Pathogenesis of anti-neutrophil cytoplasmic antibody-associated vasculitis
-
 and
and

Abstract
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) encompasses a group of potentially life-threatening disorders characterized by necrotizing small vessel vasculitis with positive serum ANCA. To date, the pathogenesis of AAV has not been fully elucidated, but remarkable progress has been achieved in the past few decades. In this review, we summarize the mechanism of AAV. The pathogenesis of AAV involves various factors. ANCA, neutrophils, and the complement system play key roles in disease initiation and progression, forming a feedback amplification loop leading to vasculitic injury. Neutrophils activated by ANCA undergo respiratory burst and degranulation, as well as releasing neutrophils extracellular traps (NETs), thus causing damage to vascular endothelial cells. Activated neutrophils could further activate the alternative complement pathway, leading to the generation of complement 5a (C5a), which amplifies the inflammatory response by priming neutrophils for ANCA-mediated overactivation. Neutrophils stimulated with C5a and ANCA could also activate the coagulation system, generate thrombin, and subsequently cause platelet activation. These events in turn augment complement alternative pathway activation. Moreover, disturbed B-cell and T-cell immune homeostasis is also involved in disease development. In-depth investigation in pathogenesis of AAV might help to offer more effective targeted therapies.
Introduction
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) encompasses a group of potentially life-threatening autoimmune disorders characterized by inappropriate recruitment of activated neutrophils with subsequent necrotizing small-vessel vasculitis (involving small intraparenchymal arteries, arterioles, capillaries, and venules) with little or no immune deposits.[1] AAV comprises granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA).[1] ANCAs against neutrophil cytoplasmic constituents, in particular proteinase 3 (PR3) and myeloperoxidase (MPO), are serological biomarkers of AAV. In most cases, MPO-ANCA is more frequent in patients with MPA, and PR3-ANCA is more frequent in patients with GPA.[2] EGPA is less commonly associated with ANCAs, notably MPO-ANCA.
AAV could manifest as multisystem disease or single-organ vasculitis. MPA and GPA can involve small vessels in almost any organ, but the most commonly involved ones are the kidneys and lungs.[1] EGPA has less overlap with MPA and GPA and is increasingly recognized as a distinct entity regarding its genetic, pathogenetic, clinical, and pathological features.
The etiology of AAV is multifaceted, including genetic and environmental predisposing factors. ANCA, neutrophils and complements are key components in the pathogenesis. In this review, we present the current understanding and recent advances in the etiology and pathogenesis of AAV.
Etiology
Although the precise etiology of AAV has yet to be fully elucidated, studies have revealed that genetic, environmental and infectious factors as well as drugs are involved in the occurrence of AAV.
Genetic Factors
Epidemiological differences have been observed in AAV, for example, the predominance of MPA with MPO-ANCA in Asian populations and the predominance of GPA with PR3-ANCA in Caucasians,[3,4] suggesting that genetic factors might be one of the causes of AAV. The strongest genetic associations with human AAV are displayed by class II major histocompatibility complex (MHC) (also known as human leukocyte antigen, HLA) genes. A genome-wide association study (GWAS) from Europe demonstrated that MPA and GPA were genetically distinct, and the strongest genetic associations were with the antigenic specificity (MPO or PR3) of ANCA instead of with the clinical manifestation (MPA or GPA).[5] PR3-ANCA-positive AAV is most strongly associated with the HLA-DP region, in particular the HLA-DPB1*0401 allele, whereas MPO-ANCA-positive AAV is highly associated with the HLA-DQ region.[5,6,7] Moreover, the HLA-DPB1*0401 genotype is associated with an increased risk of relapse in PR3-ANCA-positive disease.[8] Studies from Japan and China have shown that HLA-DRB1*0901, DQB1*0303, and DRB1*1101 are associated with MPA.[9,10] Further research revealed that MPO-HLA class II complexes could be detected both in neutrophils from patients with MPA and healthy donor-derived neutrophils stimulated with cytokines, indicating that MPO-HLA-DR complexes may be involved in the pathogenesis of MPA.[11]
In the non-MHC genes, the PTPN22 variant is associated with disease susceptibility to AAV because this gain-of-function variant results in suppression of interleukin-10 (IL-10, an immunosuppressive cytokine).[12] Variants of PRTN3 (encoding PR3), SERPINA1 (encoding α1-antitrypsin, a main inhibitor of PR3) and SEMA6A (encoding semaphorin 6a) are associated with GPA- or PR3-ANCA-positive AAV.[5,6,7] In the BACH2 gene, a rare variant which could reduce luciferase gene expression in endothelial cells is associated with MPO-ANCA-positive AAV.[7]
Epigenetic factors, such as DNA methylation, are also linked with AAV by implicating in the regulation of PRTN3 and MPO gene expression.[13,14] Patients with active disease have hypomethylation of PRTN3 and MPO and upregulated gene expression, whereas DNA methylation increases during remission with downregulated gene expression.[14]
Environmental Factors
Accumulating evidence suggests that environmental factors participate in the development of AAV. A group of geoepidemiological triggers have been identified, including latitude-dependent and seasonality factors such as UV radiation. In the 1990s, an increasing positive rate of ANCA was observed among practitioners in the mining and construction industries.
One of the most commonly reported substances associated with the occurrence of AAV is silica.[15] The role of silica is further supported by the increasing frequency of AAV observed following 3 large earthquakes in Asia.[16,17,18] Silica-induced apoptosis of macrophages and polymorphonuclear leukocytes might act as a trigger of AAV, notably MPO-ANCA-positive MPA.[15,19]
Infectious Factors
Bacterial infections have been implicated in the initiation and relapse of AAV, especially GPA. Among these bacteria, S. aureus seems to have the strongest association with GPA.[20,21] Particular virulence genes (presence or absence) of S. aureus isolates from AAV patients contribute to disease progression and/or relapse.[22] S. aureus can also produce toxic shock syndrome toxin 1, which supplies homologous peptides of complementary PR3 (cPR3) and leads to the generation of both anti-PR3 and anti-cPR3 antibodies, exacerbating GPA.[23] Moreover, proinflammatory stimuli of infectious origin, such as bacterial lipopolysaccharide (LPS), could act synergistically with ANCA to cause vasculitis.[24]
Kain et al. first reported that ANCA targeting human lysosomal membrane protein-2 (LAMP-2) could be detected in most active ANCA-associated necrotizing crescentic glomerulonephritis (NCGN).[25] As a lesser known ANCA antigen, LAMP-2 is homologous to the bacterial adhesion protein FimH. FimH is found in gram-negative bacteria, so fimbriated bacteria might induce autoantibodies to human LAMP-2 through molecular mimicry and contribute to the development of NCGN.[26] In contrast, Roth et al. reported a lower prevalence of anti-LAMP-2 antibodies in AAV. Furthermore, these antibodies failed to induce glomerulonephritis in WKY rats.[27] Therefore, the relevance of anti-LAMP-2 antibodies in AAV is still controversial.
