
Synthesis and antitumor activities of piperazine- and cyclen-conjugated dehydroabietylamine derivatives
-
Xinbin Yang
 ,
Xiaolin Qin
,
Xiaolin Qin

Abstract
A series of piperazine- and cyclen-conjugated dehydroabietylamine derivatives were synthesized and characterized by 1H NMR, 13C NMR, and HRMS. The in vitro antitumor activities of conjugates 10–13 against MCF-7 and HepG-2 tumor cell lines were evaluated using CCK-8 assay. The results show that the synthesized compounds cause a dose-dependent inhibition of cell proliferation and display different antitumor activities with the IC50 values ranging from 23.56 to 78.92 μm. Moreover, the antitumor activity of conjugate 10 against the MCF-7 cell line is superior to that of the positive control 5-fluorouracil. In addition, flow cytometric assay revealed that the representative conjugate 10 could induce apoptosis in MCF-7 tumor cells in a dose-dependent manner.
Introduction
Cancer remains the primary cause of death due to the lack of effective drugs [1]. Natural compounds have played an important role in anticancer drug discovery, where the fraction of the drugs derived from natural products amounts to 60% [2]. Dehydroabietylamine is an abietane diterpenic amine that is obtained as a part of a mixture of amines derived from rosin. Recent studies have found that dehydroabietylamine derivatives demonstrate broad biological activities, such as antibacterial, antiinflammatory, antioxidative, and antitumor activities [3–12]. These results arouse our interest in screening for new potential antitumor drugs by the introduction of various functional groups to the dehydroabietylamine skeleton.
A number of piperazine and 1,4,7,10-tetraazacyclododecane (cyclen) heterocyclic derivatives as chemotherapeutic drugs have attracted considerable attention during the past decade. Studies have shown that the introduction of piperazine and cyclen moieties can modulate the physicochemical properties and enhance the bioactivity of the compounds [13–21]. However, the studies on the synthesis and antitumor activities of heterocyclic derivatives derived from dehydroabietylamine have not been reported. Our present work is to design and synthesize a series of piperazine- and cyclen-conjugated dehydroabietylamine derivatives and to evaluate in vitro antitumor activities of these conjugates against HepG-2 and MCF-7 cells. Furthermore, the apoptotic effect induced by the representative conjugates is also investigated by flow cytometry.
Results and discussion
The synthetic route to the target conjugates 10–13 is shown in Scheme 1. The reaction between tri-Boc-protected cyclen 1 or Boc-protected piperazine 2 and chloracetyl chloride afforded the respective products 3 and 4. Compound 6 was obtained by the reaction between 3 and 5a in the presence of KOH. Compounds 7–9 were synthesized using a similar methodology. The target compounds 10–13 were obtained by deprotection of Boc group in HCl-ethanol solution. All these products exhibit good water solubility. The structures of all synthesized conjugates were confirmed by 1H NMR, 13C NMR, and HRMS.
The in vitro antitumor activities of the conjugates 10–13 were evaluated by means of CCK-8 assay against MCF-7 and HepG-2 tumor cell lines. The inhibition rates of cell viability with different concentrations of conjugates are shown in Figure 1, and the IC50 values are given in Table 1. It can be seen that the treatment with increasing doses of all conjugates causes a dose-dependent inhibition of cell proliferation. As evident from the obtained results, conjugate 10 with cyclen moiety is the most potent cytotoxic agent against MCF-7 and HepG-2 cells among the tested conjugates. Compound 10 displays a prominent inhibitory activity by almost 100% at 40 μm, with the IC50 value of 25.42 μm for MCF-7 cell and 27.05 μm for HepG-2 cell. Furthermore, the effect of conjugate 10 against the MCF-7 tumor cell line is slightly superior to that of the positive control 5-fluorouracil (5-FU), as indicated by the IC50 values. In addition, conjugate 12 with piperazine moiety shows selectivity for HepG-2 over MCF-7 with the corresponding IC50 values of 23.56 and 62.55 μm. Meanwhile, conjugates 11 and 13 containing benzene ring as part of the R group exhibit only mild cytotoxic activities against MCF-7 and HepG-2 cells.
