 6 | 107 | 30–60 °C |
aThe kobs value at 60 °C estimated from the activation parameters.
The observed first-order rate constants at 60 °C (kobs333 in Table 2) indicate that the conversion rate for the indole-binding Pd(II) species also depends on the O-donor properties; for the complexes of the ligands with a lower pKa value of the conjugate acid of O-donor exhibited a faster rate of conversion to the indole-binding species in the order iepp-MeO < iepp < iepp-NO2 < iepc. The plot of the activation energy vs. the pKa value of the conjugate acid of oxygen donor is shown in Fig. 6. This result clearly indicates that the barrier of the conversion correlates with the O-donor ability. In other words, the barrier correlates with the bond strength of Pd–O, the order of which is MeO-phenol < NO2-phenol < COO–. The rate-determining step of the conversion from the phenolate complex to the indole-binding complex may involve the Pd–O bond cleavage besides the C–H bond cleavage process.
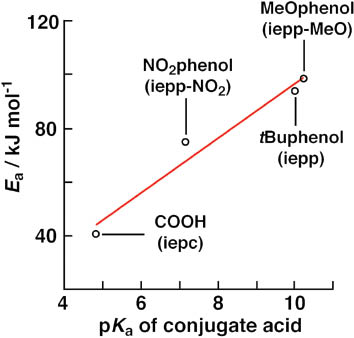
Activation energies (Ea) as a function of the pKa values of conjugate acids of the ligand O-donor groups.
It is generally agreed that cyclopalladation reactions of aryl compounds proceed by an electrophilic substitution by Pd(II) ions [9, 42–44, 47–53]. The electrophilicity of the Pd(II) center to the indole moiety should increase with the decreasing pKa value of the ligand OH group, and our experiments have shown that the reaction rates are in the order iepp-MeO-O < iepp-O < iepp-NO2-O < iepc-O (Table 2), which is in agreement with the decreasing order of the pKa values of the corresponding ligands. On the other hand, the formation ratio of the indole-binding complex to the O-donor complex was higher for the complexes of the ligands with a higher pKa value in the order iepp-MeO-O > iepp-O > iepp-NO2-O > iepc-O. Further, the addition of a base enhanced the formation of the indole-binding complex, which indicates that basic conditions are favorable for the present cyclopalladation [37]. Thus, the O-donor to C-donor conversion reaction may be explained by a mechanism involving a five-coordinate intermediate [36, 37, 42–44].
Cyclopalladation of Pd(II) complexes with 2N-donor ligands with a pendent indole ring: low concentration-favored cyclopalladation
As mentioned in the above section, the complexes of 2N1O-donor ligands having both a phenol and an indole moiety are interconvertible in solution between the Pd(II)–phenolate and Pd(II)–indole complexes [35]. The conversion kinetics of the similar geometry depend on the aromatic N-donors and the O-donor properties [36, 37], and especially the pKa value of the O-donor is very important for conversion to the indole-binding species [37]. The results may suggest that the role of exogenous donors, such as solvent molecules, is important for formation of the Pd–C bond via Pd–O intermediate. In order to understand the mechanisms of the interconversion between the Pd(II)–phenolate and Pd(II)–indole complexes in detail, we synthesized the Pd(II) complex of a simple 2N-donor ligand containing a pendent indole moiety and investigated conversion of the dichloro complex to the indole-binding complex in DMSO. From the detailed reaction analyses, we concluded that conversion to the indole-binding complex depends on the concentration of its solution, that is, a low concentration-favored the Pd–C bond formation (Scheme 1) [38].
The Pd(II) complex of a 2N-donor ligand, [Pd(L)Cl2] (1), where L is 3-(2-pyridylmethylamino)ethylindole (L), has a square-planar geometry with the pyridine and amine nitrogen atoms and two chloride ions (Fig. 7). The indole-binding species of the ligand L could be also isolated as [Pd(L-H–1)Cl] (2) by addition of 1 equiv of triethylamine [38], and was supported by X-ray crystal structure and physicochemical measurements. Complex 2 exhibited the characteristic UV-vis absorption band for the indole-binding species at 307 nm, which is in good agreement with the other indole-binding species [54]. The conversion from 1 to 2 could not be detected in DMSO-d6 (0.1 M solution) at 60 °C, which was different from the observation on the complexes of the previous 2N1O donor-ligands.