Drugs
Certain drugs can induce ANCAs or even AAV, such as propylthiouracil (PTU), minocycline, D-penicillamine, hydralazine, sulfasalazine and anti-TNF agents.[28,29] Among these agents, the most often implicated drug is PTU.[29] ANCA expression ranges from 4% to 46% in patients treated with PTU, and multiple target antigens including MPO, PR3, cathepsin G, lactoferrin, neutrophil elastase and azurocidin have been detected. Gao et al. reported that only 27.3% (6/22) of these ANCA-positive patients had clinical vasculitis (All four patients with MPO-ANCA and two of the three patients with PR3-ANCA).[30,31] The interaction between PTU and MPO has been suggested to contribute to the development of vasculitis. After repeated exposure to PTU, MPO might undergo structural changes and make itself a neoantigen.[32] In addition, PTU can be transformed into cytotoxic metabolites by neutrophil-derived MPO in the presence of hydrogen peroxidase.[33] The converted drug and its cytotoxic metabolites are immunogenic and then stimulate T cells to participate in B-cell production of ANCA.[34] In a rat model of MPO-ANCA-mediated AAV, PTU treatment induces the generation of neutrophil extracellular traps (NETs),[35] which could enhance inflammation in AAV.
Pathogenesis
To date, the pathogenesis of AAV has not been fully elucidated, but increasing evidence shows that the interaction among neutrophils, ANCA, the complement system and the coagulation system is of vital importance in the development of AAV. (Figure 1)
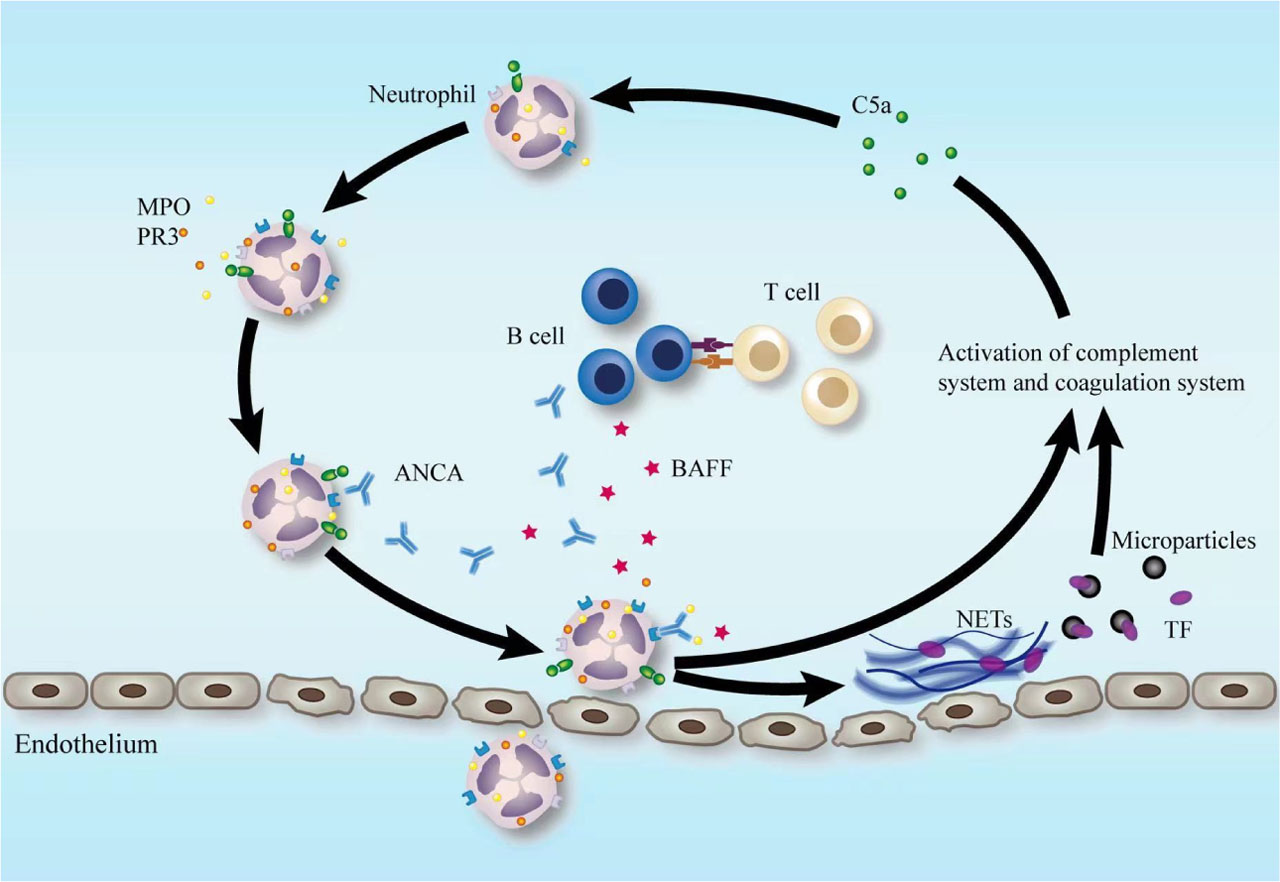
Common pathogenetic events in AAV. Priming of neutrophils by cytokines, such as C5a, leads to upregulation of the expression of ANCA target antigen on the cell surface. The primed neutrophils are further activated by ANCA, resulting in respiratory burst and degranulation, as well as the release of NETs, thus causing damage to vascular endothelial cells. Activated neutrophils could further activate the complement alternative pathway, resulting in the generation of C5a, which amplifies the inflammatory response by priming neutrophils for ANCA-mediated activation. TF-bearing microparticles are released by activated neutrophils as well, activating the coagulation system and producing thrombin. Thrombin causes activation of platelets, which in turn can further amplify the alternative complement pathway. Disturbed B-cell and T-cell immune homeostasis is also critically involved in disease development. ANCA, anti-neutrophil cytoplasmic antibody; AAV, ANCA-associated vasculitis; C5a, complement 5a; TF, tissue factor; NETs, neutrophil extracellular traps; MPO, myeloperoxidase; PR3, particular proteinase 3; BAFF, B-cell activating factor.
ANCA
Pathogenicity of ANCA
ANCAs were first reported in pauci-immune NCGN patients in 1982[36] and are the most important serological biomarkers of AAV. IgG-ANCA is the most common isotype, but IgA-ANCA and IgM-ANCA have also been reported.[37,38] Clinical and experimental data evidence that ANCAs are crucial in the pathogenesis of AAV.
