Syntheses, structures, and properties of zinc(II) and copper(II) coordination polymers with imidazole-containing ligands
-
Long Tang
 ,
Ye Guo
,
Ye Guo

Abstract
The reactions of Zn(NO3)2 · 6H2O/CuCl2 · 2H2O and 1,4-bis(1-imidazolyl)benzene (bib)/1,4-bis(1-imidazol-1-ylmethyl)benzene (biyb), carried out under hydrothermal conditions, produced two new coordination polymers [Zn(bib)(HCOO)2 · H2O] (1) and [Cu(biyb)Cl2] (2), respectively, which were characterized by elemental analysis, infrared spectroscopy, thermogravimetric analyses, and single-crystal X-ray diffraction. In complex 1, the ZnII centers are bridged by bib ligands to form a chain, where the adjacent chains are condensed by strong intermolecular O–H · · · O hydrogen-bonding and aromatic π-π stacking interactions to generate a 3D supramolecular structure. In 2, the CuII centers are bridged by biyb ligands to form, again, chains through C–H · · · Cl hydrogen bonds, and these adjacent chains with different orientations are joined to generate a 3D supramolecular structure.
1 Introduction
Design and construction of coordination polymers (CPs) have received much attention among chemists, not only owing to their various structural motifs but also to their potential applications in the areas of ion exchange, adsorption, catalysis, chirality, luminescence, nonlinear optical materials, and magnetism [1], [2], [3], [4]. Coordination polymers with supramolecular interactions are also one of the current research fields. The self-assembly of molecular building blocks in the realm of supramolecular chemistry, through several types of noncovalent interactions such as π-π stacking, hydrogen-bonding, halogen-halogen, sulfur-sulfur interactions, and others, has been regarded as more than academic curiosity [5], [6], [7], [8]. Thus, the noncovalent assemblies, especially hydrogen-bonding and π-π stacking connectivities, are among the most explored and best investigated supramolecular architectures [9], [10].
However, the self-assembly of metal-organic coordination polymers is highly influenced by many factors such as the coordination properties of the metal ions and the length and flexibility/rigidity of the ligands. In the past years, many rigid bridging ligands such as 4,4ʹ-bipyridine and 1,4-bis(1-imidazolyl)benzene (bib), 1,4-bis(4-pyridyl) benzene, and 2,4,6-tri(4-pyridyl)-1,3,5-triazine as well as nonrigid ligands such as 1,2-bis(4-pyridyl)ethane, 1,3-bis(4-pyridyl)propane, 1,4-bis(4-pyridyl) methyl-benzene, 1,3,5-tri(imidazol-1-ylmethyl)benzene, and 1,4-bis(imidazol-1-ylmethyl)-benzene have been used to construct novel metal-organic coordination polymers with transition-metal ions [11], [12], [13], [14]. Because of the strong coordinating ability of imidazole, research on imidazole-containing ligands has seen drastic development [15]. In our own research strategy, imidazole-containing rigid or nonrigid ligands have been employed in the construction of coordination polymers. Here we report two 3D supramolecular architectures, namely [Zn(bib)(HCOO)2·H2O] (1) and [Cu(biyb)Cl2] (2) [biyb=1,4-bis(1-imidazol-1-ylmethyl)benzene)]. The syntheses, crystal structures, and properties of the two compounds are presented and discussed in this article.
2 Experimental
Elemental analyses were performed on a Perkin-Elmer 2400 elemental analyzer in the USA. IR spectra were recorded on a Shimadzu FTIR-8400s spectrometer using KBr pellets in Japan. A Netzsch STA 449C thermogravimetric analyzer was used for the thermoanalyses in Germany. The fluorescence spectra were recorded using a Hitachi F-4500 fluorescence spectrophotometer at room temperature in Japan. The magnetic susceptibilities were obtained on microcrystalline powder samples using a Quantum Design MPMS SQUID magnetometer in the USA. X-ray powder diffraction (XRPD) was carried out on a Shimadzu XRD-7000 analyzer in Japan.