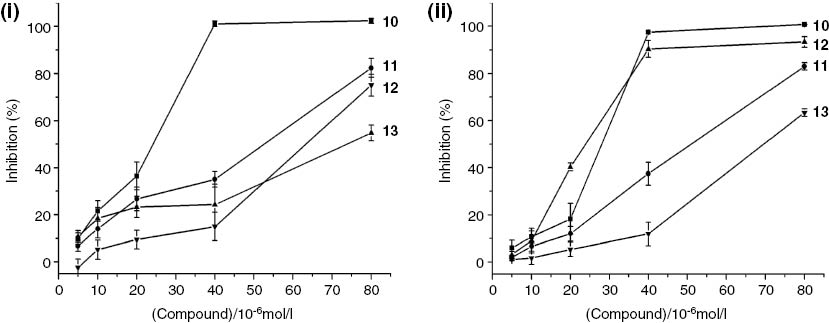
Antiproliferation effect of conjugates 10, 11, 12, and 13 against MCF-7 (i) and HepG-2 (ii) tumor cells.
Cells were plated and incubated with the indicated concentrations of 10, 11, 12, and 13 (5, 10, 20, 40, and 80 μm). After 24 h of treatment, cell proliferation was measured by the CCK-8 assay. Data represent the means±SD of triplicate experiments.
The IC50 values of the conjugates 10–13 and 5-FU against tumor cell lines.
| Cell line | IC50value (μm) | ||||
|---|---|---|---|---|---|
| 10 | 11 | 12 | 13 | 5-FU | |
| MCF-7 | 25.42±2.04 | 49.61±3.26 | 62.55±4.01 | 78.92±2.68 | 27.12±1.95 |
| HepG-2 | 27.05±1.88 | 52.63±2.94 | 23.56±1.45 | 71.96±2.33 | 6.78±0.65 |
The effect of conjugate 10 on apoptosis in MCF-7 cell was investigated. Apoptosis assay may provide preliminary information about the mechanism of growth inhibition of tumor cells. The apoptosis ratios (including the early and late apoptosis rates) induced by conjugate 10 in MCF-7 cell lines were determined by flow cytometry. The results are given in Figure 2. The apoptotic rate was 4.10% of the total number of cells in the control group. The percentage of apoptotic cells were increased to 10.46, 45.65, and 53.75% by the treatment with 10, 20, and 40 μm conjugate 10, respectively. The results indicate that apoptosis induction of the conjugate 10 in MCF-7 tumor cells changes in a dose-dependent manner.
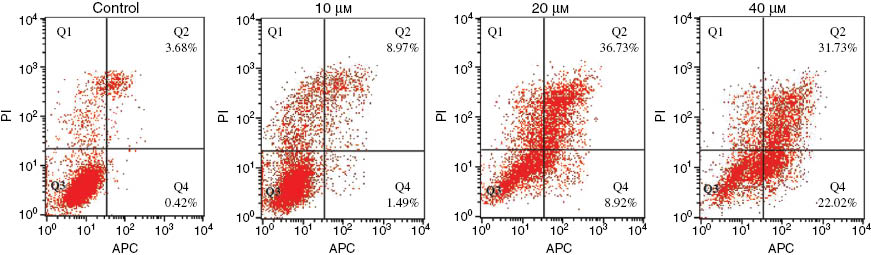
Apoptosis ratio of conjugate 10 as detected by Annexin V-APC/PI double-staining assay of MCF-7 cells.
The MCF-7 cells were treated with 10, 20, and 40 μm of conjugate 10 for 24 h. Four quadrant images were observed by flow cytometric analysis: the Q1 area represents damaged cells appearing in the process of cell collection, the Q2 region shows necrotic cells and later-period apoptotic cells, the normal cells are located in the Q3 area, and the Q4 area shows early apoptotic cells.
Conclusion
A series of cyclic polyamine-dehydroabietylamine conjugates 10–13 were synthesized and characterized. The in vitro antitumor activities of these compounds against HepG-2 and MCF-7 cells were evaluated. The effect of conjugate 10 against the MCF-7 cells was slightly superior to that of the positive control 5-FU. In addition, flow cytometric assay indicated that the representative conjugate 10 induces apoptosis in MCF-7 cells in a dose-dependent manner. These results encourage us to synthesize additional new dehydroabietylamine derivatives with the expected more potent antitumor activity.
Experimental
All reagents were purchased from commercial sources and used without further purification. The HepG-2 (liver hepatocellular carcinoma cell) and MCF-7 (human breast adeno-carcinoma cell) were obtained from ATCC. High resolution mass spectrometry (HRMS) data were recorded on a Bruker Daltonics Bio TOF instrument. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were measured on a Varian INOVA-400 spectrometer. Flow cytometry was performed using a BD FASAria Cell Sorter.