In 2004, Bansal and colleagues first reported that transplacental transfer of MPO-ANCA IgG from the mother to the neonate resulted in pulmonary hemorrhage and glomerulonephritis, providing direct evidence for the pathogenicity of ANCA.[39] The in vivo pathogenicity of ANCA was also demonstrated indirectly by the efficacy of plasma exchange for severe renal vasculitis.[40] Rituximab is a chimeric murine/human monoclonal antibody against CD20 (targeting B cells), and randomized controlled clinical trials have shown rituximab efficacy and safety in inducing remission of AAV,[41,42] suggesting a pathogenic effect of ANCA produced by mature B cells.
The strongest supporting evidence for ANCA pathogenicity comes from compelling animal models. In 2002, Xiao et al. developed a passive immune model.[43] MPO knockout (Mpo-/-) mice were immunized with mouse MPO. Transfer of anti-MPO IgG or MPO-reacting splenocytes from these mice to Rag2-/- mice (lacking functioning T lymphocytes and B lymphocytes) or wild-type mice successfully induced pauci-immune NCGN and, in some recipient mice, vasculitis in the lung and other organs. This animal model clearly demonstrates the pathogenicity of ANCA. Proinflammatory stimuli and ANCA act synergistically to induce vasculitic disease. Huugen et al. revealed that the disease severity of MPO-ANCA-induced NCGN could be enhanced in the presence of bacterial LPS.[24] Kuligowski et al. further demonstrated that low-dose anti-MPO antibody could bind to neutrophils in mice and mediate their adhesion to glomerular endothelial cells (GEnCs) in the presence of LPS, whereas high-dose anti-MPO antibody can directly mediate the adhesion between neutrophils and GEnCs independent of LPS.[44] In 2005, Little et al. established a novel rat model by immunizing WKY rats with human MPO.[45] These rats developed small vessel vasculitis with MPO-ANCA directed against leukocytes. These antibodies enhanced leukocyte adhesion and transmigration in response to the chemokine Groalpha (CXCL1). Passive transfer of immunoglobulin from these rats to naive rats conferred enhanced adhesion and transmigration responses in the recipients. Furthermore, MPO-immunized rats and recipients of ANCA-positive immunoglobulin developed extensive microvascular lesions in response to CXCL1, thus providing in vivo evidence for the capacity of ANCA to enhance leukocyte-endothelial interactions and cause microvascular damage.
In contrast, well-designed PR3-ANCA-induced AAV animal models are still lacking. The lower homology of human and mouse PR3 may contribute to the difficulty in developing GPA animal models.[46] A model of PR3-ANCA-induced lesions developed in PR3-neutrophil elastase-deficient mice. In this model, the passive transfer of murine anti-mouse PR3 resulted in stronger localized cutaneous inflammation, and perivascular infiltrates were observed around small vessels at intradermal injection sites of TNF-α.[47] In 2012, Little et al.[48] injected human hematopoietic stem cells into irradiated NOD-scid-IL2γ-/- mice and then treated the chimeric mice with human PR3-ANCA-IgG. Finally, mild pauci-immune proliferative glomerulonephritis developed, with infiltration of mouse and human leukocytes. In aggregate, these animal studies have documented that both MPO-ANCA and PR3-ANCA are capable of causing vasculitis.
The mechanism by which ANCAs cause vasculitis has been explained as follows. Small amounts of proinflammatory cytokines (e.g., IL-1β and TNF-α generated during infection) at low concentrations unable to cause full neutrophil activation are capable of inducing the translocation of ANCA target antigens (namely, PR3 and MPO) to the cell surface (priming of neutrophils).[49] ANCAs bind to their autoantigens by the F(ab’)2 portion, while the Fc portion binds to Fcγ receptors (FcγR) on neutrophils.[50] Fc–FcγR engagement results in activation of neutrophils, including respiratory bursts with the release of reactive oxygen species (ROS), degranulation, phagocytosis, upregulation of adhesion molecules, and abnormal inflammatory cytokine production accompanied by lytic enzymes, which initiate vascular endothelial injury.[44,51] ANCA antigens (PR3 and MPO) released from activated neutrophils and monocytes also cause cell damage.[52,53,54,55] Excessive neutrophil activation results in the subsequent generation of NETs, which are lattice-like structures containing extruded DNA, histones, and neutrophil granule proteins such as PR3 and MPO. NETs can augment the autoimmune response, activate the alternative complement pathway and directly enhance tissue injury.[51,56,57,58] In addition to the FcγR–mediated mechanism, the ANCA F(ab’)2 portion plays a role in ROS production and transcription of cytokine genes in normal human monocytes and neutrophils.[59] Microarray gene chip analysis has shown that the ANCA F(ab’)2 portion and ANCA IgG stimulate the transcription of a distinct subset of genes,[60] suggesting that ANCA F(ab’)2 portions are capable of low-level monocyte and neutrophil activation.
ANCA Production
As mentioned above, the pathogenicity of ANCA has been well demonstrated by clinical, animal model, and in vitro experimental evidence. However, there is less understanding of the inciting events in the breakdown of tolerance and the production of ANCA. Jennette et al. proposed the hypothetical theory of autoantigen complementarity.[23,51] This theory is based on evidence that proteins transcribed and translated from the sense strand of DNA bind to proteins that are transcribed and translated from the complementary (antisense) strand of DNA. Initiating ANCA formation entails an initial reaction to complementary peptides and results in production of antibodies not only to complementary(antisense) peptide, but also to the immunogen's sense peptide.[23,51] For example, S. aureus can supply homologous peptides of cPR3, leading to the generation of PR3- and cPR3-ANCAs.[25,26]
The overactivation of neutrophils induced by ANCAs results in NETs formation. NETs will, in turn, augment the production of ANCA by presenting PR3 and MPO antigens to dendritic cells and activating autoreactive B cells.[61]
Neutrophils
Neutrophils are of great importance in the pathogenesis of AAV. The in vitro evidence for pathogenic roles of overactivated neutrophils and NETs has been discussed in the above section of “Pathogenicity of ANCA”. As for evidence from in vivo studies, Xiao et al. reported that depletion of neutrophils in mice can prevent MPO-ANCA-induced necrotizing crescentic nephritis.[62] Later, Schreiber et al.[63] immunized Mpo−/− mice with mouse MPO and exposed them to irradiation. Then, a transplant of Mpo+/+ bone marrow (BM) was performed. Engraftment of Mpo+/+ BM cells in MPO-ANCA-positive Mpo−/− mice led to pauci-immune NCGN. In contrast, transfer of Mpo−/− BM cells into irradiated Mpo+/+ mice did not result in vasculitis. Anti-MPO IgG was applied intravenously to irradiated Mpo−/− mice with Mpo+/+ BM transplantation or irradiated Mpo+/+ mice with Mpo−/− BM transplantation. Only chimeric Mpo−/− mice with circulating MPO-positive neutrophils (with Mpo+/+ BM transplantation) developed NCGN, thereby indicating that BM-derived cells, in particular neutrophils, are crucial for the induction of AAV.
