Non-Herlitz junctional epidermolysis bullosa in a Native American newborn
-
Ayah A. Ibrahim

 ,
Macken Yrun-Duffy
,
Macken Yrun-Duffy

Abstract
This case report details the presentation, diagnosis, and management of a newborn Native American male with non-Herlitz junctional epidermolysis bullosa (JEB), a rare diagnosis specifically in the Native American population. Genetic analysis revealed a homozygous mutation in the COL17A1 gene. The management involved multidisciplinary care and highlighted the challenges in treatment, including pain management, wound care, and ethical considerations surrounding adoption within Indigenous communities. This case highlights the importance of tailored interventions and the need for further research into the genetic diversity and prevalence of epidermolysis bullosa (EB) among the Native American population.
The “butterfly wing effect” is a term utilized to describe skin fragility in those at the mercy of epidermolysis bullosa (EB). As an umbrella term, EB was first described in 1886 as keratolytic lesions of the skin leading to the sloughing of epithelialized surfaces with incredible ease [1], 2]. Currently, the term is utilized to describe over 20 different potential genetic mutations that lead to impaired intraepidermal and dermal-epidermal proteins, mostly keratin five and 14, collagen, laminin, integrin, plectin, and kindlin proteins [3]. Dysfunction of these proteins leads to impaired adhesion of squamous epithelial cells, which compromises the strength and cohesion of the skin and other squamous epithelial tissues, such as the gastrointestinal, oral cavity, and respiratory tracts, among others [3].
EB is broadly classified into three major categories based on the level at which blistering occurs: epidermolytic (EB simplex), in which blistering occurs within the basal layer of the epidermis; lucidolytic (junctional epidermolysis bullosa [JEB]), involving blistering within the lamina lucida of the basement membrane; and dermolytic (dystrophic EB), in which blistering occurs below the basement membrane in the upper dermis [3], 4]. Each of these categories is further subclassified based on the specific mutated protein that leads to functional impairment in cell adhesion.
Here, we present a case of a newborn Native American boy diagnosed with non-Herlitz junctional epidermolysis bullosa (nH-JEB), a subtype of JEB. nH-JEB is primarily caused by mutations in the LAMA3, LAMB3, and LAMC2 genes that encode laminin-332, a protein crucial for epidermal-dermal adhesion [4]. When laminin-332 is defective or absent, the structural integrity between the epidermis and dermis is compromised, leading to skin fragility and blistering [4]. Diagnosing these mutations accurately through genetic testing is critical, because traditional diagnostic methods like skin biopsies can sometimes result in misdiagnosis, particularly when overlapping features of more common EB subtypes are present.
Currently, the standard of care for EB focuses on symptom management, including wound care, infection prevention, pain management, and nutritional support [5]. However, advances in treatment approaches for nH-JEB with emerging therapies targeting laminin-332 gene mutations, such as gene therapy and protein replacement therapy, are being investigated as a potential long-term treatment [5].
This case also highlights the genetic diversity of EB, particularly within the Native American populations, where research into the prevalence and spectrum of genetic mutations remains limited. Misdiagnosis of rarer EB forms, such as nH-JEB, can delay appropriate treatment and have long-term impacts on patient outcomes. Therefore, this case serves as a reminder of the importance of precise genetic diagnosis and the need for further study into the unique genetic profiles within the Native American population.
Case description
A full-term Native American neonate was delivered in September 2017 through spontaneous vaginal delivery at 39 weeks gestation to an 18-year-old G3P2002 Native American mother. The mother received prenatal care from an unknown provider and reported an uncomplicated pregnancy. Her Group B Streptococcus status was undocumented, but she received one prophylactic dose of intravenous (IV) penicillin G before delivery. She denied the use of alcohol, illicit drugs, and tobacco, and her admission urine drug screen was negative. Additionally, umbilical cord analysis showed no evidence of prenatal drug use. Maternal tests for HIV, Hepatitis B, and Hepatitis C were nonreactive. No family history of autoimmune or dermatological conditions was reported. The mother’s previous pregnancies were all uncomplicated vaginal deliveries without postnatal issues. The biological father of all three children is also of full Native American heritage, belonging to a different federally recognized tribe than the mother.