Preparation of compound 5b
The ethanol solution (30 mL) of dehydroabietylamine (5a, 1.45 g, 5 mmol) and p-hydroxybenzaldehyde (0.61 g, 5 mmol) was heated under reflux for 4 h. After cooling, sodium borohydride (0.185 g, 5 mmol) was added and the mixture was stirred at room temperature for 12 h. Then, the mixture was concentrated under reduced pressure and quenched with water (10 mL). The aqueous phase was extracted with ethyl acetate (3×30 mL). The solvent was removed under reduced pressure and the residue was purified by column chromatography eluting with petroleum ether/ethyl acetate, 3:1, to give product 5b as a white solid: yield 72%; mp 143–145°C; 1H NMR (CDCl3): δ 0.95 (s, 3H), 1.21 (s, 3H), 1.26 (d, 6H, J = 12 Hz), 1.35–1.79 (m, 8H), 2.24–2.31 (m, 2H), 2.49 (d, H, J = 10.8 Hz), 2.80–2.84 (m, 3H), 3.63–3.72 (m, 2H), 6.72 (d, 2H, J = 8.0 Hz), 6.88 (s, 1H), 6.98 (d, 1H, J = 8.0 Hz), 7.14 (d, 2H, J = 7.0 Hz), 7.17 (d, H, J = 7.0 Hz); 13C NMR (CDCl3): δ 154.8, 147.5, 145.4, 134.8, 132.3, 129.4, 126.8, 124.3, 123.8, 115.3, 60.8, 54.1, 45.5, 38.4, 37.4, 36.9, 36.2, 33.4, 30.3, 25.4, 24.0, 19.3, 18.8, 18.7. ESI-HRMS. Anal. Calcd for C27H38NO ([M+H]+): m/z 392.2953. Found: m/z 392.2956.
Preparation of compounds 3 and 4
Compound 3 was prepared according to the literature [22] by the reaction of 1 with chloracetyl chloride in the presence of Et3N at 0°C. Purification by silica gel column chromatography eluting with petroleum ether/ethyl acetate, 1:1, gave product 3 as a white solid: yield 65%; 1H NMR (CDCl3): δ 1.46–1.49 (s, 27H, Boc-H), 3.38–3.56 (m, 16H, CH2), 4.06 (s, 2H); MS-ESI: m/z 548 (M+).
Compound 4 was synthesized using a similar procedure: yield 77%; 1H NMR (CDCl3): δ 1.47 (s, 9H, Boc-H), 3.43–3.61 (m, 8H, CH2), 4.05 (s, 2H); ESI-MS: m/z 262 (M+).
General procedure for the preparation of compounds 6–9
The tetrahydrofuran (THF) solution (30 mL) of 3 or 4(0.5 mmol), 5a or 5b (0.5 mmol), and KOH (0.084 g, 1.5 mmol) was stirred at 60°C for 10 h. After cooling, the mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with petroleum ether/ethyl acetate, 1:1, to give desired product 6–9 as a white solid.
Compound 6
This compound was synthesized from 3 and 5a: yield 75%; mp 108–110°C; 1H NMR (CDCl3): δ 0.95 (s, 3H), 1.21 (s, 3H), 1.26 (d, 6H, J = 12 Hz), 1.35–1.75 (m, 35H), 2.17–2.31 (m, 2H), 2.50 (d, H, J = 10.8 Hz), 2.79–2.87 (m, 3H), 3.38–3.47 (m, 18H), 6.86 (s, 1H), 6.98 (d, 1H, J = 8.0 Hz), 7.17 (d, H, J = 8.0 Hz); 13C NMR (CDCl3): δ 170.0, 155.4, 147.4, 145.3, 134.8, 126.7, 124.3, 123.7, 80.4, 62.1, 51.7, 51.5, 49.7, 49.6, 45.5, 38.4, 37.4, 37.0, 36.1, 33.4, 30.3, 28.5, 25.3, 24.0, 19.3, 19.1, 18.8. ESI-HRMS. Calcd for C45H76N5O7 ([M+H]+): m/z 798.5745. Found: m/z 798.5736.