Complements
In the past decade, the complement system has emerged to play an important role in the development of AAV, and C5a is considered to be a key neutrophil-activating chemokine.[64] Activation of neutrophils could further activate the complement system, in particular via the alternative pathway,[65] forming an amplification loop.
Evidence from Animal Studies
The pathogenetic role of complement activation was first verified by Xiao et al. in a mouse model of MPO-ANCA vasculitis.[65] In this model, wild-type mice with complement 3 (C3) depletion failed to develop vasculitis. Further investigation showed that factor-B-deficient and C5-deficient mice were completely protected from AAV, while disease development was comparable in C4-deficient and wild-type mice, suggesting that activation of the complement alternative pathway plays an important role in the pathogenesis of vasculitis.[65] In subsequent studies, C5a and C5a receptor (C5aR, also known as CD88) were proven to be the key players. Using the same mouse model, NCGN was markedly ameliorated or completely abolished by treating the mice with anti-C5 antibodies or C5aR-deficient BM transplants.[66,67] Knockout of C5aR ameliorated the disease in mice, whereas knockout of C5a-like receptor 2 (C5L2) exerted the opposite effect, suggesting that C5aR/CD88 enhances and C5L2 suppresses inflammation in AAV.[68] In contrast, C6 is not required for disease development in mice.[68] In summary, among the end-products of complement activation, C5a, but not C5b–9, has a crucial role in the pathogenesis of AAV.
Evidence from Human Studies
The results of human studies are consistent with those of animal studies. Chen et al. reported that patients with renal C3c deposition had more severe renal involvement.[69] C3d and factor B (marker of the alternative complement pathway) colocalized with C5b–9 in active glomerular lesions,[70] suggesting that alternative complement pathway activation is involved in human AAV. In AAV patients with renal involvement, low serum C3 levels are also associated with unfavorable outcomes.[71] Moreover, the levels of various complement activation products, in particular, Bb in urine or plasma, were higher in patients with active AAV than in patients in remission.[72]
Complement factor H (CFH) is a key regulator of the alternative complement pathway. Chen et al. found that circulating CFH levels in active AAV patients were lower than those in patients in remission and were associated with disease activity and renal outcome of AAV patients.[73] More importantly, biofunction of CFH was impaired in AAV patients.[74] One possible mechanism is that MPO binds to CFH, and the MPO-CFH interaction influences the regulatory activity of CFH.[75] In addition, modifications of CFH tyrosine residues could be detected in active AAV patients and might be relevant to the deficient activity of CFH in inhibiting neutrophil activation.[76] These findings indicate that regulators of the alternative complement pathway potentially contribute to the development of AAV.
Although all evidence for the role of the alternative complement pathway emanates from MPO-ANCA-mediated disease, notably in animal models, clinical trials suggest that this also applies to PR3-ANCA-mediated disease. In the phase III study (ADVOCATE trial)[77] involving patients with AAV, 331 patients were included, and 166 of them were assigned to receive avacopan (a small molecule inhibitor of C5aR). The first primary end point was remission at week 26 and no glucocorticoid use in the previous 4 weeks. The second primary end point was sustained remission at both weeks 26 and 52. Remission at week 26 (the first primary end point) was observed in 120 of 166 patients (72.3%) receiving avacopan and in 115 of 164 patients (70.1%) receiving prednisone (P < 0.001 for noninferiority, P = 0.24 for superiority). Sustained remission at week 52 (the second primary end point) was observed in 109 of 166 patients (65.7%) receiving avacopan and in 90 of 164 patients (54.9%) receiving prednisone (P < 0.001 for noninferiority, P = 0.007 for superiority). Among the 166 patients treated with avacopan, 94 (56.6%) were MPO-ANCA positive, and 72 (43.4%) were PR3-ANCA positive.
Evidence from in Vitro Studies
In vitro studies have demonstrated that C5a contributes to the process of neutrophil priming, which is crucial for ANCA-induced neutrophil activation.[64] Blocking C5aR could significantly downregulate neutrophil activation. In animal studies, C5L2 suppresses inflammation in AAV.[68] In contrast, Chen et al. found that the inflammatory response of C5a-primed neutrophils in vitro could be mitigated by blocking human C5L2.[78] Conclusions about the pathogenesis of AAV from animal models or in vitro studies must be tempered because they may not accurately replicate human disease. The role of C5L2 in AAV development needs further investigation.
Two key molecules, namely, high mobility group box 1 (HMGB1) and sphingosine-1-phosphate (S1P), are important downstream effector molecules in the C5a-neutrophil interaction. HMGB1 locates in the nucleus and acts as a proinflammatory mediator when released extracellularly.[79] HMGB1 increases the translocation of ANCA antigens, including MPO and PR3. During the C5a priming process, HMGB1 is released from the cytoplasm into the extracellular space and further enhances the effects of ANCA-induced neutrophil activation. In addition, HMGB1 can potentiate ANCA-induced NETs formation when interacting with RAGE, TLR2 and TLR4 in an NADPH oxidase-dependent manner.[80] The serum HMGB1 level correlates with the disease activity of AAV.[81,82] S1P is a bioactive sphingolipid metabolite released from neutrophils and can upregulate the expression of C5aR on the surface of neutrophils. S1P triggered by C5a-primed neutrophils could further activate neutrophils, forming a vicious cycle.[83] Blocking the S1P receptor will attenuate C5a-induced neutrophil activation by ANCA.[83]
Coagulation System
Patients with AAV are in a hypercoagulable state, with an increased risk of developing venous thromboembolic events.[84] In recent years, increasing evidence has suggested that coagulation system activation may also take part in AAV development. Certain serine proteases from the coagulation cascade, plasmin and thrombin in particular, can directly activate C3 and C5 independent of the traditional C3/C5 convertase.[85] C5a, in turn, can induce the expression of tissue factor (TF) in endothelial cells and neutrophils, thus triggering the extrinsic coagulation system.[86,87,88] This interaction between the coagulation and complement systems may contribute to the pathogenesis of glomerular capillary tuft infarction as well as the increase in thromboembolic events in AAV.