The infant’s birth measurements were as follows: weight: 6 lbs, 15.7 oz (3,168 g); head circumference: 13.58 inches (34.5 cm); and length: 20.47 inches (52 cm). The APGAR (Appearance, Pulse, Grimace, Activity and Respiration) scores at 1 and 5 min were eight and nine, respectively. The initial vital signs upon assessment revealed a heart rate of 141 beats per minute, a temperature of 98.1 F (36.7 °C), blood pressure of 72/41 mmHg, and respirations at 54 breaths per minute.
At delivery, the infant exhibited diffuse desquamation with erythema and peeling of the skin on the hands and feet (Figure 1) along with oral mucosal lesions with sloughing of the skin. However, the skin covering his head, trunk, upper arms, and legs were intact. The rest of the physical examination was unremarkable. Due to potential nutritional deficiencies, sepsis concerns, and potential bacterial skin infections, oral feeding was withheld, an umbilical vein catheter was inserted, and IV total parenteral nutrition with 10 % dextrose, ampicillin, and gentamycin at appropriate dosages was initiated. Baseline complete blood count and blood cultures were drawn, and the wounds were dressed in petroleum jelly gauze.
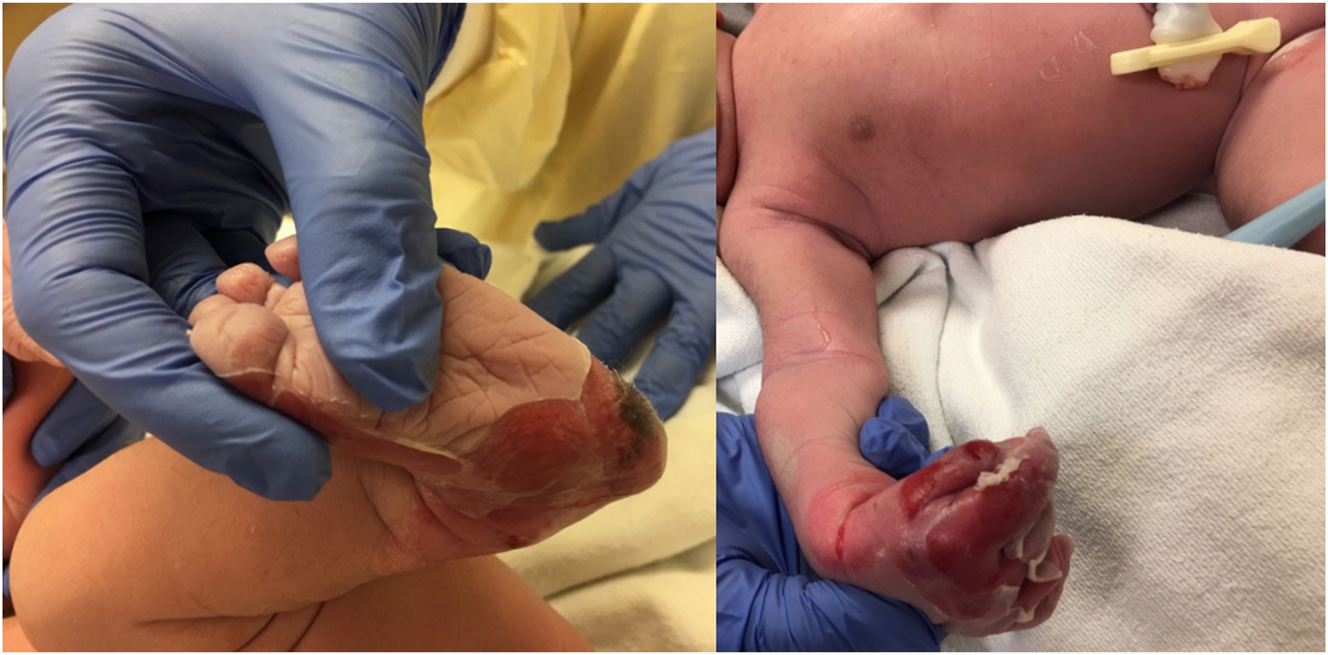
Images depicting the infant’s status postdelivery, exhibiting peeling of the skin on his hands and feet.