2.1 Synthesis and crystallization of [Zn(bib)(HCOO)2·H2O] (1)
All reagents and solvents were obtained from commercially available sources and of analytical grade. A mixture of Zn(NO3)2·6H2O (0.0297 g, 0.1 mmol), bib (0.0210 g, 0.1 mmol), dimethylformamide (DMF, 2.0 mL), formic acid (2.0 mL), and H2O (4.0 mL) was stirred and heated in a 20-mL Teflon-lined autoclave at T=373 K for 5 days, and then slowly cooled down to room temperature for crystallization. Colorless block crystals were obtained. Yield: 47% (based on Zn). Elemental analysis C14H14N4O5Zn (%): Calcd. C 43.83, H 3.68, N 14.60; found C 43.69, H 3.56, N 14.51. IR (KBr, cm−1): 3442 (vs), 3138 (s), 1618 (vs), 1423 (s), 1234 (s), 1145 (s), 1068 (s), 958(m), 839 (s), 769 (w), 646 (vs).
2.2 Synthesis and crystallization of [Cu(biyb)Cl2] (2)
A mixture of CuCl2·2H2O (0.0170 g, 0.1 mmol), biyb (0.0238 g, 0.1 mmol), DMF (2.0 mL), formic acid (2.0 mL), and H2O (4.0 mL) was stirred and heated in a 20-mL Teflon-lined autoclave at 373 K for 5 days, and then slowly cooled down to room temperature for crystallization. Blue block crystals were obtained. Yield: 45% (based on Cu). Elemental analysis C14H14N4Cl2Cu (%): Calcd. C 45.11, H 3.78, N 15.03; found C 45.02, H 3.76, N 14.94. IR (KBr, cm−1): 1516 (s), 1441 (m), 1276 (m), 1231 (vs), 1102 (vs), 1093 (s), 936 (m), 834 (m), 754 (s), 665 (vs).
2.3 Crystal structure determinations
Diffraction intensities for complexes 1 and 2 were collected at T=296(2) K on a Bruker SMART APEX-II CCD diffractometer employing graphite-monochromatized MoKα radiation (λ=0.71073 Å). The structure was solved by Direct Methods and refined by full-matrix least-squares on F2 using the programs Shelxt-2014 [16] and Shelxl-2014 [17], respectively. Non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed in the calculated positions. H atoms bonded to C atoms were treated using a riding model approximation, with C–H=0.93/0.97 Å and Uiso(H)=1.2Ueq(C) for aromatic H atoms. Water H atoms were refined in a riding mode with the constraint O–H=0.81Å and with Uiso(H)=1.5Ueq(O). The crystallographic data for compounds 1 and 2 are listed in Table 1, and selected bond lengths and angles are listed in Table 2.
Crystal data and structure refinement for compounds 1 and 2.
| Formula | C14H14N4O5Zn | C14H14N4Cl2Cu |
| Mr | 383.66 | 372.73 |
| Cryst. size, mm3 | 0.21×0.19×0.12 | 0.21×0.17×0.15 |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | P2/c (#13) | Pbcn (#60) |
| a, Å | 10.328(5) | 11.7269(13) |
| b, Å | 5.219(3) | 9.4281(11) |
| c, Å | 16.060(7) | 15.0953(16) |
| β, deg | 117.29(2) | 90 |
| V, Å3 | 769.3(7) | 1669.0(3) |
| Z | 2 | 4 |
| Dcalcd, g cm−3 | 1.66 | 1.48 |
| F(000), e | 392 | 756 |
| Radiation; λ, Å | MoKα; 0.71073 | MoKα; 0.71073 |
| μ(Mo Kα), cm−1 | 16.3 | 16.3 |
| hkl range | ±12, –4→+6, ±19 | ±15, –12→+10, –18→+20 |
| θ range, deg | 2.695–24.982 | 2.772–28.290 |
| Refl. total/unique/Rint | 4612/1354/0.0126 | 9637/2065/0.0179 |
| Param. Refined | 110 | 96 |
| R(F)/wR(F2)a (all refl.) | 0.0235/0.0666 | 0.0302/0.0908 |
| GoF (F2)b | 1.088 | 1.033 |
| Δρfin (max/min), e Å−3 | 0.36/–0.29 | 0.54/–0.30 |
aR1=||Fo|−|Fc||/Σ|Fo|, wR2=[Σw(Fo2−Fc2)2/Σw(Fo2)2]1/2, w=[σ2(Fo2)+(AP)2+BP]−1, where P=(Max(Fo2, 0)+2Fc2)/3. bGoF=[Σw(Fo2−Fc2)2/(nobs−nparam)]1/2.