In addition, the role of complement and platelet interactions in the pathogenesis of AAV was evidenced by Chen's group.[89] In patients with active AAV, platelet counts and platelet-derived microparticles are usually elevated and correlate with disease activity.[90,91] Plasma from active AAV patients could activate platelets, and this could be inhibited by thrombin or protease-activated receptor (PAR) antagonists, indicating that this activation is mediated by the thrombin-PARs pathway.[89] These activated platelets could further amplify complement alternative pathway activation.[89]
B Cells and T Cells
B Cells
ANCA is a major participator in AAV development, and B cells contribute to damaging autoantibody production. B cells can also act as antigen-presenting cells and interact with T cells. Abnormal activation of B cells was observed in AAV patients and was associated with disease severity or poor outcomes.[92,93,94] Clinical trials have shown that depletion of B cells with rituximab is a useful treatment for AAV.[41,42] All evidence suggests that B cells have a vital role in the pathogenesis of AAV.
Loss of B-cell tolerance results in the emergence of autoreactive B cells and plasma cells. The cause of the loss of immune tolerance is not known. One possible explanation is the hypothetical theory of autoantigen complementarity.[23,51] ANCA formation entails an initial response to complementary peptides produced by the host or by the invading microorganism and induces anti-idiotypic antibodies of autoantigen epitopes. In addition, B-cell activating factor (BAFF, also known as BLyS) is of vital importance in B-cell homeostasis and interaction with activated T cells. Elevated BAFF concentrations correlate with ANCA titers and disease activity.[95,96] In AAV patients, activated neutrophils could release BAFF and improve the survival of B cells.[97] Overproduction of BAFF may be one of the causes for the presence of autoreactive B cells.
T Cells
T cells have been evidenced to be implicated in the pathogenic process. Such a role is suggested by the presence of CD4+ T cells in AAV lesions[98,99,100] and by some correlation of the levels of soluble markers of T-cell activation with disease activity.[100,101] Depletion of CD4+ T cells could significantly attenuate the disease severity of AAV in mice.[102] CD4+ T helper (TH) cells can be classified as TH1 or TH2 cells. TH1 cells exert cellular immune responses and phagocyte-dependent inflammation, while TH2 cells participate according to their cytokine profile and function. Different cytokine patterns are associated with different functions. In general, TH2 cells evoke B-cell antibody production (particularly of the IgE class) and eosinophil accumulation, but inhibit functions of phagocytic cells. It is suggested that a shift from the TH1 to TH2 response has a crucial role in the development and progression from localized to generalized GPA.[103] Regulatory T (Treg) cells in AAV patients seem defective in their ability to suppress the proliferation of effector cells in cytokine production.[104] The percentage of T cells secreting IL-17 (TH17) increases in the periphery in active AAV.[105] Moreover, the transcriptional characteristics of CD8+ T cells are associated with the frequency of recurrence in AAV patients.[106]
The cause of the loss of T-cell tolerance and disturbance of T-cell immune homeostasis is not well known. B cells can act as antigen-presenting cells to T cells; thus, the state of T cells in AAV might partially depend on B-cell function.[107] In addition, T cells responsive to cPR3 have been detected in PR3-ANCA vasculitis,[108] suggesting the role of complementary peptides in T-cell autoimmunity. TH1 cells, TH17 cells and CD8+ T cells could recognize ANCA antigens released by activated neutrophils and provide help to B cells for the production ANCA or granulomatous formation.[2,19]
In summary, priming of neutrophils by cytokines, including C5a, leads to upregulation of the expression of ANCA target antigen on the cell surface. The primed neutrophils are further activated by ANCA, resulting in respiratory burst and degranulation, as well as releasing of NETs, thus causing damage to vascular endothelial cells. Activated neutrophils could further activate the complement alternative pathway, resulting in the generation of C5a, which amplifies the inflammatory response by priming neutrophils for ANCA-mediated activation. TF-bearing microparticles are released by activated neutrophils as well, activating the coagulation system and producing thrombin. Thrombin causes activation of platelets, which in turn can further amplify the alternative complement pathway. Disturbed B-cell and T-cell immune homeostasis is also critically involved in disease development.
Conclusion
The development of AAV involves various factors. ANCA, neutrophils, and the complement system are key players that form a positive feedback loop and lead to the pathogenesis of AAV. In-depth exploration of the pathogenesis of AAV might help to offer more effective targeted therapies in the future.
Funding
This study is supported by National High Level Hospital Clinical Research Funding (Multi-center Clinical Research Project of Peking University First Hospital [grant number 2022CR52]), Capital's Funds for Health Improvement and Research [grant number 2020-2-4073], National Natural Science Fund [grant numbers 81870477, 81870478, 82000668], CAMS Innovation Fund for Medical Sciences (2019-I2M-5-046).
Author contributions
Sun XJ and Li ZY contributed the central idea. Sun XJ drafted the manuscript. Li ZY and Chen M contributed to the revisions.
Informed Consent
Not applicable.
Ethical Statement
Not applicable.
Conflict of Interest
The authors declare no conflict of interest.
References
[1] Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65:1–11.Search in Google Scholar
[2] Kitching AR, Anders HJ, Basu N, et al. ANCA-associated vasculitis. Nat Rev Dis Primers. 2020;6:71.Search in Google Scholar
[3] Pearce FA, Craven A, Merkel PA, et al. Global ethnic and geographic differences in the clinical presentations of anti-neutrophil cytoplasm antibody-associated vasculitis. Rheumatology (Oxford). 2017;56:1962–1969.Search in Google Scholar
[4] Li ZY, Chang DY, Zhao MH, et al. Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibody-associated vasculitis: a study of 439 cases in a single Chinese center. Arthritis Rheumatol. 2014;66:1920–1926.Search in Google Scholar
[5] Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. 2012;367:214–223.Search in Google Scholar
[6] Xie G, Roshandel D, Sherva R, et al. Association of granulomatosis with polyangiitis (Wegener's) with HLA-DPB1*04 and SEMA6A gene variants: evidence from genome-wide analysis. Arthritis Rheum. 2013;65:2457–2468.Search in Google Scholar