Within 24 h postdelivery, the baby exhibited significant skin sloughing on both legs below the knees, particularly severe on the feet. Bullae were present on the bilateral hands, right elbow and shoulder, bilateral buttocks, and upper lumbar area, in addition to discoloration and sloughing around the nail beds. Additionally, blistering occurred from the electrocardiogram (EKG) leads on the chest. Skin sloughing occurred around the lips, with blister formation in the oral mucosa and new bullae continuing to emerge within hours. Due to extreme discomfort, a titrated fentanyl IV drip was started for pain management. On the third day of life, due to his worsening condition, he was transferred from the delivery facility to an EB tertiary center, where he remained for one month for intensive and comprehensive management.
The subsequent course at the tertiary EB center primarily focused on pain management and wound care while consulting with nutritional, dermatological, genetic, and surgical specialists. Although infectious disease was not consulted, neonatal pemphigus was in the differential. However, a genetic consult revealed a homozygous mutation in the COL17A1 gene, which encodes collagen type XVII alpha-1 chain, confirming the specific diagnosis of nH-JEB, also known as generalized intermediate JEB. A sepsis workup was initiated due to elevated procalcitonin levels (10.58 ng/mL) and a fever of 38.3 °C. Both lumbar puncture and blood cultures returned negative, but urine cultures were positive for Enterococcus. Consequently, a peripherally inserted central catheter (PICC) line was placed for antibiotic and fluid administration.
Although nutritional deficiencies were not an initial concern, a gastrostomy tube (G-tube) was proactively inserted to ensure adequate caloric intake due to feeding challenges posed by the oral mucosal lesions. Analgesics, including morphine and methadone, were administered for wound dressing changes. Renal ultrasound, chest X-ray, and upper gastrointestinal series were obtained, revealing normal renal anatomy and patchy perihilar atelectasis on chest X-ray without signs of pneumonia. On the 31st day of admission, at approximately 34 days of life, the PICC line was removed, and the patient was discharged with G-tube feedings to establish care with their community pediatrician.
By three months of age, concerns began to arise about his well-being. He was not gaining weight as expected, and he was showing signs of failure to thrive and poor hygiene. His mother struggled to keep up with the daily care required, particularly with managing the dressing changes. As a result, the infant was hospitalized and subsequently referred to Child Protective Services. He entered foster care, where his weight and condition rapidly improved. The condition of his skin lesions at eight months of age is shown in Figure 2. By 11 months, the infant began tolerating solids and liquids by mouth; however, the G-tube remained in place for medication administration and supplemental feeding due to difficulties associated with the oral ulcers.