Selected bond lengths (Å) and bond angles (deg) for 1 and 2.a
| Compound 1 | Compound 2 | ||
|---|---|---|---|
| Zn1–O1 | 1.9540(17) | Cu1–N1 | 1.9420(16) |
| Zn1–N1 | 2.0045(16) | Cu1–Cl1 | 2.2634(5) |
| O1#1–Zn1–O1 | 100.85(11) | N1–Cu1–N1#1 | 144.10(11) |
| O1–Zn1–N1#1 | 113.02(8) | N1–Cu1–Cl1 | 97.40(5) |
| O1–Zn1–N1 | 108.93(7) | N1#1–Cu1–Cl1 | 96.92(6) |
| N1#1–Zn1–N1 | 111.67(10) | Cl1–Cu1–Cl1#1 | 132.29(3) |
aSymmetry code for 1: #1 1−x, y, 1/2−z; for 2: #1 2−x, y, 3/2−z.
CCDC 1936085 (1) and 1936086 (2) contain the supplementary crystallographic data for this article. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
3 Results and discussion
3.1 Crystal and molecular structure of compound 1
Single-crystal X-ray structure determination revealed that compound 1 crystallizes in the space group P2/c with Z=2. It consists of one ZnII ion, two formate anions, one bib ligand, and one free water molecule. The ZnII atoms reside on crystallographic twofold axes. Each ZnII center is four-coordinated by two oxygen atoms from two formate anions and two nitrogen atoms from two bib ligands, yielding a distorted ZnO2N2 tetrahedral geometry (Fig. 1). The Zn–O/N bond lengths are 1.9540(17) Å/2.0045(17) Å. The O/N–Zn–O/N bond angles are in the range 100.85(11)–113.02(8)° (Table 2). In compound 1, the adjacent ZnII centers are bridged by bib ligands to form a chain along the crystallographic a-axis, with the Zn···Zn distance of 13.522(3) Å, where all formate anions bristle out from two sides of the chain (Fig. 2). Hydrogen-bonding and aromatic π-π stacking interactions play the major role in packing the molecules into a higher dimensional network. So, through intermolecular O–H···O hydrogen bonds between the free water and a carboxylate O atom of the formate anion [O3–H3A···O2 distance: 2.840(3) Å, angle: 175.79(4)°], the adjacent chains are joined to generate a layer structure (Fig. 3). Further, through aromatic π-π stacking interactions between phenyl and imidazole ring of the bib ligands (centroid-to-centroid distance: 3.755(3) Å), the adjacent layers generate a 3D supramolecular structure (Fig. 4).

Coordination environment of Zn(II) in compound 1. (Symmetry code: #1 1 – x, y, 1/2−z.)

Chain in crystals of 1.

Layer structure in crystals of 1.

3D structure in crystals of 1.
3.2 Crystal and molecular structure of compound 2
Compound 2 crystallizes in the space group Pbcn with Z=4. The asymmetric unit of 2 contains one crystallographically unique CuII ion, one biyb ligand, and two Cl atoms. As in complex 1, the metal atom resides on a crystallographic twofold axis. Each CuII center is four-coordinated by two nitrogen atoms from two biyb ligands and two Cl atoms, yielding a distorted CuN2Cl2 square planar coordination [18] (Fig. 5). The Cu–N bond length is 1.9420(16) Å and the Cu–Cl bond length is 2.2634(5) Å. The Cl/N–Cu–N/Cl bond angles are in the range 96.92(6)–144.10(11)° (Table 2). In compound 2, Each CuII ion is linked by two different μ2-bridging biyb ligands with its two neighboring CuII ions to form a chain. Interestingly, these chains have two orientations in space (Fig. 6). So, through intermolecular C–H···Cl hydrogen bonds between the Cl atom and a CH2 unit of the biyb ligand [C4–H4B···Cl1 distance: 3.626(2) Å, angle: 132.5(2)°], these adjacent chains with different orientations are joined to generate a 3D supramolecular structure (Fig. 7).