![Fig. 7
Formation reactions and molecular structures of Pd(II) complexes with a 2N-donor ligand (L) having an indole moiety; [Pd(L)Cl2] (1) and [Pd(L-H–1)Cl] (2).](/document/doi/10.1515/pac-2014-5020/asset/graphic/j_pac-2014-5020_fig_009.jpg)
Formation reactions and molecular structures of Pd(II) complexes with a 2N-donor ligand (L) having an indole moiety; [Pd(L)Cl2] (1) and [Pd(L-H–1)Cl] (2).
However, at a lower concentration of 1 (< ca. 10 mM), some proton signals of new species (2) appeared with concomitant disappearance of the signals of 1 even at room temperature. The results in Fig. 8 clearly show the concentration dependent conversion from 1 to 2 without any decomposition. The low concentration-favored conversion of [Pd(L)Cl2] (1) to [Pd(L-H–1)Cl] (2) in DMSO can be expressed by the following equation:

1H NMR spectra in DMSO-d6 at room temperature (a) and the absorption spectra in DMSO at 25 °C (b) at various initial concentrations (CPd) of complex 1. The filled and open circles in (a) denote the signals of complexes 1 and 2, respectively.

The equilibrium constant K = [2][H+][Cl–]/[1] could be determined to be K = 5 × 10–8 M2 from the 1H NMR spectral data (cf. Fig. 8a). This K value was also estimated to be in the order of 10–8 M2 from the UV-vis spectral data (cf. Fig. 8b) [38].
The initial rates analyses [41, 55] for this conversion by dilution of 1 revealed that the initial rate (v) linearly depended on both the initial concentration of 1 (CPd) and the inverse of the concentration of Cl– (1/CCl), i.e., v = kCPd/CCl, but the v was independent of the proton concentration [38]. The results suggest that the reversible dissociation of a coordinated Cl– ion from 1 is involved in the forward reaction via a solvated complex, such as [Pd(L)Cl(DMSO)]+ [40, 56–60] as shown in Scheme 2.
![Scheme 2
Proposed conversion mechanism of Pd(II) complexes [Pd(L)Cl2] (1) and [Pd(L-H–1)Cl] (2) without base in DMSO. The rate constants were estimated by the kinetic analyses for the 2-to-1 conversion under the pseudo first-order conditions (CHCl′ >> CPd).](/document/doi/10.1515/pac-2014-5020/asset/graphic/j_pac-2014-5020_fig_012.jpg)
Proposed conversion mechanism of Pd(II) complexes [Pd(L)Cl2] (1) and [Pd(L-H–1)Cl] (2) without base in DMSO. The rate constants were estimated by the kinetic analyses for the 2-to-1 conversion under the pseudo first-order conditions (CHCl′ >> CPd).
The conversion of 2 to 1, that is, the backward reaction in eq. 2 could be kinetically analyzed under the pseudo first-order conditions; [H+] and [Cl–] (= CHCl′) are much higher than CPd: the decrease of 2 was expressed by –d[2]/dt = kobs[2] at CHCl′ >> CPd, where kobs is the conditional pseudo first-order rate constant. The dependence of kobs on CHCl′ was successfully analyzed by applying the steady-state approximation [40, 41] for the intermediate I in the reverse reaction mechanism of the 1-to-2 conversion (Scheme 2) [38]. Thus, the 2-to-1 conversion was also found to proceed via the solvated intermediate, [Pd(L)Cl(DMSO)]+.
The reaction mechanism of the low concentration-favored 1-to-2 interconversion involves the solvated intermediate [Pd(L)Cl(DMSO)]+ (Scheme 2). On the contrary, no intermediate was involved in the phenolate-to-indole conversion in Pd(II) complexes of 2N1O-donor ligands, which intramolecularly proceeded via a five-coordinated transition state [36, 37]. It is noted that the 1-to-2 conversion is initiated by the Cl– interchange with DMSO in 1. This reaction step does not occur in all the phenolate-to-indole conversion system of the complexes of 2N1O-ligands mentioned in the above section [35–37]. The reaction step of the [Pd(L)Cl(DMSO)]+ intermediate to give 2 (k2 and k–2 paths in Scheme 2) may correspond to “overall” phenolate-to-indole conversion of the 2N1O-donor ligand complexes, because both these reactions involve three steps; C–H bond cleavage, Pd–C bond formation, and the dissociation of one coordinated ligand. The rate constant k2 for the indole-binding process of [Pd(L)Cl(DMSO)]+ was estimated to be >> 7.6 × 10–3 s–1 (Scheme 2), which is much larger than the rate constant for the phenolate-to-indole conversion system [36, 37]. The difference can be ascribed to the difference of the pKa values, that is, the proton affinity of DMSO is much lower than that of the phenolate moiety.