Compound 7
This compound was synthesized from 3 and 5b: yield 68%; mp 99–101°C; 1H NMR (CDCl3): δ 0.95 (s, 3H), 1.21 (s, 3H), 1.26 (d, 6H, J = 12 Hz), 1.34–1.73 (m, 35H), 2.17–2.27 (m, 2H), 2.50 (d, H, J = 10.8 Hz), 2.80–2.85 (m, 3H), 3.34–3.57 (m, 16H), 3.71–3.73 (m, 2H), 4.64 (s, 2H), 6.87 (d, 2H, J = 4.4 Hz), 6.90 (s, 1H), 6.98 (d, 1H, J = 8.0 Hz), 7.14 (d, H, J = 7.2 Hz), 7.21 (d, 2H, J = 6.0 Hz); 13C NMR (CDCl3): δ 170.1, 157.0, 155.6, 147.5, 145.4, 134.8, 132.3, 129.2, 126.7, 124.3, 123.7, 114.6, 80.5, 67.0, 60.6, 51.4, 50.4, 49.9, 49.6, 45.3, 38.5, 37.4, 36.9, 36.2, 33.4, 30.9, 28.5, 25.4, 24.0, 19.3, 18.8,18.7. ESI-HRMS. Calcd for C52H82N5O8 ([M+H]+): m/z 904.6163. Found: m/z 904.6166.
Compound 8
This compound was synthesized from 4 and 5a: yield 80%; mp 117–119°C; 1HNMR (CDCl3): δ 0.93 (s, 3H), 1.18 (s, 3H), 1.26 (d, 6H, J = 12 Hz), 1.38–1.77 (m, 17H), 2.27–2.31 (m, 2H), 2.60 (d, H, J = 10.8 Hz), 2.81–2.87 (m, 3H), 3.02–3.20 (m, 8H), 3.45–3.51 (m, 2H), 6.86 (s, 1H), 6.95 (d, 1H, J = 8.0 Hz), 7.17 (d, H, J = 8.0 Hz); 13C NMR (CDCl3): δ 170.0, 154.5, 147.4, 145.4, 134.7, 126.7, 124.2, 123.7, 80.3, 62.1, 51.9, 45.5, 44.4, 41.6, 39.5, 38.4, 37.4, 36.2, 33.4, 30.2, 28.3, 25.6, 24.0, 19.1, 18.8, 18.4. ESI-HRMS. Anal. Calcd for C31H50N3O3([M+H]+): m/z 512.3852. Found: m/z 512.3851.
Compound 9
This compound was synthesized from 4 and 5b: yield 78%; mp 112–114°C; 1HNMR (CDCl3): δ 0.90 (s, 3H), 1.21 (s, 3H), 1.26 (d, 6H, J = 12 Hz), 1.37–1.65 (m, 17H), 2.17–2.28 (m, 2H), 2.51 (d, H, J = 12 Hz), 2.82–2.86 (m, 3H), 3.40–3.57 (m, 8H), 3.68–3.70 (m, 2H), 4.68 (s, 2H), 6.87 (d, 2H, J = 8.4 Hz), 6.90 (s, 1H), 6.98 (d, 1H, J = 8.0 Hz), 7.17 (d, H, J = 8.0 Hz), 7.24 (d, 2H, J = 8.4 Hz); 13C NMR (CDCl3): δ 166.8, 156.5, 154.5, 147.5, 145.4, 134.8, 132.2, 129.3, 126.7, 124.2, 123.7, 114.3, 80.3, 68.0, 60.8, 54.0, 45.3, 45.2, 42.0, 38.5, 37.4, 37.0, 36.2, 33.4, 30.3, 28.3, 25.3, 23.9, 19.3, 18.8, 18.7. ESI-HRMS. Anal. Calcd for C38H56N3O4 ([M+H]+): m/z 618.4271. Found: m/z 618.4274.
General procedure for the preparation of compounds 10–13
To a stirred solution of 6, 7, 8, or 9 (0.3 mmol) in ethanol (10 mL) at room temperature was slowly added 5 mL of 3 m HCl in ethanol solution. After stirring overnight, the reaction mixture was concentrated under reduced pressure. The residue was washed by anhydrous ether to furnish a hydrochloride salt of 10, 11, 12, or 13 as a white powder.