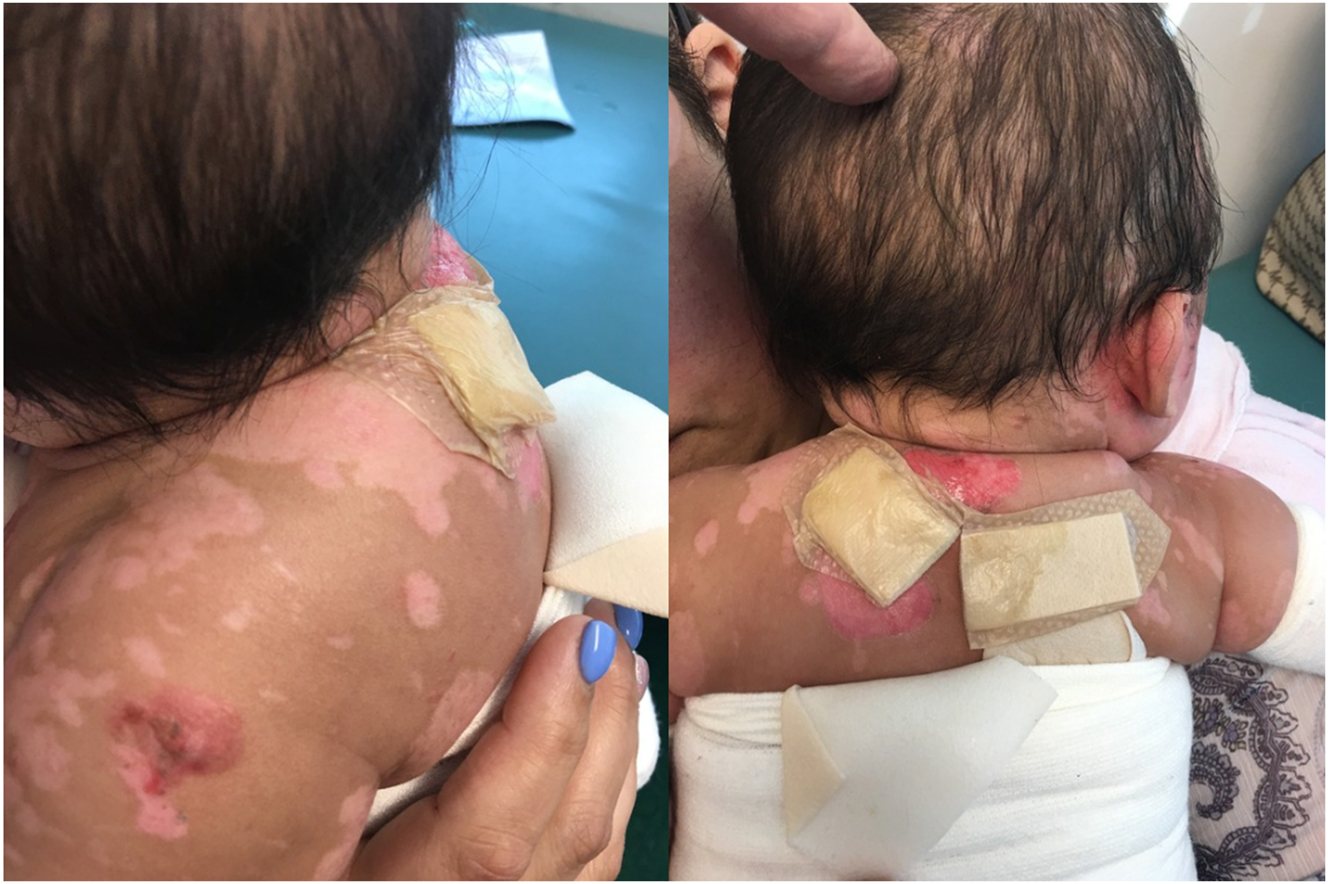
Images showing the state of the child’s skin lesions at eight months of age.
Given the parents’ inability to provide the necessary care, ethical considerations arose regarding the adoption process. A Native American foster caregiver was preferred; however, despite efforts, no suitable families were identified in either tribal community. Therefore, a non-Native American family with medical training was selected to foster and adopt the child. At 19 months, the toddler demonstrated tolerance for dressing changes with morphine as needed. He successfully transitioned to a regular diet with supplemental formula and achieved appropriate growth and developmental milestones.
Discussion
JEB is a rare autosomal recessive and severe inherited skin disorder characterized by blistering and fragility of the skin and mucous membranes due to defects in connective tissue [6]. The condition is caused by gene mutations in LAMA3, LAMB3, LAMC2, or COL17A1, impacting basement membrane formation. Most nH-JEB patients present with a mutation in COL17A1, which encodes type XVII collagen [7], 8]. nH-JEB is the milder subtype [9], while the Herlitz type (H-JEB) is more severe and results from homozygous mutations in LAMA3, LAMB3, or LAMC2, affecting laminin-322. H-JEB is characterized by widespread mucocutaneous blistering present at birth and is associated with early mortality [8]. Though nH-JEB is less severe, it remains rare, affecting approximately two individuals per million. The prognosis is poor for both subtypes of JEB, with 40–50 % of patients dying within the first year and 48 % of those with nH-JEB surviving beyond age 15 [6].
EB is a complex disorder that requires a multidisciplinary approach due to its multisystemic impact with no definitive treatment or cure. The hallmark of nH-JEB is fragile skin that blisters easily, necessitating specialized wound care to manage chronic wounds and prevent infection. A team that includes dermatologists, wound care specialists, and nutritionists is necessary to develop personalized care plans. These plans often involve the use of nonadhesive dressings, infection-control measures, and nutritional strategies to support wound healing. However, due to the rare and complex nature of nH-JEB, there is no consensus on the preferred type of dressing [10].
Patients with JEB also face increased metabolic demands, and combined with swallowing difficulties, this can result in decreased food intake. This, in turn, leads to failure to thrive, delayed puberty, anemia, and a cascade of other complications that further impair wound healing [10], 11]. Therefore, collaboration with nutritionists and gastroenterologists is essential to manage food intake and complications. Meanwhile, physical and occupational therapists play a vital role in helping patients maintain mobility and prevent joint contractures through adaptive exercises.