This concentration-dependent conversion can be assigned to the concentration-dependent 1-to-2 conversion consisting of two successive thermodynamically controlled processes. Both processes prefer lower concentrations of dissociated free ions such as Cl– and H+, which is just the case of lowering the concentration of the precursor complex 1. This is the reason why the 1-to-2 conversion is low concentration-favored.
Summary
This review is focused on characterization of Pd(II) complexes having a pendent indole moiety and their conversion mechanism to indole-binding species. The conversion between the phenolate and indole-binding complexes is dependent on some important factors. One of them is the N-donor property of the ligands and especially the steric requirement of the N-donor, which can cause distortion of the Pd(II) complexes from the square-planar structure, and both these factors affect the kinetics of conversions. On the other hand, the pKa value of the O-donor is also important especially for deprotonation of the O–H bond and protonation of the coordinated O-donor concomitant with the indole C–H bond cleavage. Further, the concentration-dependent Pd–C bond formation in the Pd(II) complex of a 2N-donor ligand was also described. It can be understood on the basis of the above fundamental results from the 2N1O-donor systems. These studies further suggest that in some cases the Pd-catalyzed cross-coupling reaction may proceed through possible formation of a solvated complex intermediate at low concentrations. Thus, the present findings may be important for understanding the reaction mechanism of the catalytic conversion for the organic substrates having Pd–C intermediate and construction of highly efficient catalytic systems.
A collection of invited papers based on presentations at the 33rd International Conference on Solution Chemistry (ICSC-33), Kyoto, Japan, 7–12 July 2013.
This work was supported in part by a Grant-in-Aid for Scientific Research (No. 21750082 to S. I. and No. 80335992 and 22550055 to Y. S.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
[1] T. W. Lyons, M. S. Sanford. Chem. Rev. 110, 1147 (2010).10.1021/cr900184eSearch in Google Scholar
[2] V. Ritleng, C. Sirlin, M. Pfeffer. Chem. Rev. 102, 1731 (2002).10.1021/cr0104330Search in Google Scholar
[3] M. Albrecht. Chem. Rev. 110, 576 (2010).10.1021/cr900279aSearch in Google Scholar
[4] N. Miyaura, A. Suzuki. Chem. Rev. 95, 2457 (1995).10.1021/cr00039a007Search in Google Scholar
[5] R. Jana, T. P. Pathak, M. S. Sigman. Chem. Rev. 111, 1417 (2011).10.1021/cr100327pSearch in Google Scholar
[6] D. J. M. Snelders, G. van Koten, R. J. M. Klein Gebbink. J. Am. Chem. Soc. 131, 11407 (2009).10.1021/ja904042hSearch in Google Scholar
[7] K. Matos, J. A. Soderquist. J. Org. Chem. 63, 461 (1998).10.1021/jo971681sSearch in Google Scholar
[8] A. J. Canty, G. van Koten. Acc. Chem. Res. 28, 406 (1995).10.1021/ar00058a002Search in Google Scholar
[9] A. D. Ryabov. Chem. Rev. 90, 403 (1990).10.1021/cr00100a004Search in Google Scholar
[10] R. H. Crabtree. Chem. Rev. 110, 575 (2010).10.1021/cr900388dSearch in Google Scholar
[11] Y. Nozaki, C. Tanford. J. Biol. Chem. 246, 2211 (1971).10.1016/S0021-9258(19)77210-XSearch in Google Scholar
[12] S. K. Burley, G. A. Petsko. Science, 229, 23 (1985).10.1126/science.3892686Search in Google Scholar