Compound 10
Yield 87%; mp 224–226°C; 1H NMR (DMSO-d6): δ 0.99 (s, 3H), 1.14 (s, 3H), 1.21 (d, 6H, J = 6.8 Hz), 1.50–1.72 (m, 8H), 2.17–2.31 (m, 2H), 2.50 (d, H, J = 10.8 Hz), 2.79–2.87 (m, 3H), 3.02–3.19 (m, 16H), 3.44–3.51 (m, 2H), 6.86 (s, 1H), 6.98 (d, 1H, J = 7.6 Hz), 7.17 (d, H, J = 8.0 Hz); 13C NMR (DMSO-d6): δ 166.9, 146.7, 145.1, 134.2, 126.3, 123.8, 123.5, 58.7, 50.8, 48.2, 46.2, 45.3, 44.4, 43.1, 37.3, 36.9, 36.0, 34.6, 32.8, 28.7, 24.9, 23.9, 18.2, 18.0. ESI-HRMS. Anal. Calcd for C30H52N5O ([M+H]+): m/z 498.4172. Found: m/z 498.4174.
Compound 11
Yield 73%; mp 244–246°C; 1H NMR (DMSO-d6): δ 0.89 (s, 3H), 1.13 (s, 3H), 1.20 (d, 6H, J = 12 Hz), 1.38–1.61 (m, 8H), 2.24–2.28 (m, 2H), 2.51 (d, H, J = 12 Hz), 2.73–2.80 (m, 3H), 3.05–3.16 (m, 16H), 3.43–3.58 (m, 2H), 4.91 (s, 2H), 6.85 (s, 1H), 6.96 (d, 1H, J = 4.0 Hz), 7.03 (d, 2H, J = 8.0 Hz), 7.13 (d, H, J = 8.0 Hz), 7.46 (d, 2H, J = 8.0 Hz); 13C NMR (DMSO-d6): δ 168.8, 158.8, 146.6, 145.2, 134.1, 131.8, 129.0, 126.2, 123.8, 123.4, 115.0, 65.7, 56.1, 50.4, 45.6, 44.6, 44.0, 42.9, 42.5, 37.4, 36.9, 35.7, 34.7, 32.8, 28.6, 24.7, 23.9, 18.1, 18.0. ESI-HRMS. Anal. Calcd for C37H58N5O2([M+H]+): m/z 604.4591. Found: m/z 604.4593.
Compound 12
Yield 75%; mp 194–196°C; 1H NMR (DMSO-d6): δ 0.99 (s, 3H), 1.14 (s, 3H), 1.21 (d, 6H, J = 12 Hz), 1.47–1.72 (m, 8H), 2.27–2.31 (m, 2H), 2.76 (d, H, J = 8.0 Hz), 2.81–2.90 (m, 3H), 3.05–3.18 (m, 8H), 3.55–3.70 (m, 2H), 6.87 (s, 1H), 6.95 (d, 1H, J = 7.2 Hz), 7.17 (d, H, J = 8.0 Hz); 13C NMR (DMSO-d6): δ 163.7, 146.7, 145.1, 134.4, 126.3, 123.8, 123.5, 58.7, 47.5, 44.2, 42.1, 41.1, 38.1, 37.3, 37.0, 36.1, 34.8, 32.9, 28.8, 25.3, 23.9, 18.6, 18.2. ESI-HRMS. Anal. Calcd for C26H42N3O ([M+H]+): m/z 412.3328. Found: m/z 412.3329.
Compound 13
Yield 70%; mp 249–251°C; 1H NMR (DMSO-d6): δ 0.90 (s, 3H), 1.13 (s, 3H), 1.26 (d, 6H, J = 12 Hz), 1.39–1.68 (m, 8H), 2.24–2.28 (m, 2H), 2.51 (d, H, J = 12 Hz), 2.73–2.80 (m, 3H), 3.10–3.32 (m, 8H), 3.65–3.70 (m, 2H), 4.92 (s, 2H), 6.85 (s, 1H), 6.94 (d, 1H, J = 4.0 Hz), 7.01 (d, 2H, J = 8.4 Hz), 7.13 (d, H, J = 8.4 Hz), 7.45 (d, 2H, J = 8.4 Hz); 13C NMR (DMSO-d6): δ 165.9, 158.5, 146.7, 145.2, 134.2, 131.8, 129.0, 126.3, 123.8, 123.5, 115.3, 65.4, 56.2, 50.5, 44.0, 42.5, 41.1, 38.1, 37.4, 36.9, 35.7, 34.7, 32.9, 28.6, 24.8, 23.9, 18.2, 18.0. ESI-HRMS. Anal. Calcd for C33H48N3O2 ([M+H]+): m/z 518.3747, Found: m/z 518.3750.