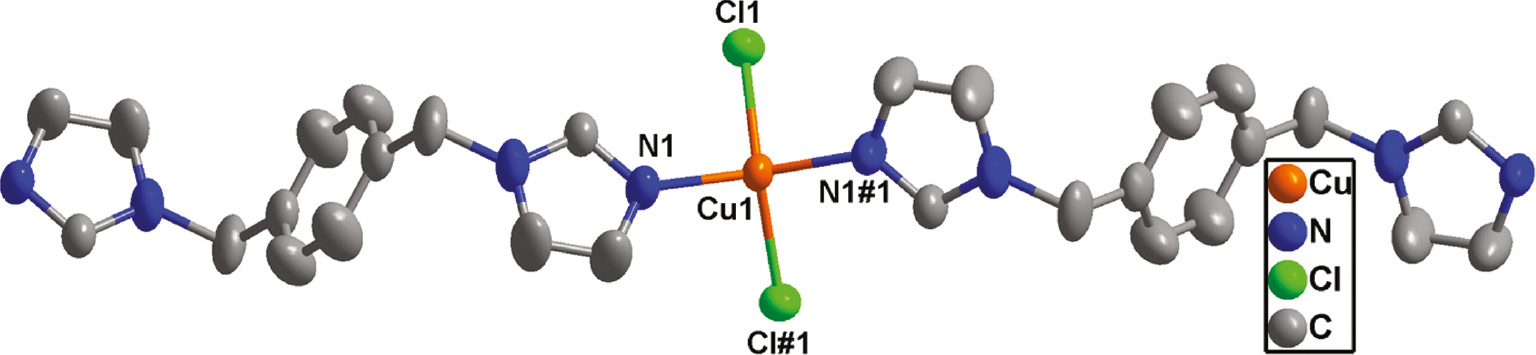
Coordination environment of Cu(II) in compound 2. (Symmetry code: #1 2−x, y, 3/2−z)
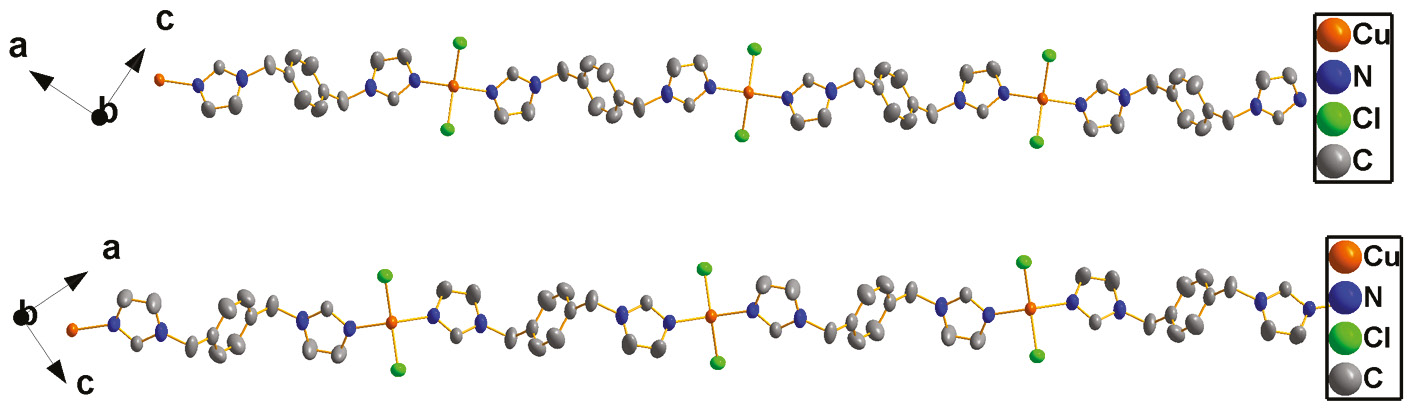
Chains in crystals of 2.

3D structure in crystals of 2.
3.3 IR spectra
In the IR spectrum of compound 1, the broad, strong bands in the frequency region near 3442 cm−1 are assigned to the characteristic stretching peaks of OH of free water molecules. The strong bands that appear around 1618 and 1423 cm−1 correspond to the asymmetric and symmetric stretching vibrations of the carboxylic group. The strong peaks that appear around 1234, 1145, and 1068 cm−1 correspond to the C=C, C–N, and C–H vibrations of the imidazole ring. In compound 2, the absorption peaks near 1441 cm−1 are assigned to the C–H vibration of the –CH2– groups. The strong bands that appear around 1231, 1102, and 1093 cm−1 correspond to the C=C, C–N, and C–H vibrations of the imidazole ring.