[13] J. W. Whittaker. Met. Ions Biol. Syst. 30, 315 (1994).10.1109/7.272257Search in Google Scholar
[14] M. S. Rogers, E. M. Tyler, N. Akyumani, C. R. Kurtis, R. K. Spooner, S. E. Deacon, S. Tamber, S. J. Firbank, K. Mahmoud, P. F. Knowles, S. E. V. Phillips, M. J. McPherson, D. M. Dooley. Biochemistry 46, 4606 (2007).10.1021/bi062139dSearch in Google Scholar
[15] J. M. Nocek, J. S. Zhou, S. De Forest, S. Priyadarshy, D. N. Beratan, J. N. Onuchic, B. M. Hoffman. Chem. Rev. 96, 2459 (1996).10.1021/cr9500444Search in Google Scholar
[16] J. Stubbe, W. A. van der Donk. Chem. Rev. 98, 705 (1998).10.1021/cr9400875Search in Google Scholar
[17] S. S. Isied. Met. Ions Biol. Syst. 27, 1 (1991).Search in Google Scholar
[18] M. Sivaraja, D. B. Goodin, M. Smith, B. M. Hoffman. Science 245, 738 (1989).10.1126/science.2549632Search in Google Scholar
[19] T. L. Poulos, R. E. Fenna. Met. Ions Biol. Syst. 30, 25 (1994).10.1007/BF02631267Search in Google Scholar
[20] Y. Shimazaki, H. Yokoyama, O. Yamauchi. Angew. Chem., Int. Ed. 38, 2401 (1999).10.1002/(SICI)1521-3773(19990816)38:16<2401::AID-ANIE2401>3.0.CO;2-VSearch in Google Scholar
[21] Y. Xue, A. V. Davis, G. Balakrishnan, J. P. Stasser, B. M. Staehlin, P. Focia, T. G. Spiro, J. E. Penner-Hahn, T. V. O’Halloran. Nat. Chem. Biol. 4, 107 (2008).10.1038/nchembio.2007.57Search in Google Scholar
[22] K. J. Franz. Nat. Chem. Biol. 4, 85 (2008).10.1038/nchembio0208-85Search in Google Scholar
[23] O. Yamauchi, M. Takani, K. Toyoda, H. Masuda. Inorg. Chem. 29, 1856 (1990).10.1021/ic00335a019Search in Google Scholar
[24] M. Takani, H. Masuda, O. Yamauchi. Inorg. Chim. Acta 235, 367 (1995).10.1016/0020-1693(95)90078-KSearch in Google Scholar
[25] M. Takani, T. Takeda, T. Yajima, O. Yamauchi. Inorg. Chem. 45, 5938 (2006).10.1021/ic060595kSearch in Google Scholar
[26] N. V. Kaminskaia, T. W. Johnson, N. M. Kostić. J. Am. Chem. Soc. 121, 8663 (1999).10.1021/ja9840246Search in Google Scholar
[27] N. V. Kaminskaia, G. M. Ullmann, D. B. Fulton, N. M. Kostić. Inorg. Chem. 39, 5004 (2000).10.1021/ic000254lSearch in Google Scholar
[28] N. V. Kaminskaia, N. M. Kostić. Inorg. Chem. 40, 2368 (2001).10.1021/ic0014033Search in Google Scholar
[29] N. M. Milović, N. M. Kostić. Met. Ions Biol. Syst. 38, 145 (2001).Search in Google Scholar
[30] Y. Shimazaki, T. Yajima, M. Takani, O. Yamauchi. Coord. Chem. Rev. 253, 479 (2009).10.1016/j.ccr.2008.04.012Search in Google Scholar
[31] B. E. Fischer, H. Sigel. J. Am. Chem. Soc. 102, 2998 (1980).10.1021/ja00529a021Search in Google Scholar
[32] L. Guogang, H. Sigel. Z. Naturforsch., B 44, 1555 (1989).10.1515/znb-1989-1215Search in Google Scholar
[33] O. Yamauchi, A. Odani, S. Hirota. Bull. Chem. Soc. Jpn. 74, 1525 (2001).10.1246/bcsj.74.1525Search in Google Scholar
[34] O. Yamauchi, A. Odani, M. Takani. J. Chem. Soc., Dalton Trans. 3411 (2002) and refs. cited therein.10.1039/B202385GSearch in Google Scholar
[35] T. Motoyama, Y. Shimazaki, T. Yajima, Y. Nakabayashi, Y. Naruta, O. Yamauchi. J. Am. Chem. Soc. 126, 7378 (2004).10.1021/ja031587vSearch in Google Scholar
[36] S. Tanooka, T. Suzuki, T. Yajima, T. Shiraiwa, S. Iwatsuki, Y. Shimazaki. Inorg. Chim. Acta 407, 41 (2013).10.1016/j.ica.2013.07.032Search in Google Scholar
[37] Y. Shimazaki, M. Tashiro, T. Motoyama, S. Iwatsuki, T. Yajima, Y. Nakabayashi, Y. Naruta, O. Yamauchi. Inorg. Chem. 44, 6044 (2005).10.1021/ic0504801Search in Google Scholar