In vitro cytotoxicity assay
Cytotoxicities of all compounds against MCF-7 and HepG-2 cell lines were determined using a cell counting kit-8 (CCK-8) assay. The cells were plated in 96-well culture plates at density of 1×104 cells per well and incubated for 24 h at 37°C in a wet atmosphere containing 5% CO2. The tested compound was dissolved in PBS and then the diluted solution was treated with the cells for 24 h at 37°C in a 5% CO2 incubator; 5-FU was used as a positive control. Then, 10 μL of a freshly diluted CCK-8 solution [5 mg/mL in phosphate buffer saline (PBS)] was added to each well for 2 h. The cell survival was evaluated by measuring the absorbance at 450 nm. The IC50 value, which indicates the inhibition growth of 50% of cells relative to non-treated control cells, was calculated as the concentration of tested compound by best fit curving estimation. All experiments were carried out in triplicate.
Assessment of cell apoptosis
The MCF-7 cells were plated in 6-well culture plates at density of 3×105 cells per well. The cells were incubated with different concentrations of compound 10 for 24 h. The cultured MCF-7 cells were washed twice with PBS (pH 7.4) and then resuspended gently in 400 μL of binding buffer. The cell solution was then stained with Annexin V-APC/PI apoptosis Kit according to the protocol of the company.
Acknowledgments
This study was financially supported by the National Science Foundation of China (No. 21172182 and 21362026).
References
[1] Son, K. H.; Oh, H. M.; Choi, S. K.; Han, D. C.; Kwon, B. M. Anti-tumor abietane diterpenes from the cones of Sequoia sempervirens. Bioorg. Med. Chem. Lett. 2005, 15, 2019–2021.10.1016/j.bmcl.2005.02.057Search in Google Scholar
[2] Newman, D. J.; Cragg, G. M. Snader, K. M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037.10.1021/np030096lSearch in Google Scholar
[3] Gonzalez, M. A. Synthetic derivatives of aromatic abietane diterpenoids and their biological activities (Mini-review). Eur. J. Med. Chem. 2014, 87, 834–842.10.1016/j.ejmech.2014.10.023Search in Google Scholar
[4] Goodson, B.; Ehrhardt, A.; Ng, S.; Nuss, J.; Johnson, K.; Giedlin, M.; Yamamoto, R.; Moos, W. H.; Krebber, A.; Ladner, M.; et al. Characterization of novel antimicrobial peptoids. Antimicrob. Agents Chemother. 1999, 43, 1429–1434.10.1128/AAC.43.6.1429Search in Google Scholar PubMed PubMed Central
[5] Wilkerson, W. W.; Galbraith, W.; DeLucca, I.; Harris, R. R. Topical antiinflammatory dehydroabietylamine derivatives. IV. Bioorg. Med. Chem. Lett. 1993, 3, 2087–2092.10.1016/S0960-894X(01)81022-2Search in Google Scholar
[6] Lu, Z.; Liu, C. X.; Yu, X.; Lin, Z. X. Synthesis and anti-free radical activities of several novel derivatives of dehydroabietylamine. Chin. J. Org. Chem. 2013, 33, 562–567.10.6023/cjoc201211020Search in Google Scholar
[7] Rao, X. P.; Z. Song, Z. Q.; He, L. Synthesis and antitumor activity of novel aminophosphonates from diterpenic dehydroabietylamine. Heteroatom Chem. 2008, 19, 512–516.10.1002/hc.20471Search in Google Scholar
[8] Lin, L. Y.; Bao, Y. L.; Chen, Y.; Sun, L. G.; Yang, X. G.; Liu, B.; Lin, Z. X.; Zhang, Y. W.; Yu, C. L.; Wu, Y.; et al. N-Benzoyl-12-nitrodehydroabietylamine-7-one, a novel dehydroabietylamine derivative, induces apoptosis and inhibits proliferation in HepG2 cells. Chem. Biol. Interact. 2012, 199, 63–73.10.1016/j.cbi.2012.06.002Search in Google Scholar