3.4 Thermogravimetric analysis
Thermogravimetric (TG) analyses were carried out to examine the thermal stability of 1 and 2 from T=20–700°C at a heating rate of 5 K·min−1 under nitrogen atmosphere (Fig. 8). The TG curve of 1 shows two weight loss steps: the first one in the range 80–120°C (obsd. 4.8%, calcd. 4.70%) is assigned to the loss of free water, and the second one of 78.4% in the temperature range 210–460°C corresponds to the decomposition of the bib ligands and formate anions (calcd. 78.26%). The TG curve of 2 shows no weight loss from 20°C until about 180°C, above which a significant weight loss of 83.1% is observed and ended at about 440°C, indicating the release the ligands (calcd. 82.95%). The final decomposition products of 1 and 2 were confirmed to be ZnO and CuO, respectively, by their PXRD patterns.
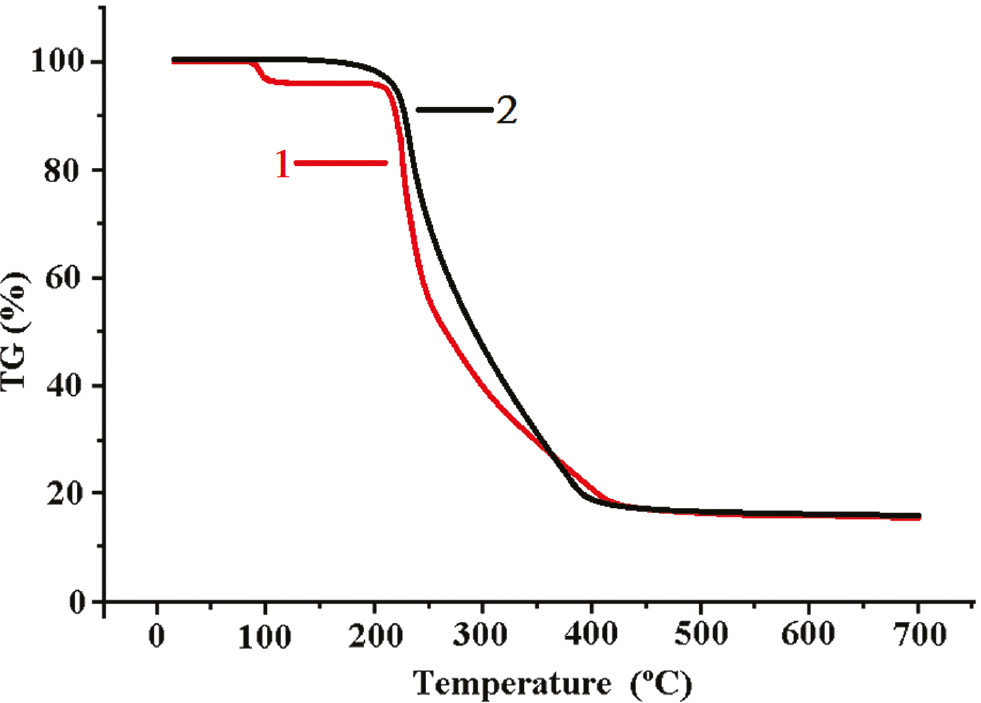
TG curves of compounds 1 and 2.
3.5 Fluorescence properties of compound 1
The luminescence properties of compound 1 and of the free bib ligand were studied in the solid state at room temperature. As shown in Fig. 9, the photoluminescence (PL) spectrum of compound 1 displays a relatively wide emission band with a maximum at 433 nm (λex=307 nm). Upon excitation at 374 nm, the free bib ligand shows an intense emission peak at 444 nm. Compared to the bib ligand, compound 1 shows a blue shift of 11 nm, probably due to the ligand-to-metal charge transfer (LMCT) [19], [20].

Emission spectra of the free bib ligand and compound 1.