Given the chronic and painful nature of EB, psychosocial support is crucial for both the patient and their families. The impact of JEB on quality of life and overall coping requires a holistic approach that addresses mental and emotional health. Studies have shown that fostering a sense of community significantly improves the quality of life for EB patients [12]. Above all, pain management remains a priority to ensure that patients can engage in activities [10]. Together, these multidisciplinary efforts aim to manage complications and enhance the patient’s overall well-being by addressing both physical and emotional needs.
Kindler syndrome, a type of EB, is the only documented case of EB in the Native American population but is isolated to individuals from the Ngobe-Bugle tribe in Panama and southern Costa Rica, unrelated to our patient, who is from a US federally recognized tribe [13]. Given our patient’s complete Native American background, it raises questions about common lineages between tribes in Central America and the United States and the prevalence of specific EB subtypes within these populations. However, there were no documented occurrences of EB found in the patient’s specific Native American tribe or any tribes in the United States. This suggests a unique genetic profile within these communities, highlighting the need for further research into EB’s genetic diversity and prevalence among Native American populations.
The Indian Child Welfare Act (ICWA) is a federal law enacted to protect the interests of Native American children and to promote the stability and security of tribal communities. When a Native American child is considered for adoption by a non-Native American family, ICWA mandates specific procedures to ensure tribal sovereignty and cultural preservation. The process involves notification of the child’s tribe and active efforts to place the child within the tribal community or with extended family before considering placement outside the tribe [14]. The primary limitation of this case is the need to ensure patient anonymity, which restricted the ability to provide detailed information regarding the patient’s specific tribal heritage. In this case, tribal governance determining the child’s best interest lay in adoption by non-Native American parents with medical expertise in nH-JEB, given the lack of experience within the Native American population. This decision highlights the importance of balancing medical needs, cultural preservation, and cross-cultural adoption processes within Indigenous communities.
Conclusions
The presented case of an nH-JEB in a Native American infant emphasizes the complex interplay between genetic predisposition, clinical management, and cultural considerations. The complex nature of EB, compounded by its rarity and diverse genetic landscape, necessitates a multidisciplinary approach to diagnosis and treatment. This case highlights the importance of tailored interventions, such as pain management, wound care, and nutritional support, in mitigating the profound impact of EB on patients and their families. Furthermore, the ethical dilemmas surrounding adoption, particularly within Indigenous communities governed by the ICWA, highlight the need for sensitivity to cultural norms and preferences while prioritizing the child’s medical needs. Moving forward, continued research into the genetic diversity and prevalence of EB among Native American populations is imperative to establish targeted interventions and improve outcomes for these individuals.
-
Research ethics: The local Institutional Review Board deemed the study exempt from review.
-
Informed consent: Informed consent was obtained from all individuals included in this study.
-
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: None declared.
-
Research funding: None declared.
-
Data availability: Not applicable.
References
1. Fine, JD, Eady, RA, Bauer, EA, Bauer, JW, Bruckner-Tuderman, L, Heagerty, A, et al.. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008;58:931–50. https://doi.org/10.1016/j.jaad.2008.02.004.Search in Google Scholar PubMed
2. Koebner, H. Hereditare anlage zur blasenbildung (epidermolysis bullosa hereditaria). Dtsch Med Wochenschr 1886;12:21–2. https://doi.org/10.1055/s-0028-1139665.Search in Google Scholar
3. Prodinger, C, Reichelt, J, Bauer, JW, Laimer, M. Epidermolysis bullosa: advances in research and treatment. Exp Dermatol 2019;28:1176–89. https://doi.org/10.1111/exd.13979.Search in Google Scholar PubMed PubMed Central