[9] Chen, Y.; Lin, Z. X.; Zhou, A. M. Synthesis and antitumor activities of a novel class of dehydroabietylamine derivatives. Nat. Prod. Res. 2012, 26, 2188–2195.10.1080/14786419.2011.648191Search in Google Scholar
[10] Xing, Y. H.; Zhang, W.; Song, J. J.; Zhang, Y. X.; Jiang, X. X.; Wang, R. Anticancer effects of a novel class rosin-derivatives with different mechanisms. Bioorg. Med. Chem. Lett. 2013, 23, 3868–3872.10.1016/j.bmcl.2013.04.069Search in Google Scholar
[11] Huang, X. C.; Wang, M.; Wang, H. S.; Chen, Z. F.; Zhang, Y.; Pan, Y. M. Synthesis and antitumor activities of novel dipeptide derivatives derived from dehydroabietic acid. Bioorg. Med. Chem. Lett. 2014, 24, 1511–1518.10.1016/j.bmcl.2014.02.001Search in Google Scholar
[12] Huang, X. C.; Jin, L.; Wang, M.; Liang, D.; Chen, Z. F.; Zhang, Y.; Pan, Y. M.; Wang, H. S. Design, synthesis and in vitro evaluation of novel dehydroabietic acid derivatives containing a dipeptide moiety as potential anticancer agents. Eur. J. Med. Chem. 2014, 89, 370–385.10.1016/j.ejmech.2014.10.060Search in Google Scholar
[13] Kong, D. Y.; Qin, C.; Meng, L. H.; Xie, Y. Y. Synthesis, structural characterization and antitumor activity evaluations of copper complex with tetraazamacrocyclic ligand. Bioorg. Med. Chem. Lett. 1999, 9, 1087–1092.10.1016/S0960-894X(99)00139-0Search in Google Scholar
[14] Cholody, W. M.; Hernandez, L.; Hassner, L.; Scudiero, D. A.; Djurickovic, D. B.; Michejda, C. J. Cholody, W. M.; Hernandez, L.; Hassner, L.; Scudiero, D. A.; et al. Bisimidazoacridones and related compounds: new antineoplastic agents with high selectivity against colon tumors. J. Med. Chem. 1995, 38, 3043–3052.10.1021/jm00016a007Search in Google Scholar
[15] Rokosz, L. L.; Huang, C. Y.; Reader, J. C.; Stauffer, T. M.; Chelsky, D.; Sigal, N. H.; Ganguly, A. K.; Baldwin, J. J. Surfing the piperazine core of tricyclic farnesyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5537–5543.10.1016/j.bmcl.2005.08.074Search in Google Scholar
[16] Chen, J. J.; Lu, M.; Jing, Y. K.; Dong, J. H. The synthesis of l-carvone and limonene derivatives with increased antiproliferative effect and activation of ERK pathway in prostate cancer cells. Bioorg. Med. Chem. 2006, 14, 6539–6547.10.1016/j.bmc.2006.06.013Search in Google Scholar
[17] Zeng, S.; Liu, W.; Nie, F. F.; Zhao, Q.; Rong, J. J.; Wang, J.; Tao, L.; Qi, Q.; Lu, N.; Li, Z. Y.; et al. LYG-202, a new flavonoid with a piperazine substitution, shows antitumor effects in vivo and in vitro. Biochem. Biophys. Res. Commun. 2009, 38, 551–556.10.1016/j.bbrc.2009.05.099Search in Google Scholar
[18] Kamal, A.; Kumar, P. P.; Seshadri, B. N.; Srinivas, O.; Kumar, M. S.; Sen, S.; Kurian, N.; Juvekar, A. S.; Zingde, S. M. Phosphonate-linked pyrrolo[2,1-c][1,4]benzodiazepine conjugates: synthesis, DNA-binding affinity and cytotoxicity. Bioorg. Med. Chem. 2008, 14, 3895–3906.10.1016/j.bmc.2008.01.040Search in Google Scholar
[19] Liu, M. C.; Yang, S. J.; Jin, L. H.; Hu, D. Y. Xue, W.; Song, B. A. Yang, S. Synthesis and cytotoxicity of novel ursolic acid derivatives containing an acyl piperazine moiety. Eur. J. Med. Chem. 2012, 58, 128–135.10.1016/j.ejmech.2012.08.048Search in Google Scholar
[20] Kong, D.; Meng, L.; Ding, J.; Xie, Y.; Huang, X. New tetraazamacrocyclic ligand with neutral pendent groups 1,4,7,10-tetrakis (2-cyanoethyl)-1,4,7,10-tetraazacyclododecane (L) and its cobalt(II), nickel(II) and copper(II) complexes: synthesis, structural characterization and antitumor activity. Polyhedron 2000, 19, 217–223.10.1016/S0277-5387(99)00369-1Search in Google Scholar