3.6 Magnetic properties of compound 2
The magnetic behavior of compound 2 was investigated over the temperature range 2–300 K in a field of 10 kOe (1 kOe=7.96×104 A m−1). The magnetic susceptibilities χMT and 1/χM vs. T plots are shown in Fig. 10. The χMT value of 2 at T=300 K is 0.386 cm3 K mol−1, which is only slightly larger than 0.375 cm3 K mol−1 expected for the spin-only Cu(II) ion. Upon cooling, the value of χMT decreases continuously down to 50 K, and then rapidly drops near 2 K. The temperature dependence of the reciprocal susceptibility (1/χM) obeys the Curie-Weiss law with θ=−1.43 K, C=0.389 cm3 K mol−1, and R=2.38×10−4 (R=∑[(χM)obs−z(χM)calcd]2/∑(χM)obs2). The value of θ indicates weak antiferromagnetic interactions between adjacent Cu(II) (S=1/2) ions.

Thermal variation of 1/χM and χMT for 2 (∆, 1/χM experimental values; ○, χMT experimental values and solid lines represent the best fit obtained from the Hamiltonian given in the text).
Funding source: National Natural Science Foundation of China
Award Identifier / Grant number: 21573189
Award Identifier / Grant number: 21503183
Award Identifier / Grant number: 21763028
Funding statement: This work was supported by the National Natural Science Foundation of China (Nos. 21573189, 21503183, and 21763028), the Research Foundation Project of Yan’an University, and the National College Students’ innovation and entrepreneurship training program (D2017009).
References
[1] T. Yamada, K. Otsubo, R. Makiura, H. Kitagawa, Chem. Soc. Rev.2013, 42, 6655–6669.10.1039/c3cs60028aSuche in Google Scholar PubMed
[2] M. H. Zeng, Z. Yin, Y. X. Tan, W. X. Zhang, Y. P. He, M. Kurmoo, J. Am. Chem. Soc.2014, 136, 4680–4688.10.1021/ja500191rSuche in Google Scholar PubMed
[3] Y. P. Wu, W. Zhou, J. Zhao, W. W. Dong, Y. Q. Lan, D. S. Li, C. H. Sun, X. H. Bu, Angew. Chem. Int. Ed.2017, 56, 13001–13005.10.1002/anie.201707238Suche in Google Scholar PubMed
[4] X. L. Wang, Y. Xiong, X. T. Sha, G. C. Liu, H. Y Lin, Cryst. Growth Des.2017, 17, 483–496.10.1021/acs.cgd.6b01299Suche in Google Scholar
[5] J. Jiang, H. Furukawa, Y. B. Zhang, O. M. Yaghi, J. Am. Chem. Soc.2016, 138, 10244–10251.10.1021/jacs.6b05261Suche in Google Scholar PubMed
[6] L. Tang, F. Fu, J. J. Wang, L. J. Gao, D. D. Chao, Z. Wang, Polyhedron2015, 88, 116–124.10.1016/j.poly.2014.12.023Suche in Google Scholar
[7] M. Du, C. P. Li, C. S. Liu, S. M. Fang, Coord. Chem. Rev.2013, 257, 1282–1305.10.1016/j.ccr.2012.10.002Suche in Google Scholar
[8] S. Kitagawa, K. Uemura, Chem. Soc. Rev.2005, 34, 109–119.10.1039/b313997mSuche in Google Scholar PubMed
[9] Y. P. Wu, G. W. Xu, W. W. Dong, J. Zhao, D. S. Li, J. Zhang, X. H. Bu, Inorg. Chem.2017, 56, 1402−1411.10.1021/acs.inorgchem.6b02476Suche in Google Scholar PubMed
[10] Y. X. Sun, Y. Y. Sun, H. Zheng, H. L. Wang, Y. Han, Y. Yang, L. Wang, CrystEngComm2016, 18, 8664–8671.10.1039/C6CE01709FSuche in Google Scholar
[11] S. A. Barnett, N. R. Champness, Coord. Chem. Rev.2003, 246, 145−168.10.1016/S0010-8545(03)00121-8Suche in Google Scholar
[12] E. Q. Gao, Y. X. Xu, C. H. Yan, CrystEngComm2004, 6, 298–302.10.1039/b411198bSuche in Google Scholar
[13] Z. X. Li, X. Chu, G. H. Cui, Y. Liu, L. Li, G. L. Xue, CrystEngComm2011, 13, 1984–1989.10.1039/C0CE00865FSuche in Google Scholar
[14] C. Liu, G. H. Cui, K. Y. Zou, J. L. Zhao, X. F. Gou, Z. X. Li, CrystEngComm2013, 15, 324–331.10.1039/C2CE26532JSuche in Google Scholar