[38] S. Iwatsuki, T. Suzuki, T. Yajima, T. Shiraiwa, O. Yamauchi, Y. Shimazaki. Inorg. Chim. Acta 377, 111 (2011).10.1016/j.ica.2011.07.045Search in Google Scholar
[39] Y. Shimazaki, T. Yajima, T. Shiraiwa, O. Yamauchi. Inorg. Chim. Acta 362, 2467 (2009).10.1016/j.ica.2008.11.006Search in Google Scholar
[40] R. B. Jordan. Reaction Mechanisms of Inorganic and Organometallic Systems, 3rd ed., Oxford University Press, Oxford (2007).10.1093/oso/9780195301007.003.0007Search in Google Scholar
[41] J. H. Espenson. Chemical Kinetics and Reaction Mechanisms, 2nd ed., McGraw-Hill, New York (2002).Search in Google Scholar
[42] T. Yagyu, S. Iwatsuki, S. Aizawa, S. Funahashi. Bull. Chem. Soc. Jpn. 71, 1857 (1998).Search in Google Scholar
[43] T. Yagyu, S. Aizawa, S. Funahashi. Bull. Chem. Soc. Jpn. 71, 619 (1998).Search in Google Scholar
[44] T. Yagyu, S. Aizawa, S. Funahashi. Chem. Lett. 1107 (1996).Search in Google Scholar
[45] K. C. Gross, R. G. Seybold. Int. J. Quantum Chem. 85, 569 (2001).10.1002/qua.1525Search in Google Scholar
[46] A. E. Martell, R. M. Smith. Critical Stability Constants, Vol. 3, Plenum Press, New York (1977).Search in Google Scholar
[47] G. W. Parshall. Acc. Chem. Res. 3, 139 (1970).10.1021/ar50028a004Search in Google Scholar
[48] M. Beller, T. H. Riermeier, S. Haber, H.-J. Kleiner, W. A. Herrmann. Chem. Ber. 129, 1259 (1996).10.1002/cber.19961291018Search in Google Scholar
[49] M. Gómez, J. Granell, M. Martinez. Eur. J. Inorg. Chem. 217 (2000).10.1002/(SICI)1099-0682(200001)2000:1<217::AID-EJIC217>3.0.CO;2-QSearch in Google Scholar
[50] A. D. Ryabov, I. K. Sakodinskaya, A. K. Yatsimirsky. J. Chem. Soc., Dalton Trans. 2629 (1985).10.1039/dt9850002629Search in Google Scholar
[51] A. J. Deeming, I. P. Rothwell. Pure. Appl. Chem. 52, 649 (1980).10.1351/pac198052030649Search in Google Scholar
[52] R. P. Thummel, Y. J. Jahng. Org. Chem. 52, 73 (1987).10.1021/jo00377a013Search in Google Scholar
[53] M. Gómez, J. Granell, M. Martinez. Organometallics 16, 2539 (1997).10.1021/om961099eSearch in Google Scholar
[54] T. Yajima, Y. Shimazaki, N. Ishigami, A. Odani, O. Yamauchi. Inorg. Chim. Acta 337, 193 (2002).10.1016/S0020-1693(02)01034-4Search in Google Scholar
[55] R. G. Wilkins. The Study of Kinetics and Mechanism of Reactions of Transition Metal Complexes, Allyn and Bacon, Boston (1974).Search in Google Scholar
[56] R. H. Crabtree. The Organometallic Chemistry of the Transition Metals, 5th ed., John Wiley, New York (2009).Search in Google Scholar
[57] D. T. Richens. Chem. Rev. 105, 1961 (2005).10.1021/cr030705uSearch in Google Scholar
[58] L. I. Elding, A.-B. Gröning. Inorg. Chim. Acta 31, 243 (1978).10.1016/S0020-1693(00)95010-2Search in Google Scholar
[59] L. I. Elding. Inorg. Chim. Acta 31, 683 (1972), and refs. therein.10.1016/S0020-1693(00)91884-XSearch in Google Scholar
[60] R. G. Pearson, H. B. Gray, F. Basolo. J. Am. Chem. Soc. 82, 787 (1960).10.1021/ja01489a006Search in Google Scholar
©2014 IUPAC & De Gruyter Berlin Boston
Articles in the same Issue
- Masthead
- Masthead
- Preface
- International Union of Pure and Applied Chemistry
- Conference paper
- Optimization of superamphiphobic layers based on candle soot
- Probing “ambivalent” snug-fit sites in the KcsA potassium channel using three-dimensional reference interaction site model (3D-RISM) theory
- Perspectives for hybrid ab initio/molecular mechanical simulations of solutions: from complex chemistry to proton-transfer reactions and interfaces