[7] Dahlqvist J, Ekman D, Sennblad B, et al. Identification and Functional Characterization of a Novel Susceptibility Locus for Small Vessel Vasculitis with MPO-ANCA. Rheumatology (Oxford). 2022;61:3461–3470.Search in Google Scholar
[8] Chen D, McInnis E, Wu E, et al. Immunological Interaction of HLA-DPB1 and Proteinase 3 in ANCA Vasculitis is Associated with Clinical Disease Activity. J Am Soc Nephrol. 2022;33:1517–1527.Search in Google Scholar
[9] Tsuchiya N, Kobayashi S, Hashimoto H, et al. Association of HLA-DRB1*0901-DQB1*0303 haplotype with microscopic polyangiitis in Japanese. Genes Immun. 2006;7:81–84.Search in Google Scholar
[10] Luo H, Chen M, Yang R, et al. The association of HLA-DRB1 alleles with antineutrophil cytoplasmic antibody-associated systemic vasculitis in Chinese patients. Hum Immunol. 2011;72:422–425.Search in Google Scholar
[11] Hiwa R, Ohmura K, Arase N, et al. Myeloperoxidase/HLA Class II Complexes Recognized by Autoantibodies in Microscopic Polyangiitis. Arthritis Rheumatol. 2017;69:2069–2080.Search in Google Scholar
[12] Cao Y, Yang J, Colby K, et al. High basal activity of the PTPN22 gain-of-function variant blunts leukocyte responsiveness negatively affecting IL-10 production in ANCA vasculitis. PLoS One. 2012;7:e42783.Search in Google Scholar
[13] Ciavatta DJ, Yang J, Preston GA, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. 2010;120:3209–3219.Search in Google Scholar
[14] Jones BE, Yang J, Muthigi A, et al. Gene-Specific DNA Methylation Changes Predict Remission in Patients with ANCA-Associated Vasculitis. J Am Soc Nephrol. 2017;28:1175–1187.Search in Google Scholar
[15] Gregorini G, Ferioli A, Donato F, et al. Association between silica exposure and necrotizing crescentic glomerulonephritis with p-ANCA and anti-MPO antibodies: a hospital-based case-control study. Adv Exp Med Biol. 1993;336:435–440.Search in Google Scholar
[16] Li J, Cui Z, Long JY, et al. The frequency of ANCA-associated vasculitis in a national database of hospitalized patients in China. Arthritis Res Ther. 2018;20:226.Search in Google Scholar
[17] Takeuchi Y, Saito A, Ojima Y, et al. The influence of the Great East Japan earthquake on microscopic polyangiitis: A retrospective observational study. PLoS One. 2017;12:e0177482.Search in Google Scholar
[18] Yashiro M, Muso E, Itoh T, et al. Significantly high incidence and high morbidity of acute renal failure with respiratory tract involvement of p-ANCA-related angitis revealed in Kobe city and the environs after the Kobe earthquake in 1995. Clin Nephrol. 1999;51:190–191.Search in Google Scholar
[19] Chen M, Kallenberg CG. ANCA-associated vasculitides--advances in pathogenesis and treatment. Nat Rev Rheumatol. 2010;6:653–664.Search in Google Scholar
[20] Stegeman CA, Tervaert JW, Sluiter WJ, et al. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med. 1994;120:12–17.Search in Google Scholar
[21] Salmela A, Rasmussen N, Tervaert JWC, et al. Chronic nasal Staphylococcus aureus carriage identifies a subset of newly diagnosed granulomatosis with polyangiitis patients with high relapse rate. Rheumatology (Oxford). 2017;56:965–972.Search in Google Scholar
[22] Glasner C, de Goffau MC, van Timmeren MM, et al. Genetic loci of Staphylococcus aureus associated with anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitides. Sci Rep. 2017;7:12211.Search in Google Scholar
[23] Pendergraft WF 3rd, Preston GA, Shah RR, et al. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004;10:72–79.Search in Google Scholar
[24] Huugen D, Xiao H, van Esch A, et al. Aggravation of anti-myeloperoxidase antibody-induced glomerulonephritis by bacterial lipopolysaccharide: role of tumor necrosis factor-alpha. Am J Pathol. 2005;167:47–58.Search in Google Scholar
[25] Kain R, Matsui K, Exner M, et al. A novel class of autoantigens of anti-neutrophil cytoplasmic antibodies in necrotizing and crescentic glomerulonephritis: the lysosomal membrane glycoprotein h-lamp-2 in neutrophil granulocytes and a related membrane protein in glomerular endothelial cells. J Exp Med. 1995;181:585–597.Search in Google Scholar
[26] Kain R, Exner M, Brandes R, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008;14:1088–1096.Search in Google Scholar
[27] Roth AJ, Brown MC, Smith RN, et al. Anti-LAMP-2 antibodies are not prevalent in patients with antineutrophil cytoplasmic autoantibody glomerulonephritis. J Am Soc Nephrol. 2012;23:545–555.Search in Google Scholar
[28] Scott J, Hartnett J, Mockler D, et al. Environmental risk factors associated with ANCA associated vasculitis: A systematic mapping review. Autoimmun Rev. 2020;19:102660.Search in Google Scholar
[29] Chen M, Gao Y, Guo XH, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nat Rev Nephrol. 2012;8:476–483.Search in Google Scholar
[30] Gao Y, Zhao MH, Guo XH, et al. The prevalence and target antigens of antithyroid drugs induced antineutrophil cytoplasmic antibodies (ANCA) in Chinese patients with hyperthyroidism. Endocr Res. 2004;30:205–213.Search in Google Scholar
[31] Zhao MH, Chen M, Gao Y, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2006;69:1477–1481.Search in Google Scholar
[32] Lee E, Hirouchi M, Hosokawa M, et al. Inactivation of peroxidases of rat bone marrow by repeated administration of propylthiouracil is accompanied by a change in the heme structure. Biochem Pharmacol. 1988;37:2151–2153.Search in Google Scholar
[33] Jiang X, Khursigara G, Rubin RL. Transformation of lupus-inducing drugs to cytotoxic products by activated neutrophils. Science. 1994;266:810–813.Search in Google Scholar
[34] von Schmiedeberg S, Goebel C, Gleichmann E, et al. Neutrophils and drug metabolism. Science. 1995;268:585–586.Search in Google Scholar
[35] Nakazawa D, Tomaru U, Suzuki A, et al. Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012;64:3779–3787.Search in Google Scholar
[36] Davies DJ, Moran JE, Niall JF, et al. Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? Br Med J (Clin Res Ed). 1982;285:606.Search in Google Scholar
[37] Jayne DR, Jones SJ, Severn A, et al. Severe pulmonary hemorrhage and systemic vasculitis in association with circulating anti-neutrophil cytoplasm antibodies of IgM class only. Clin Nephrol. 1989;32:101–106.Search in Google Scholar
[38] Kelley JM, Monach PA, Ji C, et al. IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci U S A. 2011;108:20736–20741.Search in Google Scholar
[39] Bansal PJ, Tobin MC. Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Ann Allergy Asthma Immunol. 2004;93:398–401.Search in Google Scholar
[40] Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180–2188.Search in Google Scholar
[41] Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211–220.Search in Google Scholar