[15] T. Wu, J. Zhang, C. Zhou, L. Wang, X. Bu, P. Feng, J. Am. Chem. Soc.2009, 131, 6111–6113.10.1021/ja901725vSuche in Google Scholar PubMed
[16] G. M. Sheldrick, Acta Crystallogr.2015, A71, 3–8.10.1107/S2053273314026370Suche in Google Scholar
[17] G. M. Sheldrick, Acta Crystallogr.2015, C71, 3–8.Suche in Google Scholar
[18] C. A. Krasinski, B. L. Solomon, F. F. Awwadi, C. P. Landee, M. M. Turnbull, J. L. Wikaira, J. Coord. Chem.2017, 70, 914−935.10.1080/00958972.2016.1278213Suche in Google Scholar
[19] Y. Q. Xu, D. Q. Yuan, B. L. Wu, L. Han, M. Y. Wu, F. L. Jiang, M. C. Hong, Cryst. Growth Des.2006, 6, 1168−1174.10.1021/cg0506774Suche in Google Scholar
[20] Y. J. Cui, Y. F. Yue, G. D. Qian, B. L. Chen, Chem. Rev.2012, 112, 1126−1162.10.1021/cr200101dSuche in Google Scholar PubMed
© 2019 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Research Articles
- Structural elucidation of two new compounds from Artemisia ordosica and their antioxidative activity
- Chemical constituents from Penianthus camerounensis Dekker (Menispermaceae)
- Zur Formylierung von Hydroxy- und Alkoxyaromaten mit Ameisensäure
- Chitosan-attached nano-Fe3O4 as a superior and retrievable heterogeneous catalyst for the synthesis of benzopyranophenazines using chitosan-attached nano-Fe3O4
- Syntheses, structures, and properties of zinc(II) and copper(II) coordination polymers with imidazole-containing ligands
- Synthesis, crystal structure and antimicrobial activities of a dinuclear silver(I) complex of bis(diphenylphosphano)methane and thiourea
- [Nb6Cl12(HIm)6](OAc)2·3MeOH – a hydrogen-bonded network of niobium cluster cations, acetate anions and methanol molecules
- Synthesis of the first nickel borate nitrate K7Ni[B18O24(OH)9](NO3)6· (H3BO3)
- Polymorphs and derivates of Sr2LiAlO4:Eu2+
- Investigation of mesitylene-solvated group 13 mixed-metal halides: syntheses and crystal structures of bis(1,3,5-trimethylbenzene)gallium(I) tetrachlorido- and tetrabromidoaluminate(III) and (1,3,5-trimethylbenzene)gallium(I) tetraiodidoaluminate(III). Variation of the gallium-π-arene bond strength
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Research Articles
- Structural elucidation of two new compounds from Artemisia ordosica and their antioxidative activity
- Chemical constituents from Penianthus camerounensis Dekker (Menispermaceae)
- Zur Formylierung von Hydroxy- und Alkoxyaromaten mit Ameisensäure
- Chitosan-attached nano-Fe3O4 as a superior and retrievable heterogeneous catalyst for the synthesis of benzopyranophenazines using chitosan-attached nano-Fe3O4
- Syntheses, structures, and properties of zinc(II) and copper(II) coordination polymers with imidazole-containing ligands
- Synthesis, crystal structure and antimicrobial activities of a dinuclear silver(I) complex of bis(diphenylphosphano)methane and thiourea
- [Nb6Cl12(HIm)6](OAc)2·3MeOH – a hydrogen-bonded network of niobium cluster cations, acetate anions and methanol molecules
- Synthesis of the first nickel borate nitrate K7Ni[B18O24(OH)9](NO3)6· (H3BO3)
- Polymorphs and derivates of Sr2LiAlO4:Eu2+
- Investigation of mesitylene-solvated group 13 mixed-metal halides: syntheses and crystal structures of bis(1,3,5-trimethylbenzene)gallium(I) tetrachlorido- and tetrabromidoaluminate(III) and (1,3,5-trimethylbenzene)gallium(I) tetraiodidoaluminate(III). Variation of the gallium-π-arene bond strength