- The complex structure of ionic liquids at an atomistic level: from “red-and-greens” to charge templates
- Amide I IR probing of core and shell hydrogen-bond structures in reverse micelles
- Kinetic studies on cyclopalladation in palladium(II) complexes containing an indole moiety
- Properties of the one-electron oxidized copper(II) salen-type complexes: relationship between electronic structures and reactivities
- Solvation of a sponge-like geometry
- What is “hypermobile” water?: detected in alkali halide, adenosine phosphate, and F-actin solutions by high-resolution microwave dielectric spectroscopy
- Reentrant condensation, liquid–liquid phase separation and crystallization in protein solutions induced by multivalent metal ions
- Emulsion-templated macroporous polymer/polymer composites with switchable stiffness
- Effective interaction between small unilamellar vesicles as probed by coarse-grained molecular dynamics simulations
- Enthalpies of solution, limiting solubilities, and partial molar heat capacities of n-alcohols in water and in trehalose crowded media
- Protonation of alkanolamines and cyclic amines in water at temperatures from 293.15 to 373.15 K
- IUPAC Technical Report
- Defining the transfer coefficient in electrochemistry: An assessment (IUPAC Technical Report)
- IUPAC Recommendations
- Definition of the transfer coefficient in electrochemistry (IUPAC Recommendations 2014)
Articles in the same Issue
- Masthead
- Masthead
- Preface
- International Union of Pure and Applied Chemistry
- Conference paper
- Optimization of superamphiphobic layers based on candle soot
- Probing “ambivalent” snug-fit sites in the KcsA potassium channel using three-dimensional reference interaction site model (3D-RISM) theory
- Perspectives for hybrid ab initio/molecular mechanical simulations of solutions: from complex chemistry to proton-transfer reactions and interfaces
- The complex structure of ionic liquids at an atomistic level: from “red-and-greens” to charge templates
- Amide I IR probing of core and shell hydrogen-bond structures in reverse micelles
- Kinetic studies on cyclopalladation in palladium(II) complexes containing an indole moiety
- Properties of the one-electron oxidized copper(II) salen-type complexes: relationship between electronic structures and reactivities
- Solvation of a sponge-like geometry
- What is “hypermobile” water?: detected in alkali halide, adenosine phosphate, and F-actin solutions by high-resolution microwave dielectric spectroscopy
- Reentrant condensation, liquid–liquid phase separation and crystallization in protein solutions induced by multivalent metal ions
- Emulsion-templated macroporous polymer/polymer composites with switchable stiffness
- Effective interaction between small unilamellar vesicles as probed by coarse-grained molecular dynamics simulations
- Enthalpies of solution, limiting solubilities, and partial molar heat capacities of n-alcohols in water and in trehalose crowded media
- Protonation of alkanolamines and cyclic amines in water at temperatures from 293.15 to 373.15 K
- IUPAC Technical Report
- Defining the transfer coefficient in electrochemistry: An assessment (IUPAC Technical Report)
- IUPAC Recommendations
- Definition of the transfer coefficient in electrochemistry (IUPAC Recommendations 2014)