4. Mariath, LM, Santin, JT, Schuler-Faccini, L, Kiszewski, AE. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol 2020;95:551–69. https://doi.org/10.1016/j.abd.2020.05.001.Search in Google Scholar PubMed PubMed Central
5. Sait, H, Srivastava, S, Saxena, D. Integrated management strategies for epidermolysis bullosa: current insights. [published correction appears in Int J Gen Med. 2022 Jun 27;15:5807–5808. doi: 10.2147/IJGM.S379410]. Int J Gen Med 2022;15:5133–44. https://doi.org/10.2147/IJGM.S342740.Search in Google Scholar PubMed PubMed Central
6. Pfendner, EG, Lucky, AW. Junctional epidermolysis bullosa. In: Adam, MP, Feldman, J, Mirzaa, GM, Pagon, RA, Wallace, SE, Amemiya, A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 2008:1993–2024 pp. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1125/ [Updated 2018 Dec 20].Search in Google Scholar
7. Laimer, M, Lanschuetzer, CM, Diem, A, Bauer, JW. Herlitz junctional epidermolysis bullosa. Dermatol Clin 2010;28:55–60. https://doi.org/10.1016/j.det.2009.10.006.Search in Google Scholar PubMed
8. Nakamura, H, Sawamura, D, Goto, M, Kida, M, Ariga, T, Sakiyama, Y, et al.. Analysis of the COL17A1 in non-Herlitz junctional epidermolysis bullosa and amelogenesis imperfecta. Int J Mol Med 2006;18:333–7. https://doi.org/10.3892/ijmm.18.2.333.Search in Google Scholar
9. Bhinder, MA, Arshad, MW, Zahoor, MY, Shehzad, W, Tariq, M, Shabbir, MI. Junctional epidermolysis bullosa (Non-Herlitz type). J Coll Physicians Surg Pak 2017;27:308–10.Search in Google Scholar
10. Denyer, J, Pillay, E, Clapham, J. Best practice guidelines for skin and wound care in epidermolysis bullosa. An International Consensus. Wounds International 2017. https://www.eb-clinet.org/fileadmin/user_upload/Media_Library/EB-CLINET/Dokumente/CPGs/International_Consensus_Best_Practice_Guidelines_Skin_and_Wound_Care_in_Epidermolysis_Bullosa.pdf.Search in Google Scholar
11. Zidorio, AP, Dutra, ES, Leão, DO, Costa, IM. Nutritional aspects of children and adolescents with epidermolysis bullosa: literature review. An Bras Dermatol 2015;90:217–23. https://doi.org/10.1590/abd1806-4841.20153206.Search in Google Scholar PubMed PubMed Central
12. Martin, K, Geuens, S, Asche, JK, Bodan, R, Browne, F, Downe, A, et al.. Psychosocial recommendations for the care of children and adults with epidermolysis bullosa and their family: evidence based guidelines. Orphanet J Rare Dis 2019;14:133. https://doi.org/10.1186/s13023-019-1086-5.Search in Google Scholar PubMed PubMed Central
13. Penagos, H, Jaen, M, Sancho, MT, Saborio, MR, Fallas, VG, Siegel, DH, et al.. Kindler syndrome in Native Americans from Panama: report of 26 cases. Arch Dermatol 2004;140:939–44. https://doi.org/10.1001/archderm.140.8.939.Search in Google Scholar PubMed
14. Indian Child Welfare Act|child Welfare information gateway. Available from: www.childwelfare.gov, https://www.childwelfare.gov/topics/tribal-child-welfare/indian-child-welfare-act/.Search in Google Scholar
© 2024 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Medical Education
- Review Article
- Trends in osteopathic medical education: a scoping review
- Musculoskeletal Medicine and Pain
- Review Article
- Osteopathic approach to injuries of the overhead thrower’s shoulder
- Neuromusculoskeletal Medicine (OMT)
- Original Article
- The effect of osteopathic manipulative treatment on chronic rhinosinusitis
- Obstetrics and Gynecology
- Clinical Practice
- Management of endometriosis: a call to multidisciplinary approach
- Pediatrics
- Case Report
- Non-Herlitz junctional epidermolysis bullosa in a Native American newborn
- Public Health and Primary Care
- Original Article
- Improving vascular access knowledge and assessment skill of hemodialysis staff
Articles in the same Issue
- Frontmatter
- Medical Education
- Review Article
- Trends in osteopathic medical education: a scoping review
- Musculoskeletal Medicine and Pain
- Review Article
- Osteopathic approach to injuries of the overhead thrower’s shoulder
- Neuromusculoskeletal Medicine (OMT)
- Original Article
- The effect of osteopathic manipulative treatment on chronic rhinosinusitis
- Obstetrics and Gynecology
- Clinical Practice
- Management of endometriosis: a call to multidisciplinary approach
- Pediatrics
- Case Report
- Non-Herlitz junctional epidermolysis bullosa in a Native American newborn
- Public Health and Primary Care
- Original Article
- Improving vascular access knowledge and assessment skill of hemodialysis staff