[21] Zhang, J. X.; Li, H. G.; Chan, C. F.; Lan, R. F.; Chan, W. L.; Law, G. L.; Wong, W. K.; Wong, K. L. A potential water-soluble ytterbium-based porphyrin–cyclen dual bio-probe for Golgi apparatus imaging and photodynamic therapy. Chem. Commun. 2012, 48, 9646–9648.10.1039/c2cc34963aSearch in Google Scholar
[22] Yang, X. B.; Feng, J.; Zhang, J.; Zhang, Z. W.; Lin, H. H.; Zhou, L. H.; Yu, X. Q. Synthesis, DNA binding and cleavage activities of the copper (II) complexes of estrogen-macrocyclic polyamine conjugates. Bioorgan. Med. Chem. 2008, 16, 3871–3877.10.1016/j.bmc.2008.01.037Search in Google Scholar
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Preliminary Communications
- Montmorillonite K10 catalyzed multi component reactions (MCR): synthesis of novel thiazolidinones as anticancer agents
- Synthesis, antibacterial, and antifungal activities of new pyrimidinone derivatives
- Research Articles
- Formation of 1-methyl[1,2,4]triazolo[4,3-a] quinazolin-5(4H)-ones by reaction of 2-hydrazinoquinazolin-4(3H)-ones with acetylacetone
- Synthesis of new 4′-(N-alkylpyrrol-2-yl)-2,2′: 6′,2″-terpyridines via N-alkylation of a pyrrole moiety
- Efficient synthesis of 3-(bromomethyl)-5-methylpyridine hydrobromide
- One-pot synthesis of 5-[1-substituted 4-acetyl-5-methyl-1H-pyrrol-2-yl)]-8-hydroxyquinolines using DABCO as green catalyst
- A new on-fluorescent sensor for Ag+ based on benzimidazole bearing bis(ethoxycarbonylmethyl)amino groups
- Synthesis of new derivatives of 10H-benzo[b]pyridazino[3,4-e][1,4]thiazines
- Efficient and convenient synthesis of pyrido [2,1-b]benzothiazole, pyrimidopyrido[2,1-b]benzothiazole and benzothiazolo[3,2-a][1,8]naphthyridine derivatives
- Synthesis of 3-benzylidene-dihydrofurochromen-2-ones: promising intermediates for biflavonoid synthesis
- Synthesis and antitumor activities of piperazine- and cyclen-conjugated dehydroabietylamine derivatives
- Synthesis, characterization, and antimicrobial evaluation of novel spiropiperidones
Articles in the same Issue
- Frontmatter
- Preliminary Communications
- Montmorillonite K10 catalyzed multi component reactions (MCR): synthesis of novel thiazolidinones as anticancer agents
- Synthesis, antibacterial, and antifungal activities of new pyrimidinone derivatives
- Research Articles
- Formation of 1-methyl[1,2,4]triazolo[4,3-a] quinazolin-5(4H)-ones by reaction of 2-hydrazinoquinazolin-4(3H)-ones with acetylacetone
- Synthesis of new 4′-(N-alkylpyrrol-2-yl)-2,2′: 6′,2″-terpyridines via N-alkylation of a pyrrole moiety
- Efficient synthesis of 3-(bromomethyl)-5-methylpyridine hydrobromide
- One-pot synthesis of 5-[1-substituted 4-acetyl-5-methyl-1H-pyrrol-2-yl)]-8-hydroxyquinolines using DABCO as green catalyst
- A new on-fluorescent sensor for Ag+ based on benzimidazole bearing bis(ethoxycarbonylmethyl)amino groups
- Synthesis of new derivatives of 10H-benzo[b]pyridazino[3,4-e][1,4]thiazines
- Efficient and convenient synthesis of pyrido [2,1-b]benzothiazole, pyrimidopyrido[2,1-b]benzothiazole and benzothiazolo[3,2-a][1,8]naphthyridine derivatives
- Synthesis of 3-benzylidene-dihydrofurochromen-2-ones: promising intermediates for biflavonoid synthesis
- Synthesis and antitumor activities of piperazine- and cyclen-conjugated dehydroabietylamine derivatives
- Synthesis, characterization, and antimicrobial evaluation of novel spiropiperidones