[42] Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232.Search in Google Scholar
[43] Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–963.Search in Google Scholar
[44] Kuligowski MP, Kwan RY, Lo C, et al. Antimyeloperoxidase antibodies rapidly induce alpha-4-integrin-dependent glomerular neutrophil adhesion. Blood. 2009;113:6485–6494.Search in Google Scholar
[45] Little MA, Smyth CL, Yadav R, et al. Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood. 2005;106:2050–2058.Search in Google Scholar
[46] Jenne DE, Frohlich L, Hummel AM, et al. Cloning and functional expression of the murine homologue of proteinase 3: implications for the design of murine models of vasculitis. FEBS Lett. 1997;408:187–190.Search in Google Scholar
[47] Pfister H, Ollert M, Frohlich LF, et al. Antineutrophil cytoplasmic autoantibodies against the murine homolog of protein-ase 3 (Wegener autoantigen) are pathogenic in vivo. Blood. 2004;104:1411–1418.Search in Google Scholar
[48] Little MA, Al-Ani B, Ren S, et al. Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PLoS One. 2012;7:e28626.Search in Google Scholar
[49] Charles LA, Caldas ML, Falk RJ, et al. Antibodies against granule proteins activate neutrophils in vitro. J Leukoc Biol. 1991;50:539–546.Search in Google Scholar
[50] Porges AJ, Redecha PB, Kimberly WT, et al. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via Fc gamma RIIa. J Immunol. 1994;153:1271–1280.Search in Google Scholar
[51] Jennette JC, Nachman PH. ANCA Glomerulonephritis and Vasculitis. Clin J Am Soc Nephrol. 2017;12:1680–1691.Search in Google Scholar
[52] Baldus S, Eiserich JP, Mani A, et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J Clin Invest. 2001;108:1759–1770.Search in Google Scholar
[53] Yang JJ, Kettritz R, Falk RJ, et al. Apoptosis of endothelial cells induced by the neutrophil serine proteases proteinase 3 and elastase. Am J Pathol. 1996;149:1617–1626.Search in Google Scholar
[54] Ballieux BE, Hiemstra PS, Klar-Mohamad N, et al. Detachment and cytolysis of human endothelial cells by proteinase 3. Eur J Immunol. 1994;24:3211–3215.Search in Google Scholar
[55] Taekema-Roelvink MEJ, C VANK, Heemskerk E, et al. Proteinase 3 interacts with a 111-kD membrane molecule of human umbilical vein endothelial cells. J Am Soc Nephrol. 2000;11:640–648.Search in Google Scholar
[56] Soderberg D, Segelmark M. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis. Front Immunol. 2016;7:256.Search in Google Scholar
[57] Kessenbrock K, Krumbholz M, Schonermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–625.Search in Google Scholar
[58] Wang H, Wang C, Zhao MH, et al. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol. 2015;181:518–527.Search in Google Scholar
[59] Kettritz R, Jennette JC, Falk RJ. Crosslinking of ANCA-antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol. 1997;8:386–394.Search in Google Scholar
[60] Yang JJ, Preston GA, Alcorta DA, et al. Expression profile of leukocyte genes activated by anti-neutrophil cytoplasmic auto-antibodies (ANCA). Kidney Int. 2002;62:1638–1649.Search in Google Scholar
[61] Sangaletti S, Tripodo C, Chiodoni C, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood. 2012;120:3007–3018.Search in Google Scholar
[62] Xiao H, Heeringa P, Liu Z, et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167:39–45.Search in Google Scholar
[63] Schreiber A, Xiao H, Falk RJ, et al. Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol. 2006;17:3355–3364.Search in Google Scholar
[64] Chen M, Jayne DRW, Zhao MH. Complement in ANCA-associated vasculitis: mechanisms and implications for management. Nat Rev Nephrol. 2017;13:359–367.Search in Google Scholar
[65] Xiao H, Schreiber A, Heeringa P, et al. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64.Search in Google Scholar
[66] Huugen D, van Esch A, Xiao H, et al. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007;71:646–654.Search in Google Scholar
[67] Schreiber A, Xiao H, Jennette JC, et al. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–298.Search in Google Scholar
[68] Xiao H, Dairaghi DJ, Powers JP, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol. 2014;25:225–231.Search in Google Scholar
[69] Chen M, Xing GQ, Yu F, et al. Complement deposition in renal histopathology of patients with ANCA-associated pauci-immune glomerulonephritis. Nephrol Dial Transplant. 2009;24:1247–1252.Search in Google Scholar
[70] Xing GQ, Chen M, Liu G, et al. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009;29:282–291.Search in Google Scholar
[71] Augusto JF, Langs V, Demiselle J, et al. Low Serum Complement C3 Levels at Diagnosis of Renal ANCA-Associated Vasculitis Is Associated with Poor Prognosis. PLoS One. 2016;11:e0158871.Search in Google Scholar
[72] Gou SJ, Yuan J, Wang C, et al. Alternative complement pathway activation products in urine and kidneys of patients with ANCA-associated GN. Clin J Am Soc Nephrol. 2013;8:1884–1891.Search in Google Scholar
[73] Chen SF, Wang FM, Li ZY, et al. Plasma complement factor H is associated with disease activity of patients with ANCA-associated vasculitis. Arthritis Res Ther. 2015;17:129.Search in Google Scholar
[74] Chen SF, Wang FM, Li ZY, et al. The functional activities of complement factor H are impaired in patients with ANCA-positive vasculitis. Clin Immunol. 2017;175:41–50.Search in Google Scholar
[75] Chen SF, Wang FM, Li ZY, et al. Myeloperoxidase influences the complement regulatory activity of complement factor H. Rheumatology (Oxford). 2018;57:2213–2224.Search in Google Scholar
[76] Chen SF, Wang FM, Li ZY, et al. Complement Factor H Inhibits Anti-Neutrophil Cytoplasmic Autoantibody-Induced Neutrophil Activation by Interacting With Neutrophils. Front Immunol. 2018;9:559.Search in Google Scholar
[77] Jayne DRW, Merkel PA, Schall TJ, et al. Avacopan for the Treatment of ANCA-Associated Vasculitis. N Engl J Med. 2021;384:599–609.Search in Google Scholar
[78] Hao J, Wang C, Yuan J, et al. A pro-inflammatory role of C5L2 in C5a-primed neutrophils for ANCA-induced activation. PLoS One. 2013;8:e66305.Search in Google Scholar
[79] Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478.Search in Google Scholar
[80] Ma YH, Ma TT, Wang C, et al. High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res Ther. 2016;18:2.Search in Google Scholar
[81] Ma TT, Wang H, Wang C, et al. Urinary levels of high mobility group box-1 are associated with disease activity in antineutrophil cytoplasmic autoantibody-associated vasculitis. PLoS One. 2015;10:e0123586.Search in Google Scholar
[82] Wang C, Gou SJ, Chang DY, et al. Association of circulating level of high mobility group box 1 with disease activity in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Care Res (Hoboken). 2013;65:1828–1834.Search in Google Scholar
[83] Hao J, Huang YM, Zhao MH, et al. The interaction between C5a and sphingosine-1-phosphate in neutrophils for antineutrophil cytoplasmic antibody mediated activation. Arthritis Res Ther. 2014;16:R142.Search in Google Scholar
[84] Ma TT, Huang YM, Wang C, et al. Coagulation and fibrinolysis index profile in patients with ANCA-associated vasculitis. PLoS One. 2014;9:e97843.Search in Google Scholar
[85] Amara U, Flierl MA, Rittirsch D, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010;185:5628–5636.Search in Google Scholar
[86] Ritis K, Doumas M, Mastellos D, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177:4794–4802.Search in Google Scholar
[87] Kambas K, Chrysanthopoulou A, Vassilopoulos D, et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis. 2014;73:1854–1863.Search in Google Scholar
[88] Huang YM, Wang H, Wang C, et al. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol. 2015;67:2780–2790.Search in Google Scholar
[89] Miao D, Li DY, Chen M, et al. Platelets are activated in ANCA-associated vasculitis via thrombin-PARs pathway and can activate the alternative complement pathway. Arthritis Res Ther. 2017;19:252.Search in Google Scholar
[90] Willeke P, Kumpers P, Schluter B, et al. Platelet counts as a biomarker in ANCA-associated vasculitis. Scand J Rheumatol. 2015;44:302–308.Search in Google Scholar
[91] Miao D, Ma TT, Chen M, et al. Platelets release proinflammatory microparticles in anti-neutrophil cytoplasmic antibody-associated vasculitis. Rheumatology (Oxford). 2019:kez044. doi: 10.1093/rheumatology/kez044.Search in Google Scholar
[92] Wang C, Li ZY, Gong Y, et al. Increased frequency of IgD-CD27(hi)CD38(hi) B cells and its association with the renal involvement in ANCA-associated vasculitis. Arthritis Res Ther. 2022;24:109.Search in Google Scholar
[93] Merino-Vico A, van Hamburg JP, Tas SW. B Lineage Cells in ANCA-Associated Vasculitis. Int J Mol Sci. 2021;23:387.Search in Google Scholar
[94] Zabinska M, Koscielska-Kasprzak K, Krajewska J, et al. Immune Cells Profiling in ANCA-Associated Vasculitis Patients-Relation to Disease Activity. Cells. 2021;10:1773.Search in Google Scholar
[95] Steinmetz OM, Velden J, Kneissler U, et al. Analysis and classification of B-cell infiltrates in lupus and ANCA-associated nephritis. Kidney Int. 2008;74:448–457.Search in Google Scholar
[96] Nagai M, Hirayama K, Ebihara I, et al. Serum levels of BAFF and APRIL in myeloperoxidase anti-neutrophil cytoplasmic autoantibody-associated renal vasculitis: association with disease activity. Nephron Clin Pract. 2011;118:c339–c345.Search in Google Scholar
[97] Holden NJ, Williams JM, Morgan MD, et al. ANCA-stimulated neutrophils release BLyS and promote B cell survival: a clinically relevant cellular process. Ann Rheum Dis. 2011;70:2229–2233.Search in Google Scholar
[98] Komocsi A, Lamprecht P, Csernok E, et al. Peripheral blood and granuloma CD4(+)CD28(−) T cells are a major source of interferon-gamma and tumor necrosis factor-alpha in Wegener's granulomatosis. Am J Pathol. 2002;160:1717–1724.Search in Google Scholar
[99] Bolton WK, Innes DJ, Jr., Sturgill BC, et al. T-cells and macrophages in rapidly progressive glomerulonephritis: clinicopathologic correlations. Kidney Int. 1987;32:869–876.Search in Google Scholar
[100] Schmitt WH, Heesen C, Csernok E, et al. Elevated serum levels of soluble interleukin-2 receptor in patients with Wegener's granulomatosis. Association with disease activity. Arthritis Rheum. 1992;35:1088–1096.Search in Google Scholar
[101] Wang G, Hansen H, Tatsis E, et al. High plasma levels of the soluble form of CD30 activation molecule reflect disease activity in patients with Wegener's granulomatosis. Am J Med. 1997;102:517–523.Search in Google Scholar
[102] Ruth AJ, Kitching AR, Kwan RY, et al. Anti-neutrophil cytoplasmic antibodies and effector CD4+ cells play nonredundant roles in anti-myeloperoxidase crescentic glomerulonephritis. J Am Soc Nephrol. 2006;17:1940–1949.Search in Google Scholar
[103] Lamprecht P, Bruhl H, Erdmann A, et al. Differences in CCR5 expression on peripheral blood CD4+CD28− T-cells and in granulomatous lesions between localized and generalized Wegener's granulomatosis. Clin Immunol. 2003;108:1–7.Search in Google Scholar
[104] Free ME, Bunch DO, McGregor JA, et al. Patients with anti-neutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum. 2013;65:1922–1933.Search in Google Scholar
[105] Nogueira E, Hamour S, Sawant D, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant. 2010;25:2209–2217.Search in Google Scholar
[106] McKinney EF, Lyons PA, Carr EJ, et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat Med. 2010;16:586–591, 1p following 91.Search in Google Scholar
[107] Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001;1:147–153.Search in Google Scholar
[108] Yang J, Bautz DJ, Lionaki S, et al. ANCA patients have T cells responsive to complementary PR-3 antigen. Kidney Int. 2008;74(9):1159–1169.Search in Google Scholar
© 2023 Xiao-Jing Sun et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Editorial
- The challenge of ocular inflammation in systemic vasculitis: How to address inequalities of care?
- Review
- Animal models for large vessel vasculitis – The unmet need
- Pathogenesis of anti-neutrophil cytoplasmic antibody-associated vasculitis
- Original Article
- Clinical and vascular features of stroke in Takayasu's arteritis: A 24-year retrospective study
- Development and internal validation of a model to predict long-term survival of ANCA associated vasculitis
- Case Report
- Intermittent fever and cough in a 56-year-old patient: Relapsing polychondritis and extranodal NK/T-cell lymphoma
- Images
- Unusual presentation of a mass and “cobble-stone like” changes in the bronchus of a patient with granulomatosis with polyangitis
- Letter to the Editor
- COVID-19 vaccine-associated autoimmune disorders: Comments
Articles in the same Issue
- Editorial
- The challenge of ocular inflammation in systemic vasculitis: How to address inequalities of care?
- Review
- Animal models for large vessel vasculitis – The unmet need
- Pathogenesis of anti-neutrophil cytoplasmic antibody-associated vasculitis
- Original Article
- Clinical and vascular features of stroke in Takayasu's arteritis: A 24-year retrospective study
- Development and internal validation of a model to predict long-term survival of ANCA associated vasculitis
- Case Report
- Intermittent fever and cough in a 56-year-old patient: Relapsing polychondritis and extranodal NK/T-cell lymphoma
- Images
- Unusual presentation of a mass and “cobble-stone like” changes in the bronchus of a patient with granulomatosis with polyangitis
- Letter to the Editor
- COVID-19 vaccine-associated autoimmune disorders: Comments